

Abstract
In previous work, a modified version of the Bayesian information criterion (mBIC) was proposed to locate multiple interacting quantitative trait loci (QTL). Simulation studies and real data analysis demonstrate good properties of the mBIC in situations where the error distribution is approximately normal. However, as with other standard techniques of QTL mapping, the performance of the mBIC strongly deteriorates when the trait distribution is heavy tailed or when the data contain a significant proportion of outliers. In the present article, we propose a suitable robust version of the mBIC that is based on ranks. We investigate the properties of the resulting method on the basis of theoretical calculations, computer simulations, and a real data analysis. Our simulation results show that for the sample sizes typically used in QTL mapping, the methods based on ranks are almost as efficient as standard techniques when the data are normal and are much better when the data come from some heavy-tailed distribution or include a proportion of outliers.
A variety of statistical methods that can be applied to locate quantitative trait loci (QTL) exist. The classical methods like single-marker t-tests (Sax 1923) and interval mapping (Lander and Botstein 1989; Haley and Knott 1992) are based on a single-QTL model and may lead to biased estimators of the QTL size and location when the trait is influenced by more than one QTL. Composite-interval mapping (CIM) (Zeng 1993) and multiple-QTL mapping (MQM) (Jansen and Stam 1994) account for multiple QTL by including additional background markers into the model. Both these methods improve the precision of locating QTL with significant main effects but they do not allow detection of epistatic QTL, which influence the trait only by interacting with other genes. The most direct approach to locate multiple, interacting QTL relies on fitting a multiple-regression model, relating the trait values to marker genotypes. This approach was adopted, for example, by Kao et al. (1999), Carlborg et al. (2000), Carlborg and Andersson (2002), Kao and Zeng (2002), Yi and Xu (2002), Yi et al. (2003), Bogdan et al. (2004), Narita and Sasaki (2004), Yi et al. (2005), and Baierl et al. (2006). The most difficult part in constructing an appropriate regression model is the decision on the number of its components (i.e., the QTL number). This decision is particularly important in cases where the trait is influenced by some linked QTL. In such situations, the estimated QTL locations may depend substantially on the number of QTL in the model. When the size of the model is underestimated, for example, two linked QTL may be easily represented as one putative QTL in the middle between two real QTL locations. The opposite situation occurs when the size of the model is overestimated, leading to the incorrect identification of spurious QTL.
Since the size of the chosen model depends on the level of significance used for including or deleting its components, the choice of the corresponding threshold value may substantially influence the results of the analysis. The corresponding problem exists also in the framework of Bayesian statistics, where the final estimates of the QTL locations depend on the prior distribution of the QTL number. In the setting of classical statistics, a systematic approach for the comparison of different models is provided by model selection criteria. Different model selection criteria serve different purposes. If the purpose of the study is the choice of markers for marker-assisted selection, then one should consider the criteria aiming at minimizing the prediction error, like, e.g., the Akaike information criterion (Akaike 1974). Note that the prediction does not suffer much from including several markers closely linked to a QTL. However, in the case when the purpose of the study is to identify real QTL, then consistent criteria, like, e.g., the Bayesian information criterion (BIC) (Schwarz 1978), seem to be a better choice.
The classical model selection criteria were developed on the basis of asymptotic arguments and assuming that the sample size is large in comparison to the size of the analyzed models. This assumption is no longer satisfied in the large-scale genome scans for QTL, where the number of markers may be comparable to the sample size. In particular, Broman (1997) and Broman and Speed (2002) report for the QTL-mapping setting that the usually conservative BIC has a strong tendency to overestimate the QTL number. This problem has been further discussed in Bogdan et al. (2004), where an appropriate modified version of the BIC (mBIC) has been proposed. The mBIC has a strong connection to Bayesian statistics and allows one to incorporate prior knowledge on the QTL number and to compare posterior probabilities of different models. In the case when prior knowledge on the QTL number is not available, Bogdan et al. (2004) propose a standard version of the mBIC, based on a fixed prior distribution for the QTL number. As shown in the Appendix of Bogdan et al. (2004), the resulting penalty solves the multiple-testing problem and allows one to keep the type I error under control under a standard model setup. In the case when the outcome of the mBIC suggests that the fixed, relatively conservative prior proposed in Bogdan et al. (2004) is inadequate, Baierl et al. (2006) propose to repeat the procedure with the prior adjusted due to the results obtained in the initial search.
Bogdan et al. (2004) and Baierl et al. (2006) report the results of extensive simulation studies demonstrating good properties of the mBIC under a wide range of possible genetic scenarios and an ideal normal error distribution. In practice, however, the distribution of the trait is rarely normal. While, due to the central limit theorem, moderate deviations from normality do not have much influence on the properties of the mBIC, we expect the criterion to lose its good properties when the error distribution is heavy tailed or when the data contain some outliers. It is widely known that these types of violations of model assumptions have a strong influence on methods based on the comparison of means and so they will also have a large, deteriorating influence on all standard methods of QTL mapping.
