

Abstract
Ageing is a complex process that negatively impacts the development of the immune system and its ability to function. The mechanisms that underlie these age-related defects are broad and range from defects in the haematopoietic bone marrow to defects in peripheral lymphocyte migration, maturation and function. The thymus is a central lymphoid organ responsible for production of naïve T cells, which play a vital role in mediating both cellular and humoral immunity. Chronic involution of the thymus gland is thought to be one of the major contributing factors to loss of immune function with increasing age. It has recently been demonstrated that thymic atrophy is mediated by a shift from a stimulatory to a suppressive cytokine microenvironment. In this review we present an overview of the morphological, cellular and biochemical changes that have been implicated in the decline of thymic and peripheral immune function with ageing. We conclude with the clinical implications of age-associated immunosenescence to vaccine development for tumours and infectious disease. A fundamental understanding of the complex mechanisms by which ageing attenuates immune function will enable translational research teams to develop new therapies and vaccines specifically aimed at overcoming these defects in immunological function in the aged.
Keywords: ageing, haematopoiesis, thymopoiesis, B cells, T cells, infectious diseases, vaccines
Introduction
Ageing is a complex process affecting a wide variety of physiological functions, including the development and maintenance of the peripheral immune system [1]. It is widely accepted that the ageing process attenuates host ability to mount a robust or effective immune response. Only recently have advances in cellular and molecular phenotyping enabled researchers to more clearly elucidate the mechanisms that underlie immunoscenescence associated with ageing.
The peripheral immune system develops from haematopoietic stem cells that originate in the bone marrow. Lymphoid progenitors (including T and B cells) emigrate from the bone marrow and migrate to specialized peripheral sites, ie thymus, spleen, lymph nodes, to further mature, differentiate and acquire appropriate self/non-self-education. Upon identification of danger or a foreign invader, innate immune cells respond by destroying infected cells (NK cells) and releasing cytokines and chemokines to recruit additional cells to fight the infection and alter host tissues, a process commonly referred to as inflammation. This innate immune response can further progress to an adaptive (antigen-specific) immune response with the recruitment of effector T and B lymphocytes. Following effective clearance of the invading pathogen, the host immune response must wane and return to a quiescent state to prevent inadvertent damage. This latter process is in part mediated by a specialized subset of T cells called regulatory T cells (Treg).
T lymphocytes become specialized in the thymus gland to play a pivotal role in conducting the adaptive immune orchestra resulting in cellular immunity (via CD8 cytotoxic T cells) and B cell-mediated humoral immunity. T cells are considered to be highly vulnerable to the ‘effects’ of ageing. A number of factors have been linked to the decline in T cell function with age; however, it appears that chronic age-induced thymic atrophy and resultant decreased output of naïve T cells is the most important factor [2].
In this review we present an overview of the morphological, cellular and biochemical changes that have been implicated in the decline of immune function with ageing. To accomplish this we have ontologically divided the review into: (a) a brief overview of the impact of ageing on bone marrow stroma and its resident stem cell populations; (b) a discussion of age-induced changes that modulate thymopoiesis; and (c) a summary of defects associated with ageing in peripheral lymphoid tissues and lymphocyte subsets. We conclude with a discussion of the clinical implications of this age-associated decline in immune function, with specific regard to tumour and infectious disease vaccine development.
Bone marrow-derived stem cells
The bone marrow contains pluripotent stem cells that mature into bone tissue (non-haematopoietic cells) and cells that form peripheral blood cells, which further develop in specialized secondary compartments into functional immune cells (haematopoietic lineages) [3]. The stromal matrix of the bone marrow compartment is composed of accessory cells, such as megakaryocytes, osteoblasts, osteoclasts, adipocytes, chondrocytes, myoblasts and fibroblasts, which nurture and drive stem cell production [3]. The haematopoietic compartment of bone marrow decreases with increasing age and is replaced by fatty adipose tissue [3], thus suggesting a very early morphological impact of ageing on immune system ontogeny.
As with other stromal tissues, the bone marrow itself produces and responds to a wide variety of cytokines and hormones. Disturbance of this cytokine milieu may in part be responsible for the age-induced changes in bone marrow morphology and haematopoietic stem cell output. Systemic growth hormone, for example, is known to significantly decrease with age and this loss has been linked to the increased adiposity and decreased cellularity of aged bone marrow [4]. In support of this, French and colleagues have demonstrated a significant boost in cellularity and decreased adiposity of bone marrow in aged mice when treated with recombinant growth hormone [5].
Current studies implicate age-induced dysregulation of cytokine and hormone networks with this loss in bone marrow stem cell output across the lifespan. Despite a general age-induced decrease in cellularity of the bone marrow, Wang et al have shown that there is surprisingly an increase in the number of bone marrow-resident macrophages with age [6]. These aged macrophages, however, have a decreased ability to secrete tumour necrosis factor (TNF), a key inflammatory cytokine [6]. Macrophage-derived TNF [7] and IL-1 are essential for stromal cell secretion of other cytokines critical to stromal integrity, such as IL-6, IL-11, M-CSF, GM-CSF and receptor activator of NFκB ligand [3]. Ageing has also been shown to dampen the secretion of IL-7 by bone marrow stromal cells [8]. IL-7 is an essential survival cytokine for developing lymphocytes. Together, these data suggest that the composition of bone marrow stroma and its ability to nurture haematopoietic precursors is substantially compromised with ageing.
Haematopoietic stem cells
Haematopoietic stem cells (HSCs) give rise to all cellular components of the immune system (lymphoid and myeloid) and are defined as lineage negative (Lin−) CD34+ cells [9]. To date, very little is known about the effects of ageing on HSC and whether ageing HSC can influence the downstream function of the immune system. Many factors impact haematopoiesis at the stem cell level, including HSC number [10], function [11–13] and the ability to home to secondary lymphoid organs [14].
Despite the observation that the haematopoietic compartment of bone marrow decreases with age, there is no specific evidence that HSC numbers decrease with age. Murine studies do, however, highlight the importance of age and host genetic background in determining HSC number and their ability to successfully engraft within bone marrow [15–18]. For example, short-lived DBA/2 mice have fewer HSCs than long-lived C57Bl/6 mice [10]. Age does not, however, appear to influence the proliferative capacity of HSCs. In vitro proliferation assays of stem cells from aged animals show initial robust proliferation rates that subsequently decline, while stem cell proliferation from young animals are initially steady, and later surpass the aged group [11–13]. The homing and engraftment ability of transplants with aged HSCs is substantially attenuated, thus more HSCs from old mice are required to reestablish an ablated immune system [14]. The full impact of ageing on these uncommitted HSCs, and how this contributes to overall immunosenescence, remains to be elucidated and is therefore a fertile area for more detailed investigation.
B cell progenitors
Some HSCs differentiate into common lymphoid progenitors (CLPs), defined by having a Lin−, Sca-1lo, c-Kitlo, IL-7Rα+ surface phenotype. These were once thought to give rise to both B and T cells, but are now thought to be CLP/early B cell precursor (EBP) cells, which progress through stages of development as pro-B cells, pre-B cells, to become immature B cells [19,20].
Ageing has been shown to result in reduced numbers of CLPs and decreased CLP proliferation rates in vivo [21]. Initial reports suggested that pro-B cells did not decrease with age [22]; however, Johnson et al have shown that while the frequency of pro-B cells decreases with age, the absolute number stays constant, while bone marrow cellularity increases [23]. Other studies report a decline in pro-B cells numbers with age [21,24]. These discrepancies could be due to inconsistent ways of defining the pro-B cell (ie cell markers) with rapidly evolving technical advances in identifying these cells and fluorescence activated cell sorting.
Conversely, pre-B cells decrease markedly with age, most likely because of a block between the pro- and pre- B cell stages of development [23,25–27]. Decreased responsiveness of developing B cells to IL-7 [24,26], decreased V-DJ recombination of immunoglobulin genes [21] and decreased expression of surrogate light chain λ5 [8] have all been observed in aged mice. These factors play a key role in the pro-to pre-B cell block. The decrease in V-DJ recombination has in part been attributed to decreased activity of the transcription factors E12 and E47, which are required for immunoglobulin heavy chain expression [21,22]. There is additional evidence that aged bone marrow stromal cells are less able to support B cell expansion [20], thought to be due to decreased IL-7 production [18,27]. Despite the discrepancy over the precise site of block in B cell development, it is clear that there is a general age-induced decline in B cell lymphopoiesis that is initiated at the stem cell level in the bone marrow.