The classical approach to reduce the influence of outlying observations on the results of regression is to use robust regression methods based on M-estimates (see, e.g., Jurečková and Sen 1996) or MM-estimates (see, e.g., Yohai 1985). In Baierl et al. (2007), robust versions of mBIC based on M-estimates were proposed. Simulation results reported in Baierl et al. (2007) show that these robust versions perform similarly to the standard mBIC when the error distribution is close to normal and have much better properties when the error distribution is heavy tailed or when the data contain some outliers. The methods based on M-estimates, however, require much more computational effort than least-squares regression. This is a disadvantage in the context of QTL mapping, where the verification of a large number of competing models is required.
An alternative solution to the problem of nonnormality of the error distribution is provided by nonparametric methods based on ranks. In the context of QTL mapping, this approach has been proposed and investigated, e.g., in Kruglyak and Lander (1995), Broman (2003), and Zou et al. (2003). A major advantage of rank-based statistics is that their distribution under the “null” hypothesis does not depend on the error distribution. Moreover, as demonstrated in Zou et al. (2003) (see also Lehmann 1975), the asymptotic efficiency of rank tests is only slightly smaller than that of the classical tests when the error distribution is normal and much higher when the error distribution is heavy tailed.
In this article, we use the idea of rank tests and propose a new version of the mBIC that is based on ranks instead of on the original trait values. For continuous error distributions and for the standard null model of no effects, we prove that the asymptotic distribution of the rank version of the mBIC is the same as the null distribution of the regular mBIC for normal errors.
Our simulation study demonstrates that this asymptotic approximation works very well already for sample sizes n ≥ 200. The results also indicate that the rank version of the mBIC performs similarly to the standard version when the error distribution is normal and much better in the case when it is heavy tailed or the data contain some proportion of outliers. A real data analysis points also at advantages of using the rank version of the mBIC.
METHODS
We start by reviewing the use of multiple regression in the case of locating QTL in a backcross population. Suppose that we observe values 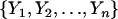 , of some quantitative trait for n individuals. Let Xij denote a variable that describes the genotype of the ith individual at marker j. In backcross populations, this variable would take one of only two values,
, of some quantitative trait for n individuals. Let Xij denote a variable that describes the genotype of the ith individual at marker j. In backcross populations, this variable would take one of only two values, 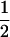 and
and 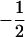 , depending on whether the individual is heterozygous or homozygous at locus j. By Nm we denote the number of available markers. To detect QTL, we look for neighboring markers, using a multiple-regression model
, depending on whether the individual is heterozygous or homozygous at locus j. By Nm we denote the number of available markers. To detect QTL, we look for neighboring markers, using a multiple-regression model
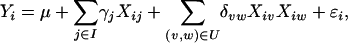 |
(1) |
where I is a certain subset of the set N = {1, … , Nm}, U is a certain subset of the Cartesian product N × N (N × N is the set of all pairs of elements of N), and ɛi is the error term. The third term in Equation 1, XivXiw, corresponds to pairwise interactions, the so-called epistatic effects. There are at most Ne = Nm(Nm − 1)/2 possible interactions.
As we do not know the QTL number or their locations, we use a model selection procedure to choose the best markers in model (1). One popular method for this purpose is the BIC proposed by Schwarz (1978). However, in the case of locating QTL, the BIC has a tendency to overestimate the QTL number (see, e.g., Broman and Speed 2002). Therefore, in Bogdan et al. (2004), a modified version of the BIC, called the mBIC, has been proposed. The mBIC allows us to take prior information on the number of QTL into account.
Let p and r denote the number of main and epistatic terms included in the regression model of the form (1). By E(P) and E(R), we denote the expected values of the corresponding prior distributions. With the mBIC, the model that minimizes the expression
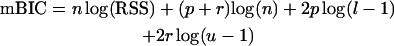 |
(2) |
is chosen. Here RSS denotes the residual sum of squares, l = Nm/E(P), and u = Ne/E(R). In the case of no prior information, Bogdan et al. (2004) suggest using
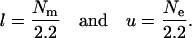 |
As shown in Bogdan et al. (2004), this choice takes the multiple-testing problem into account and guarantees that the overall type I error does not exceed 0.08 for a sample of size 200 and >30 markers when the error term is normal; i.e., ɛi ∼ N(0, σ2). Due to the consistency of the mBIC, the type I error decreases when the sample size increases.
The mBIC criterion has been designed under the assumption of normal errors. Therefore it works well, i.e., has a high power and is consistent, as long as the error term is close to normally distributed. However, we expect the properties of the mBIC to strongly deteriorate when the error terms come from a distribution with heavy tails or when the data contain a certain proportion of outliers. A typical solution in the situation where the data are not normal is to use a nonparametric method based on ranks; see, e.g., Kruglyak and Lander (1995) and Zou et al. (2003). In both articles, the authors demonstrated that the loss in efficiency of rank-based QTL mapping is very small compared to classical methods in the case when the error term comes from a normal distribution. On the other hand, a nonparametric method can be much more efficient when the errors come from a distribution with heavy tails. Motivated by these findings we define a new version of the mBIC based on ranks.
Rank-based model selection and the modified BIC:
When applying rank-based model selection, one exchanges the trait values by their ranks. A major advantage of using ranks is that the distribution of the test statistic under the null hypothesis of no QTL does not depend on the distribution of the error terms. Using ranks strongly reduces the influence of heavy tails and outlying observations.