T cell progenitors
The bone marrow also supports the generation of T cell progenitors that migrate to the thymus to complete their education and differentiation. While CLPs (Lin−, c-Kitlo, Sca-1lo, IL-7Rα+) have not been specifically identified in the thymus, a CLP-derived progenitor, CLP-2 (Lin−, CD19−, B220+, c-Kitneg/lo, CD44hi, hCD25+, IL-7Rα+), was identified in human CD25 transgenic mice to efficiently home to the thymus gland [21], suggesting that CLP-2 cells are committed T cell progenitors. Other data suggest that T cell progenitors are derived not from CLPs but from early T-lineage progenitors (ETPs), defined by surface expression of (Lin−, CD44+, c-Kithi, IL-7Rαneg/lo) [19]. ETP cells are functionally different from CLP/CLP-2 cells because they are unresponsive to IL-7, due to a lack of IL-7R. Mice deficient in ikaros (a transcription factor known to be required for CLPs) were found to have no identifiable CLPs, resulting in arrested B cell, but normal levels of ETP and T cell, development [19]. This strongly supports the notion that T cells arise from this distinct ETP stem cell subset.
Studies in aged mice have shown a reduction in the number and proliferation of ETP cells, and increased ETP apoptosis [28]. In addition to the ability of growth hormone to stimulate aged bone marrow stroma [29], it can also have a positive effect on ETP and stimulate thymopoiesis in aged mice, bone marrow transplant mice, and bone marrow colonized fetal thymus organ culture [30,31]. The degree to which ETP migration contributes to the attenuation of thymopoiesis with age has yet to be fully elucidated, and further research in this area is needed to establish a direct link.
Thymus gland
Thymopoiesis
Thymocyte progenitor cells enter the thymus and begin their differentiation and education process as CD3−, CD4−, CD8− cells [32–34]. Double-negative CD4−, CD8− populations progress from DN I (CD44+, CD25−) to DN II (CD44+, CD25+) to DN III (CD44−, CD25+) to DN IV (CD44−, CD25−) before progressing to double-positive CD3+, CD4+, CD8+ thymocytes. The thymocytes rearrange their T cell receptor (TCR) genes at this stage, and undergo positive and negative selection. During this process, cells down-regulate either the CD4 or the CD8 molecule and become single positive (CD4 or CD8) naïve T cells, which are exported to the periphery. The overall process of T cell maturation and education is orchestrated by cytokines, hormones, corticosteroids and epithelial cells, dendritic cells, macrophages and fibroblasts that make up the thymic stroma [33,35–37]. Production of broadly reactive T cells by the thymus and maintenance of a diverse peripheral T cell repertoire are critical to the robustness of the human immune system.
Thymic atrophy
As an individual ages, the thymus involutes and the output of new T cells falls significantly [38–40]. In 1985, Steinman et al elegantly demonstrated that thymic function gradually starts decreasing from year one of life [38,39]. The observation of dual components of the human thymus, the true thymic epithelial space, in which thymopoiesis occurs, and the non-epithelial non-thymopoietic perivascular space [38,39], was critical to the current understanding of thymic atrophy. The expansion of the perivascular space (adipocytes, peripheral lymphocytes, stroma) with age results in a shift in the ratio of true thymic epithelial space to perivascular space. The thymic epithelial space shrinks to less than 10% of the total thymus tissue by 70 years of age. When extrapolated, Steinman's data suggest that the thymus would cease to produce new T cells at approximately 105 years of age (Figure 1) [41].
Figure 1.
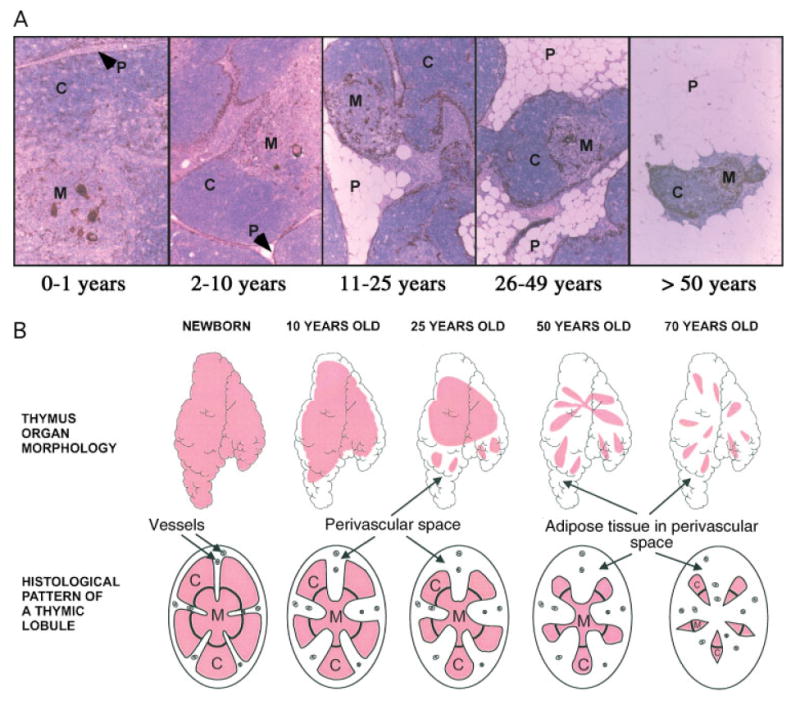
The human thymus across the lifespan. [A] Representative views of human thymus morphology throughout ageing. All tissue was formalin-fixed, paraffin-embedded and sections stained with haematoxylin and eosin and anti-keratin antibody [brown] to determine the percentage thymic epithelial space [each panel, ×25]. C, cortex; M, medulla; P, perivascular space. [B] Graphical depiction of the impact of age on human thymus morphology. Thymic epithelial space, pink; perivascular space, white. Reprinted with permission from reference [45]
There are several ways to monitor thymic atrophy. Kong et al initially showed that the presence of excised episomes of T cell receptor DNA identified recently produced T cells in the chicken thymus, as this excision event only occurs with TCR gene rearrangement [42]. Douek et al adapted this technique for human TCR DNA, and showed that signal joint T cell receptor excision circles (sjTREC) could be quantified by real-time PCR and used as a molecular marker of newly produced (naïve) recent thymic emigrants (RTEs) [43]. Recently, we have adapted these techniques to monitor sjTREC produced in mice [44]. Using this sensitive molecular technique, we have documented attenuation of thymopoiesis at the molecular level in healthy thymus tissue throughout normal ageing in humans (Figure 2A) [45] and in mice (Figure 3A) [44]. The impact of thymic attenuation with increasing age is also reflected in the quantity of peripheral naïve T cells in humans (Figure 2B) [45] and mice (Figure 3B) [44].
Figure 2.
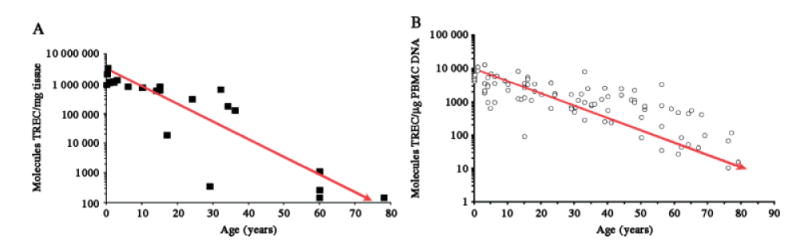
Impact of ageing on human thymic output and peripheral naïve T cell pool. [A] Molecules of sjTREC/mg whole human thymus tissue. [B] Molecules of sjTREC/μg isolated peripheral blood mononuclear cell DNA [n = 100 donors, aged 6 months–80 years]
Figure 3.