Let 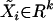 denote the row vector of regressors, i.e., of markers Xij and interactions XivXiw in the model (1) for the ith individual. The k-dimensional column vector of corresponding regression parameters γj and δvw is denoted by β. Consider a regression model
denote the row vector of regressors, i.e., of markers Xij and interactions XivXiw in the model (1) for the ith individual. The k-dimensional column vector of corresponding regression parameters γj and δvw is denoted by β. Consider a regression model 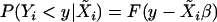 , where F is an unknown distribution. We want to test the hypothesis of no QTL evidence, H0: β = 0, on the basis of the sample consisting of n individuals. For this problem, Zou et al. (2003) used the Wilcoxon score statistic both in the simple- and in the multiple-regression case. Let
, where F is an unknown distribution. We want to test the hypothesis of no QTL evidence, H0: β = 0, on the basis of the sample consisting of n individuals. For this problem, Zou et al. (2003) used the Wilcoxon score statistic both in the simple- and in the multiple-regression case. Let 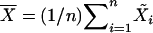 be a vector of regressor means, and let Ri denote the rank of the ith observation. The Wilcoxon score statistic takes the form
be a vector of regressor means, and let Ri denote the rank of the ith observation. The Wilcoxon score statistic takes the form 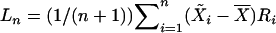 . Let
. Let 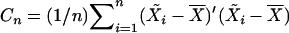 be the estimate of the covariance matrix of the regressor variables, and let
be the estimate of the covariance matrix of the regressor variables, and let 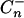 be its generalized inverse. The Ln-statistic has an important asymptotic property: when the covariance matrix between regressor variables is positive definite then under the null hypothesis of no
be its generalized inverse. The Ln-statistic has an important asymptotic property: when the covariance matrix between regressor variables is positive definite then under the null hypothesis of no 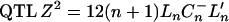 has asymptotically a chi-square distribution with k d.f. (see, e.g., Puri and Sen 1985). As is shown in the appendix, there is a connection between the Z2-statistic and the robust version of the BIC.
has asymptotically a chi-square distribution with k d.f. (see, e.g., Puri and Sen 1985). As is shown in the appendix, there is a connection between the Z2-statistic and the robust version of the BIC.
Our proposed construction of the robust (r)BIC, the rank version of the mBIC, is very simple. Let X denote a standard design matrix, with the ith row 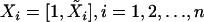 . After substituting the trait values by their ranks, we calculate the rank residual sum of squares,
. After substituting the trait values by their ranks, we calculate the rank residual sum of squares, 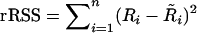 , where
, where 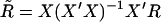 . By replacing RSS with rRSS in Equation 2, we obtain
. By replacing RSS with rRSS in Equation 2, we obtain
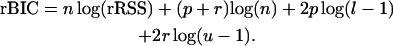 |
(3) |
Remark 1. Lemma 1 below shows that independent of the distribution of the error term, the asymptotic null distribution of rBIC is the same as the distribution of mBIC when the error term is normal. Therefore to keep the type I error at the desired level, the penalty coefficients l and u are chosen in the same way as for mBIC [see (2)].
For the previously defined model we have the following proposition.
Proposition 1. If the trait distribution F is continuous, rBIC is distribution free under the null hypothesis H0: β = 0.
The result follows from the fact that the distribution of the vector of ranks R1, … , Rn is independent of the distribution F under the hypothesis H0: β = 0, if F is continuous (see, e.g., Lehmann 1975 or Hájek et al. 1999). As the rBIC depends on the observations only through their ranks, it is distribution free.
By simulations and theoretical calculations, Bogdan et al. (2004) and Baierl et al. (2006) showed that the mBIC criterion controls the type I error when the error terms come from a normal distribution. Let us denote by RSSk the residual sum of squares related to a given model with k regressors, and let RSS0 be the residual sum of squares for the null model with no QTL. Note that the mBIC prefers a model with k regressors over the null model if 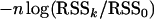 is greater than the penalty specified on the right-hand side of Equation 2. Note also that under the null hypothesis of no QTL
is greater than the penalty specified on the right-hand side of Equation 2. Note also that under the null hypothesis of no QTL 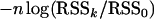 has asymptotically a chi-square distribution with k d.f. In the following lemma, we show that the asymptotic null distribution does not change when the trait values are replaced by their ranks. The result does not depend on the distribution of the error term and suggests that the type I error of the rBIC will be close to that of the mBIC.
has asymptotically a chi-square distribution with k d.f. In the following lemma, we show that the asymptotic null distribution does not change when the trait values are replaced by their ranks. The result does not depend on the distribution of the error term and suggests that the type I error of the rBIC will be close to that of the mBIC.
Lemma 1. Consider a regression model 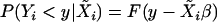 , where F is continuous and
, where F is continuous and 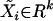 is the vector of regressors. Let rRSSk denote the corresponding rank residual sum of squares and let rRSS0 denote the rank residual sum of squares under the null model of no QTL. If the matrix of covariances between regressor variables is positive definite, then the expression
is the vector of regressors. Let rRSSk denote the corresponding rank residual sum of squares and let rRSS0 denote the rank residual sum of squares under the null model of no QTL. If the matrix of covariances between regressor variables is positive definite, then the expression
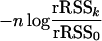 |
has asymptotically a χ2(k) distribution under the null hypothesis H0: β = 0.
A proof is given in the appendix.
Remark 2. In the case of QTL mapping, the covariance matrix between regressor variables is positive definite, if the recombination fraction between any pair of markers is larger than zero.