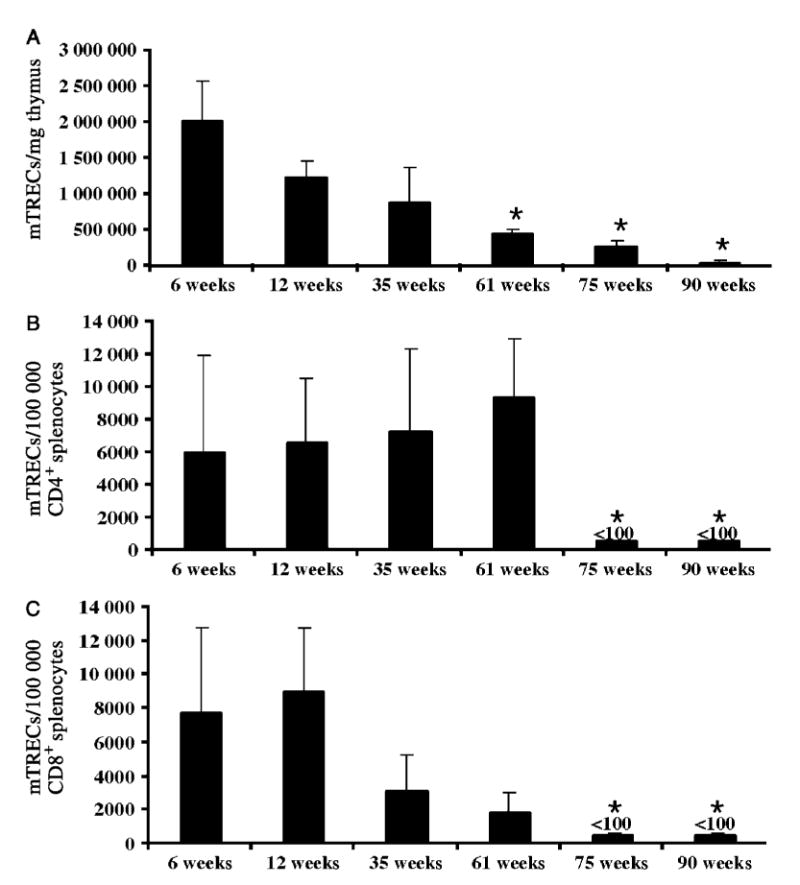
Impact of ageing on BALB/c mouse thymic output and splenic naïve T cell pools. [A] Molecules of mTREC/mg thymus tissue during ageing. Molecules of mTREC in naïve splenic CD4 [B] and CD8 [C] T cells in mice the age range 6–90 weeks [n = 3/group]. Data are mean ± SEM of three mice/group. *p < 0.05 compared to 6 week-old mice [44,45]
In vivo labelling of RTEs has recently been accomplished by Fink and colleagues, using transgenic mouse technology [46]. The gene for green fluorescent protein was introduced under the control of the recombinase activating gene-2 (RAG2) promotor to label naïve T cells and monitor thymic output in mice [46]. This in vivo labelling model confirmed our mouse sjTREC studies [44] and demonstrated that the CD4 : CD8 RTE diminishes with age, that RTE maturation is suboptimal in aged mice and that functional activation of RTE is decreased in aged mice [46].
We and others have also demonstrated that while the thymopoietic area of the human thymus decreases with age, the thymopoietic potential per cell, as measured by sjTRECs [47] or by TCR ligation-mediated polymerase chain reaction [3], remains constant at least until approximately 50 years of age [43,45,47–50]. Taken together, these observations of thymopoietic potential in aged thymus tissue provide promise that future therapeutic regeneration of the true thymic epithelium space could potentially restore normal thymus output in the aged.
Mechanisms of age-induced thymic involution
Thymic atrophy observed with increasing age is speculated to result from: ageing of the T cell progenitor population [51]; loss of self-peptide expressing thymic epithelium [TE] [52]; defects in rearrangement of TCRβ genes. [53]; and ageing of the thymic microenvironment with loss of trophic cytokines, such as IL-7 [54]. Our recent studies [45,50] and the work of others [55] have demonstrated that aged thymuses do not have a block in TCRβ rearrangements. No evidence for loss of TE peptides has been found in either mice or humans. As in bone marrow and peripheral sites, cytokines within the thymus are crucial for thymopoiesis. Moreover, biological pathways that control the thymic cytokine microenvironment represent candidate targets for therapies to modify age-induced thymic involution.
Thymic epithelial cells produce a number of colony-stimulating factors and haematopoietic cytokines such as IL-1, IL-3, IL-6, IL-7, transforming growth factor (TGF)β, oncostatin M (OSM) and leukaemia inhibitory factor (LIF) [45,56–59]. Intrathymic and systemic production of these potent cytokines regulates the complex process of thymopoiesis, and thus all are candidate targets for therapies aimed at promoting thymopoiesis. For example, IL-7 treatment can stimulate both thymopoiesis and peripheral T cell expansion, which leads to more rapid and successful T cell immune reconstitution following stem-cell transplantation in young mice [60,61]. IL-7 is necessary for thymopoiesis, promoting the survival of DN thymocytes by maintaining the anti-apoptotic protein Bcl-2 [62,63] and inducing V-DJ recombination [64]. We have recently reported that human IL-7 administration to young mice (aged 6–8 weeks) minimally increased thymopoiesis and peripheral T cell expansion [48].