Remark 3. In the case of a one-QTL model, there is a close relationship between 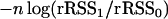 and the Wilcoxon rank statistics Ws, namely
and the Wilcoxon rank statistics Ws, namely
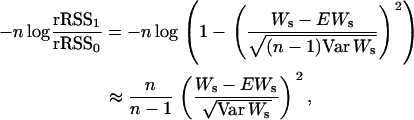 |
where EWs and Var Ws are the mean and the variance of Ws. In this case, Lemma 1 confirms a well-known result on the asymptotic normality of Ws.
If F is discrete, ties usually arise among the observed values. A discrete F may be due to a genuinely discrete quantitative trait or due to a limited measurement precision. For tied (equal) observations ranks are not well defined, and therefore some modifications are necessary to apply a rank-based statistic. One of the methods to handle such a situation is to use midranks. Let d1, … , de be the number of observations tied at the smallest value, the second smallest, and so on. The corresponding ranks are 1, 2, … , d1 in the first group, d1 + 1, d1 + 2, … , d1 + d2 in the second, etc. To each observation in group t, we assign the average of the dt ranks in this group. These averages are called midranks. Conditional on the quantities e, d1, d2, … , de, the distribution of the midranks is independent of F (Lehmann 1975). Hence rank statistics and in particular the rBIC will still be conditionally distribution free, if we use midranks. Thus the performance should again not depend on the error-term distribution.
In the next section, we investigate the performance of the rBIC for a model with additive and epistatic terms under different error distributions.
SIMULATIONS
To investigate the performance of the rBIC, we performed computer simulations. To simulate the distribution of marker genotypes, we used the Haldane model with no interference.
We consider two setups. Setup 1 involves two chromosomes each of length 100 cM with markers equally spaced every 10 cM. In this setting, we consider both the null model involving no effects and a three-QTL model involving one main (chromosome 1 at 20 cM, effect size 0.55) and one epistatic effect (chromosome 2 at 20 and at 70 cM, effect size 1.2). Setup 1 has also been considered in Baierl et al. (2007).
In the second setup, we consider three chromosomes each of length 100 cM with seven, eight, and seven markers, respectively, distributed randomly across the chromosome. The distances between the markers range from 1 to 29 cM with a mean distance of 15.79 cM. To narrow these intervals and to enable a location of QTL at a finer scale, we used regression interval mapping according to Haley and Knott (1992). This method relies on imputing putative QTL between markers and replacing their missing genotypes by expected values, calculated on the basis of neighboring markers. Using this approach, we imputed additional marker genotypes to reduce the intervals between adjacent markers to a maximum of 10 cM. The second setup is considered under the null model of no effects and an alternative model involving three main and three epistatic effects. The locations and sizes of the main QTL effects are as follows: QTL1 is on chromosome 1 at 20 cM with γ1 = 0.8, QTL2 is on chromosome 2 at 20 cM with γ2 = 0.7, and QTL3 is on chromosome 3 at 1 cM with γ3 = 0.6. The epistatic effects are specified as follows: interaction 1 involves QTL1 and QTL3 with δ1 = 1.6, interaction 2 involves QTL2 and a new QTL on chromosome 3 at 75 cM with δ2 = 1.4, and interaction 3 involves two new QTL, both on chromosome 1 at 27 and 60 cM, respectively, with δ3 = 1.2. The locations of the QTL, markers, and imputed positions are shown in Figure 1.
Figure 1.—

Summary of simulation setup 2. The marker locations and imputed positions are indicated by long and short vertical bars, respectively. QTL are indicated by ×'s. The exact positions and effect sizes are given in the Simulations section.
Setup 1 involves backcross populations of size 200, whereas in setup 2 population sizes of both 200 and 500 are considered. Both the mBIC and the rBIC criterion are applied in the standard form with l = Nm/2.2 and u = Ne/2.2. To solve the problem of searching over a large class of possible models, we follow Bogdan et al. (2004) and use a forward selection. At every step of this procedure a new regression model is built by adding an explanatory variable that leads to the largest decrease in RSS. The forward selection strategy is terminated after 30 steps, resulting in 31 regression models. Then we use mBIC or rBIC to chose the “best” of these models.
The simulation results are based on 3000 replications.
To investigate the robustness of our proposed criterion, we consider five different error term distributions, which are defined according to Baierl et al. (2007):
Normal: 1.11 × N(0, 1).
Laplace: 1.08 × Laplace(0, 1).
Cauchy: Cauchy(0, 0.75).
Tukey's gross error model: 1.081 × Tukey(0.95, 100, 1).
χ2 with 6 d.f. centered around the mean: 0.342 × (
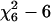 ).
).
In Tukey's gross error model, the error distribution is a mixture of two normal distributions leading to a certain percentage of outliers. More specifically, Tukey(α, τ, σ) = λ × N(0, σ2) + (1 − λ) × N(0, τ × σ2), where λ ∼ Binomial(1, α).
A main effect is assumed to be correctly identified, if at least one of the chosen markers is within 15 cM of the true QTL. Every additionally selected marker within this range is counted as a false positive. An epistatic effect is assumed to be correctly identified, if both markers of the chosen interaction term are within 15 cM of the respective QTL.
The application of a ±15-cM detection window is motivated by the fact that for n = 200, the standard deviation of the estimates of QTL location is close to 10 cM, if their magnitudes are similar to our simulated effect sizes. In the case of the Cauchy distribution, the standard error of QTL localization reaches even 15 cM and in this case a 15-cM detection window is somewhat restrictive, leading to an underestimation of the power and an overestimation of the false discovery rate. The observed errors related to QTL location are inherent to any QTL-mapping procedure in a backcross population. They result from a strong correlation between neighboring markers and from propagating the QTL signal over all linked markers. A discussion of this phenomenon in the case of standard interval mapping can be found in Bogdan and Doerge (2005).
The false discovery rate (FDR) (Benjamini and Hochberg 1995) is estimated as
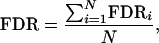 |
where N is the number of replications of the simulation experiment and
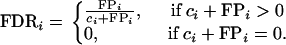 |
Here ci stands for the number of correctly identified terms, both main and epistatic, at replication i, and FPi is the number of false positives that appear at replication i. Under the null model, the false discovery rate is equivalent to the multiple type I (or familywise) error of detecting at least one incorrect effect.
RESULTS AND DISCUSSION
For setup 1, the type I errors under the null model of no effect are summarized in Table 1. The differences between the results for the mBIC and the rBIC depend on the error term distribution and are small in most cases (Cauchy error is an exception). According to Proposition 1, the distribution of the rBIC under the null hypothesis does not depend on the error distribution and the slight differences in type I error observed for different error distributions are due to random simulation errors.
TABLE 1.
Type I errors under the null model (no QTL) for setup 1
| Error distribution
|
|||||
|---|---|---|---|---|---|
| Normal | Laplace | Cauchy | Tukey | Chi2 | |
| mBIC | 0.052 | 0.050 | 0.196 | 0.070 | 0.051 |
| rBIC | 0.052 | 0.058 | 0.045 | 0.048 | 0.050 |
In the context of setup 1, we compare the power and FDR of our proposed rank-based method to the M-estimates investigated by Baierl et al. (2007) as well as to the classical BIC. In regression, M-estimates are obtained by minimizing more general measures of distance instead of the residual sum of squares. In Baierl et al. (2007), the following three contrast functions have been considered as a measure of distance: Huber's, the Bisquare, and Hampel's contrast function. Their simulations indicate that the use of the above-mentioned robust contrast functions leads to much better results than those obtained by least-squares regression in cases when the error terms come from a heavy-tailed distribution. In the normal case, both methods work comparably.
The results for the first setup in the case of two effects are presented in Figures 2 (average percentage of correctly identified effects) and 3 (FDR). (Exact numbers can be found in Table 2.) In the case of nonnormal distributions, the percentage of correctly identified effects for the rank method is comparable to the best values obtained by M-estimates. None of the M-estimators performs significantly better for every type of error term. In Figure 3 on the other hand, we observe that our rank method leads to slightly higher FDRs. Overall, the rank method works well compared to other robust methods of QTL detection. In addition, the method is very simple to use and computationally less demanding than M-estimates.
Figure 2.—

Percentage of correctly identified main and epistatic effects taken from Baierl et al. (2007) (shaded bars) and for the rank-based method (horizontal solid lines).
Figure 3.—

False discovery rates from Baierl et al. (2007) (shaded bars) and for the rank-based method (horizontal solid lines).
TABLE 2.
Results for setup 1 (two-effects model)
| mBIC
|
rBIC
|
|||
|---|---|---|---|---|
| Error | FDR | % corr | FDR | % corr |
| 1. Normal | 0.149 | 0.469 | 0.146 | 0.448 |
| 2. Laplace | 0.120 | 0.209 | 0.137 | 0.329 |
| 3. Cauchy | 0.189 | 0.036 | 0.142 | 0.222 |
| 4. Tukey | 0.087 | 0.067 | 0.152 | 0.368 |
| 5. χ2 | 0.142 | 0.403 | 0.156 | 0.508 |
% corr, percentage of correctly identified effects.
Note that the original BIC criterion leads to a considerably higher percentage of correct identification but also (see Figure 3) to extremely high false discovery rates.
Next we consider the second setup, which is more realistic in terms of the marker distribution. Our simulations indicate that the type I error is smaller in most cases for the rBIC than for the mBIC (see Table 3). The largest differences are observed for the Cauchy and Tukey error distributions.
TABLE 3.
Type I errors under the null model (no QTL) for setup 2
| Error distribution
|
||||||
|---|---|---|---|---|---|---|
| Normal | Laplace | Cauchy | Tukey | χ2 | ||
| 200 | mBIC | 0.031 | 0.035 | 0.099 | 0.054 | 0.033 |
| 200 | rBIC | 0.033 | 0.034 | 0.037 | 0.029 | 0.031 |
| 500 | mBIC | 0.019 | 0.016 | 0.088 | 0.028 | 0.015 |
| 500 | rBIC | 0.020 | 0.022 | 0.023 | 0.017 | 0.014 |
For the six-effect model in setup 2 and a 15-cM identification window, the FDR for the rBIC ranges from 12 to 14% for n = 200 and from 3 to 9% for n = 500 (see Table 4). The relatively large FDR values for n = 200 are caused by a large standard deviation of the estimates of QTL location. Our additional simulations demonstrated that this standard error reaches 15 cM for our simulated QTL and both sample sizes when the error term is Cauchy distributed. Thus a significant proportion of “false positives” is due to correctly identified but imprecisely localized QTL. When applying a more liberal ±30-cM detection window we recorded the FDR at a level of 3–8% for n = 200 and at a level of 0.5–1.5% for n = 500.