Until recently, the existing paradigm was that age-induced thymic atrophy was solely due to the loss of trophic cytokines such as IL-7 [65]. We have observed, however, that intrathymic IL-7 steady-state mRNA levels remain constant with age, which is not consistent with this notion [37]. In contrast, we have reported significant elevations in RNA transcripts for cytokines such as LIF, IL-6 and OSM in aged human thymus tissue [37]. We have also observed increases in these IL-6 gene family cytokines in aged mouse thymus tissue (unpublished observation). Moreover, we have reported that LIF is a key mediator of endotoxin-induced thymic atrophy in a corticosteroid-dependent manner [66]. Thus, we hypothesized an alternative model of thymic involution, in which cytokines are actively induced in the thymus that suppress thymopoiesis with ageing (Figure 4).
Figure 4.
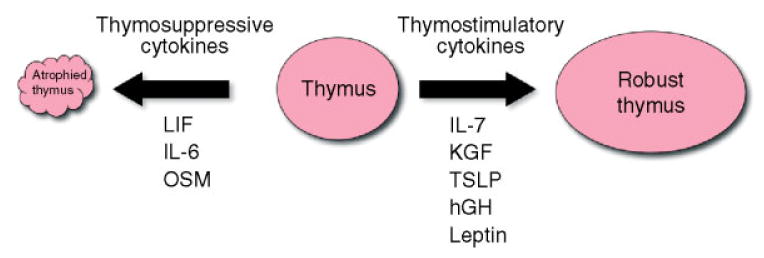
Schematic of cytokine stimuli that impact thymopoiesis
To test this hypothesis, we exogenously administered these cytokines over 3 days to young mice and showed a rapid involution of the thymus and loss of thymopoiesis [37]. These findings support a paradigm shift in our understanding of age-induced thymic involution. We propose that thymic atrophy and decreased thymopoiesis is an active process mediated by the up-regulation of thymosuppressive cytokines, which results in an observed decrease in peripheral naïve T cells with ageing (Figure 3).
In addition to age-related thymic atrophy, chemotherapy, irradiation prior to transplant, septic shock and other acute stress can also lead to thymic atrophy, providing additional experimental models to elucidate mechanisms underlying age-induced thymic atrophy. Oberholzer et al have shown that intrathymic injection of adenovirus-expressing IL-10 prevented sepsis-induced thymocyte apoptosis and thymic atrophy [67,68]. Leptin-deficient (ob/ob) and leptin receptor deficient (db/db) mice have chronic thymic atrophy [69,70], suggesting a key regulatory role for leptin in thymopoiesis. We and others have shown that leptin protects against bacterial endotoxin-induced thymic atrophy [71,72]. It has also been demonstrated that keratinocyte growth factor (KGF) and thymic stromal lymphopoietin (TSLP), which is known to stimulate thymus stromal cells, can promote thymopoiesis in mice following chemotherapy-induced thymic atrophy [73–75]. Growth hormone has been shown to have a stimulatory effect on thymopoiesis in old mice and ablative/bone marrow transplant mice [30,31]. Together, these studies suggest that coupling thymostimulatory regimens such as IL-10, leptin, KGF and TSLP with inhibition of thymosuppressive cytokines may be efficacious in aged models of immune reconstitution and function.
Peripheral lymphoid tissues
B cell progenitors undergo maturation and differentiation in secondary lymphoid tissues, such as the spleen and lymph nodes. These specialized lymphoid tissues are uniquely designed to filter the blood/lymph for antigens and provide a highly organized structure for T and B cells to interact with one another and critical antigen presenting accessory cells (ie dendritic cells, macrophages) of the immune system. The arteries of the spleen are surrounded by T lymphocytes forming the periarteriolar lymphoid sheath (PALS). Primary lymphoid follicles populated by B cells are adjacent to the PALS. Other cell types, such as T cells, dendritic cells and macrophages, make up the marginal zone. Collectively, these lymphocyte-rich areas constitute the white pulp. The remainder of the organ is comprised of red pulp, where macrophages and red blood cells reside in the sinusoidal space [7]. Similarly, lymph nodes are designed to filter lymphatic fluid drained from tissue for foreign antigens. The outer region of the lymph node, the cortex, contains B cell-rich primary follicles. Primary follicles expand into secondary follicles, or germinal centres, upon antigen recognition [7].