TABLE 4.
Results for setup 2 (six-effect model)
|
n = 200
|
n = 500
|
|||||||
|---|---|---|---|---|---|---|---|---|
| mBIC
|
rBIC
|
mBIC
|
rBIC
|
|||||
| Error | FDR | % corr | FDR | % corr | FDR | % corr | FDR | % corr |
| 1. Normal | 0.122 | 0.407 | 0.124 | 0.370 | 0.034 | 0.875 | 0.036 | 0.856 |
| 2. Laplace | 0.139 | 0.179 | 0.137 | 0.232 | 0.067 | 0.630 | 0.054 | 0.723 |
| 3. Cauchy | 0.095 | 0.008 | 0.128 | 0.136 | 0.080 | 0.007 | 0.087 | 0.508 |
| 4. Tukey | 0.102 | 0.051 | 0.137 | 0.292 | 0.118 | 0.165 | 0.044 | 0.789 |
| 5. χ2 | 0.138 | 0.354 | 0.129 | 0.382 | 0.040 | 0.841 | 0.034 | 0.865 |
% corr, percentage of correctly identified effects.
Results included in Table 4 demonstrate that the FDR for the rBIC is comparable to or smaller than the FDR for the standard mBIC independently of the error distribution. We also observe that the rBIC is slightly less efficient than the mBIC, if the error distribution is normal. The corresponding loss of power is equal to 4 percentage points for n = 200 (from 41 to 37%) and to 2 percentage points for n = 500 (from 88 to 86%). For all other investigated error distributions, the rBIC has a larger power than the mBIC. A particularly large difference occurs for the Tukey distribution, where the power of the rBIC is 79% compared to 16% for the mBIC when n = 500. For the Cauchy distribution the mBIC completely fails (the power is <1%), whereas the power of the rBIC for n = 500 is 51%. Note that both the Tukey and the Cauchy distribution lead to a certain proportion of outliers.
Overall the results confirm that the rank-based method works comparably as well as the mBIC for normal errors, but much better when outliers are present.
Real data analysis:
To verify the performance of the rBIC in the case of real data, we reanalyze a data set from Mähler et al. (2002). The data concern colitis susceptibility strains that carry a deficient IL-10 gene that is important in limiting the immune response against intestinal antigens. We consider their data from a backcross to the less susceptible B6 strain. We analyze two quantitative traits, MidPC1 and CecumPC1, which are the first principal components of four scores measuring the severity and type of lesions on middle colon and cecum, respectively. As demonstrated in Figures 4 and 5, the distribution of the trait strongly deviates from the normal distribution in both cases.
Figure 4.—

Distribution of MidPC1 in the set of 203 individuals from the backcross B6 population.
Figure 5.—

Distribution of CecumPC1 in the set of 203 individuals from the backcross B6 population.
The data set contains 203 individuals and 12 markers on 9 chromosomes that were selected from a preliminary genome scan on 40 individuals and 67 markers spread across all 20 chromosomes.
Mähler et al. (2002) report one main effect on chromosome 12 for the trait MidPC1 (D12Mit214) and one suggestive QTL for CecumPC1 on chromosome 13. They do not detect any interactions.
Due to missing trait or genotype information, we removed 16 (CecumPC1) and 15 observations (MidPC1) from the data set before applying our method. In addition, we excluded marker D17Mit88, which had missing genotypes for 62 individuals. Imputation of missing genotype data was not feasible because of the low marker density.
The considered traits are summaries of discrete measures (scores). Of 187 observations for CecumPC1, there are 32 different trait values, 45% of the observations fall within 1 of 4 most frequent values, and the most numerous group contains 13% of the observations. There are also only 19 different values for MidPC1. Among the 188 observations of this trait, 42% are equal to the most frequent value and 11% to the second-most frequent. To derive ranks for individuals with identical trait values, midranks as discussed in methods were calculated.
Since we do not have any prior information, we use the standard versions of the mBIC and the rBIC with l = Nm/2.2 and u = Ne/2.2.
Applying both the mBIC and the rBIC to the MidPC1 data set, we find two effects, one main and one epistatic. The main effect found by our approach is the same as in Mähler et al. (2002). However, we also detected an epistatic effect between markers on chromosomes 4 and 7 that considerably improves the fit of the model to the data. The fraction of the variance explained by the model, the R2, increases from 0.0768 for the one-effect model to 0.1397 for the model that also includes the interaction term.
For the second trait, CecumPC1, the mBIC does not find any effects. When using the rBIC on the other hand, we get one main effect on chromosome 5 (D5Mit205). This effect is different from the one that was suggested by Mähler et al. (2002). This difference is explained in Figure 6, which shows the relationship between regular t-test statistics and Wilcoxon rank statistics, for each of the considered 11 markers. The plot demonstrates that values of the Wilcoxon statistic are strongly correlated with values of the t-statistic. However, the ranking of markers according to the Wilcoxon statistic differs from the ranking due to t-test results. In particular marker 9, identified by Mähler et al. (2002) and having the largest absolute value of the t-statistic, has a smaller value of the Wilcoxon rank statistic than marker 3, identified by rBIC. The respective P-values of the Wilcoxon test are 0.0089 for marker 9 and 0.0026 for marker 3, which supports the choice of marker 3. For the t-test, the P-values are 0.0061 for marker 9 and 0.0066 for marker 3. After correcting for multiple testing, none of the t-statistics were significant and none of these effects were detected by the regular mBIC criterion. The marker D5Mit205 (marker 3) was also detected by the robust version of mBIC proposed in Baierl et al. (2007), which additionally detects effect no. 9.
Figure 6.—

Absolute values of the t-test statistics vs. absolute values of the Wilcoxon statistics, for the 11 markers used in the analysis of the CecumPC1.
Summary:
We defined a new version of the mBIC based on ranks. In our approach, we followed Kruglyak and Lander (1995) and Zou et al. (2003), who proposed rank-based methods for QTL mapping and demonstrated their good properties.
Our results show that the rank version of the mBIC performs very well, at least when n ≥ 200. This phenomenon is in accordance with asymptotic limit theorems on the distribution of rank statistics, given, for example, in Puri and Sen (1985) or Hájek et al. (1999). The classical central limit theorem can be used to explain the relatively good performance of the original mBIC under the chi-square or the Laplace error distribution. However, in the case when the error distribution is heavy tailed or when the data contain some proportion of outliers, the rank version of the mBIC performs much better than the standard version. Our results also suggest that the rank version of the mBIC performs comparably to robust versions of this criterion based on M-estimates but is much easier to handle computationally.
The main purpose of our simulations was the comparison of standard and rank-based (robust) regression. The simulations were performed under an ideal situation of no missing genotype data or genotype errors. However, the simulations for setup 2, which used Haley and Knott (1992) interval mapping to fill the genotypes of putative QTL, demonstrate that the rBIC can perform very well when some missing genotype data are replaced with their expected values. Some additional simulations demonstrating a good performance of the mBIC under missing genotype data can be found in Baierl et al. (2006). We also expect that the main conclusions resulting from the comparison of the standard and rank-based regression would hold in the case of the genotyping errors, since both these methods of data analysis would be similarly affected. The nonnormal error distribution causes additional problems for standard methods of QTL mapping, which can be solved by applying rank-based regression.
To obtain best results the mBIC and the rBIC should be used with an all-subsets model selection. The application of forward selection in our simulation study was motivated by the computational constraints related to the large number of replications needed to estimate the power and FDR of our procedure. According to the results reported in Broman (1997) and Broman and Speed (2002) and our experience, forward selection performs relatively well in the context of QTL mapping, even though it has a slight tendency to include some extraneous markers. We believe that the estimates of power obtained from our simulation study are good indicators of the performance of the rBIC under an all-subsets model selection, while the FDR might be slightly overestimated.
This article is focused on backcross populations and the standard version of the mBIC and the rBIC. However, we expect to see similar patterns when the methods are used for different experimental designs (see, e.g., Baierl et al. 2006) or when prior knowledge is used to modify the penalty.
The methods described in this article have been implemented in Matlab and are available at http://www.im.pwr.wroc.pl/∼mzak/rBIC.
Acknowledgments
We thank Paweł Koteja for helpful discussions and two anonymous referees for helpful suggestions.
APPENDIX: PROOF OF LEMMA 1
We show that when the covariance matrix of regressor variables is positive definite then under the null hypothesis of no QTL, H0: β = 0, the statistic
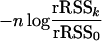 |
(A1) |
has an asymptotic χ2(k) distribution. For the multiple-regression model, we have that
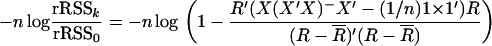 |
We compare the right-hand side expression under the logarithm with the statistic
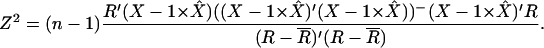 |
Here 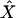 denotes the 1 × (k + 1) vector of column averages. According to Puri and Sen (1985) Z2 has an asymptotic chi-square distribution with k d.f.
denotes the 1 × (k + 1) vector of column averages. According to Puri and Sen (1985) Z2 has an asymptotic chi-square distribution with k d.f.
To compare (A1) with Z2 we at first show that
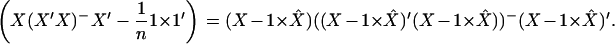 |
Multiplying both sides by X′ from the left and by X from the right and using the properties of the generalized inverse matrix we get
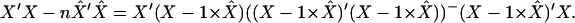 |
(A2) |
On the right-hand side, we now substitute the first and last X by 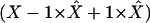 .
.
It is easy to check that the expression becomes 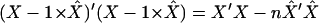 after these transformations, which is exactly the left side of (A2). Thus
after these transformations, which is exactly the left side of (A2). Thus
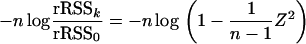 |
As n tends to infinity, the expression 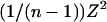 tends to 0, and we can use the approximation log(1 + x) ≈ x to obtain
tends to 0, and we can use the approximation log(1 + x) ≈ x to obtain
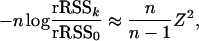 |
which is asymptotically χ2(k) distributed.