Age-associated architectural changes exist within these secondary lymphoid tissues. Studies of aged human spleens have demonstrated a decrease in arterial vessels and increase in stromal cells over lymphocytes compared to younger spleens [76]. Interestingly, total splenic weight increases with age due to fibroblast infiltration [77]. Studies in rat models have also shown a decrease in cortex lymphocyte cellularity and a reduction of germinal centres with age [78]. Similarly, studies of lymph node morphology in aged humans revealed a slow decline in paracortical and medullary zones, an increase in adipose tissue and less evidence of germinal centre formation in the elderly [79]. These changes indicate a decreased ability to provide the proper environment for immune responses to take place, but whether this has a direct impact on immunosenescence remains unknown.
The composition of the lymphocytes within the spleen and lymph nodes, and particularly the proper formation of the germinal centre, is known to change significantly with age. Connoy et al found age-specific changes in the composition of splenocytes, including increased frequency of B cells and memory CD4 T cells, decreased γ/δ T cells and naïve CD4 T cells, increased expression of CD28 on T cells and increased expression of a senescence marker, p16INK4a, on B cells and CD8+ T cells [80]. Lazuardi et al found age-induced changes in human lymph nodes, including a decrease in CD8 T cells, naïve T cells, IgM-expressing B cells, and significantly smaller germinal centres [81]. These observations are indicative of age-related changes to peripheral B and T cells and reflect normal changes to the immune system as it encounters more and more antigen with age.
Peripheral B lymphocytes
Peripheral B cell number does not decline with age, yet the quality of the humoral immune response declines and is characterized by lower antibody responses, decreased high-affinity antibodies and decreased IgG isotype class switching [82]. Secondary B cell development occurs in the germinal centres, where clonal expansion, somatic hypermutation and isotype switching occur. This process is highly dependent on T cells and follicular dendritic cells. The homing of immature B cells to the secondary lymphoid organs, which contain germinal centres, is decreased across the lifespan [83,84]. Defective homing decreases the chance that an antigen will be recognized by its antigen-specific B cell and may reduce the pool of naïve B cells. While the B cell compartment in lymphoid organs becomes increasingly composed of memory cells with age [83], this loss of naïve B cells in the periphery may dampen an individual's ability to respond to novel antigens as they age.
B cell proliferation potential declines in aged mice [85], perhaps due to declining B cell activation, reduced B7 expression [86] and defective surface Ig/B cell receptor signalling [87]. Interestingly, proliferative capacity can be restored to aged B cells by treatment with IL-2 or IL-4, highlighting the role of T helper cells in B cell activation [85]. Aged B cells have defective somatic hypermutation which decreases the ability of B cells to increase their receptor affinity [84]. Contributing factors to this deficiency include the loss of cell-cell interactions and cytokines needed for this process to occur, due to the reported attenuation of dendritic cell function age [88]. Together, these age-related defects affect secondary B cell development within the germinal centre.
Peripheral T lymphocytes
With ageing there is a shift in the ratio of naïve to memory T cells in the periphery. This results in the expansion of a T cell memory compartment to maintain peripheral T cell homeostasis. A recent model proposed by Haynes and Eaton [89,90] outlines the effects of ageing on the naïve T cell compartment and humoral responses. Using a CD4 TCR transgenic mouse specific for pigeon cytochrome c, they were able to track a population of T cells throughout the ageing process. Following in vitro or in vivo stimulation with pigeon cytochrome c, naïve T cells from aged mice exhibited reduced activation, differentiation and cytokine production [89,90]. Specifically, Th1 memory cells derived from aged naïve cells produced much less IL-2, and Th2 memory cells derived from aged naïve cells produced much less IL-4 and IL-5 than memory cells generated from young naïve cells. These differences in aged versus young T cells were abrogated by the addition of exogenous IL-2. Taken together with observations from other studies showing that aged CD4+ T cells have decreased CD40L [91], a critical co-stimulatory ligand for T-B cell interactions, and that IL-2 enhances CD40L expression [92], Haynes and Eaton suggested that impaired T-B cell interactions due to decreased IL-2 production by aged T cells significantly contributes to the impaired humoral (B cell) responses in the aged [89,90]. Other studies support these findings, reporting that aged CD4+ T helper cells provide inadequate assistance in germinal centres and promote low-affinity antibody production [93]. In addition to impaired naïve CD4+ T cell responses, memory T cell activation has also been shown to be attenuated, showing reduced signalling capacity and proliferation [94,95]. Upon closer examination, memory T cell function in aged mice seems to be dependent upon when the response was initiated, since memory cells produced early in life are normal [89].