References
- Akaike, H., 1974. A new look at the statistical model identification. IEEE Trans. Automat. Control 19: 716–723. [Google Scholar]
- Baierl, A., M. Bogdan, F. Frommlet and A. Futschik, 2006. On locating multiple interacting quantitative trait loci in intercross designs. Genetics 173: 1693–1703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baierl, A., A. Futschik, M. Bogdan and P. Biecek, 2007. Locating multiple interacting quantitative trait loci using robust model selection. Comput. Stat. Data Anal. (http://www.sciencedirect.com/science/journal/01679473) (in press). [DOI] [PMC free article] [PubMed]
- Benjamini, Y., and Y. Hochberg, 1995. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. R. Stat. Soc. B 57: 289–300. [Google Scholar]
- Bogdan, M., and R. W. Doerge, 2005. Biased estimators of quantitative trait locus heritability and location in interval mapping. Heredity 95: 476–484. [DOI] [PubMed] [Google Scholar]
- Bogdan, M., J. K. Ghosh and R. W. Doerge, 2004. Modifying the Schwarz Bayesian information criterion to locate multiple interacting quantitative trait loci. Genetics 167: 989–999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broman, K. W., 1997. Identifying quantitative trait loci in experimental crosses. Ph.D. Dissertation, University of California, Berkeley, CA.
- Broman, K. W., 2003. Mapping quantitative trait loci in the case of a spike in the phenotype distribution. Genetics 163: 1169–1175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broman, K. W., and T. P. Speed, 2002. A model selection approach for the identification of quantitative trait loci in experimental crosses. J. R. Stat. Soc. B 64: 641–656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlborg, Ö., and L. Andersson, 2002. The use of randomisation testing for detection of multiple epistatic QTL. Genet. Res. 79: 175–184. [DOI] [PubMed] [Google Scholar]
- Carlborg, Ö., L. Andersson and B. Kinghorn, 2000. The use of a genetic algorithm for simultaneous mapping of multiple interacting quantitative trait loci. Genetics 155: 2003–2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hájek, J., Z. Šidák and P. K. Sen, 1999. Theory of Rank Tests. Academic Press, New York/London/San Diego.
- Haley, C. S., and S. A. Knott, 1992. A simple regression method for mapping quantitative traits in line crosses using flanking markers. Heredity 69: 315–324. [DOI] [PubMed] [Google Scholar]
- Jansen, R. C., and P. Stam, 1994. High resolution of quantitative traits into multiple loci via interval mapping. Genetics 136: 1447–1455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jurečková, J., and P. K Sen, 1996. Robust Statistical Procedures: Asymptotics and Interrelations. Wiley, New York.
- Kao, C. H., and Z-B. Zeng, 2002. Modeling epistasis of quantitative trait loci using Cockerham's model. Genetics 160: 1243–1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kao, C. H., Z-B. Zeng and R. D. Teasdale, 1999. Multiple interval mapping for quantitative trait loci. Genetics 152: 1203–1216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kruglyak, L., and E. S. Lander, 1995. A nonparametric approach for mapping quantitative trait loci. Genetics 139: 1421–1428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lander, E. S., and D. Botstein, 1989. Mapping Mendelian factors underlying quantitative traits using RFLP linkage maps. Genetics 121: 185–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehmann, E. L., 1975. Nonparametrics: Statistical Methods Based on Ranks. Holden-Day, San Francisco.
- Mähler, M., C. Most, S. Schmidtke, J. P. Sunberg, R. Li et al., 2002. Genetics of colitis susceptibility in IL-10-deficient mice: backcross versus F2 result contrasted by principal component analysis. Genomics 80(3): 274–282. [DOI] [PubMed] [Google Scholar]
- Narita, A., and Y. Sasaki, 2004. Detection of multiple QTL with epistatic effects under a mixed inheritance model in an outbred population. Genet. Sel. Evol. 36: 415–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puri, M. L., and P. K. Sen, 1985. Nonparametric Methods in General Linear Models. Wiley, New York.
- Sax, K., 1923. The association of size differences with seed-coat pattern and pigmentation in Phaseolus vulgaris. Genetics 8: 552–560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarz, G., 1978. Estimating the dimension of a model. Ann. Stat. 6: 461–464. [Google Scholar]
- Yi, N., and S. Xu, 2002. Mapping quantitative trait loci with epistatic effects. Genet. Res. 79: 185–198. [DOI] [PubMed] [Google Scholar]
- Yi, N., S. Xu and D. B. Allison, 2003. Bayesian model choice and search strategies for mapping interacting quantitative trait loci. Genetics 165: 867–883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi, N., B. S. Yandell, G. A. Churchill, D. B. Allison, E. J. Eisen et al., 2005. Bayesian model selection for genome-wide epistatic QTL analysis. Genetics 170: 1333–1344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yohai, V. J., 1985. High breakdown-point and high efficiency robust estimates for regression. Ann. Stat. 15: 642–656. [Google Scholar]
- Zeng, Z.-B., 1993. Theoretical basis of separation of multiple linked gene effects on mapping quantitative trait loci. Proc. Natl. Acad. Sci. USA 90: 10972–10976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou, F., B. S. Yandell and J. P. Fine, 2003. Rank based statistical methodologies for QTL mapping. Genetics 165: 1599–1605. [DOI] [PMC free article] [PubMed] [Google Scholar]