While CD4 T helper cell function is required for optimal humoral responses, CD8 cytotoxic effector T lymphocytes (CTLs) are essential for viral clearance. Efforos and Walford demonstrated that peak cytolytic T cell responses are delayed in old mice compared to young mice following in vivo inoculation with influenza virus [96]. The intensity of the peak CTL response in these aged mice was 30% of that in young mice. Similar results have also been document for anti-influenza CTL responses in humans [97].
More recently, mechanistic studies show that a decreased frequency and number of influenza-virus-specific CD8+ T cells are major factors in the ability to clear infection [98]. In these studies, CTL activity strongly correlated with number of virus-specific CD8 T cells, regardless of age, and not intrinsic cytolytic defects of these T cells. This indicates that a defect in the expansion or proliferation of virus-specific CD8+ T cells is a major contributing factor during ageing. The decrease in virus-specific CD8 T cell number in this study also correlated with decreased numbers of IFNγ -producing cells with age. Studies performed with an alternative E55+ retrovirus infection model also showed decreased proliferative capacity of the CD8 T cell population as a function of age [99]. Collectively, these data show that the proliferative and IFNγ secretion of the CTL response to viral infection is compromised in ageing. It is clear that diminished CD4 and CD8 T cell function make a significant direct contribution to the senescence of the immune system associated with ageing.
Regulatory T cells (Tregs)
In recent years, the vital role of CD4+, CD25+, Foxp3+ regulatory T cells (Tregs) in the maintenance of immune homeostasis has been described [100]. Tregs play a key role in controlling the host immune response to prevent excessive collateral damage to host tissues [101–103]. Tregs are also critical for protecting the host from self-reactive lymphocytes that have escaped the rigorous selection processes designed to delete autoreactive immunocompetent cells. Two critically different types of CD4+ Treg cells are naturally occurring CD4+, CD25+, Foxp3+ cells derived directly from the thymus (nTregs), and CD4+, CD25+, Foxp3+ cells that are derived from peripheral CD4+, CD25−, Foxp3− cells that become anergic and gain suppressive capabilities following exposure to conditions such as low antigen dose, oral tolerance, immunotherapy or chronic antigen exposure (iTregs) [104]. It has been suggested that a decrease in Treg numbers or function could result in autoimmune disease or rejection of a transplant, while an excess (or normal level) of Tregs might contribute to poor responses to cancer, vaccines and infectious diseases [101–104].
A clinical demonstration of the important role of Tregs in human health is the X-linked disease IPEX [105]. IPEX is characterized by immune dysregulation, polyenocrinopathy and enteropathy attributed to a Foxp3 gene mutation and severe deficiency in Tregs [105]. Foxp3 is a transcription factor which mediates the Treg cell lineage commitment and function [106]. Prior to the identification of Foxp3 as a definitive nuclear marker of Treg cells, researchers characterized Tregs as CD4+, CD25+, a population of cells that may also include recently activated T cells. For the purpose of this review, we define Tregs as CD4+, CD25+, keeping in mind that additional markers are often used in the identification of these cells, which probably underlies some of the conflicting data regarding this cell population and their function with ageing.
The waning output of T cells and general decline in thymic function with age indicates that the production of thymus-derived Tregs would also be disturbed in aged persons. Indeed, the prevalence of Tregs appears to fluctuate with age; 2.3–9.5% of umbilical cord blood (earliest postnatal peripheral blood source) CD4+ cells are Tregs, while in healthy adults (aged 20–60 years) an average of 0.6–8.7% of CD4+ T cells are CD4+, CD25+ Tregs [107,108]. In elderly adults (65+ years) it is unclear whether the Treg pool is significantly different from that in younger adults, as some authors have shown that they have an increase in Tregs compared to young adults, while others demonstrated no correlation between age and Treg prevalence [109–112]. Characterization of Tregs from young and elderly donors consisting of healthy and non-healthy people revealed that those with poor health (of either age group) had significantly more Tregs than their healthy counterparts, with the young, non-healthy subjects having over twice the number of Tregs than the young, healthy group [112]. This study also reported that among the healthy subjects, aged donors had significantly higher numbers of Tregs than the young.
Whether or not advanced age is associated with a decline in the function, or suppressive capacity, of Tregs is also controversial. The ability of CD4+, CD25+ cells from C57BL/6 mice of various ages to suppress CD4+, CD25− (traditional CD4 effector) cells in vitro was not affected by age [113]. This study also reaffirmed the dogma that CD4 effector cells from aged mice show suboptimal proliferation in response to stimulation [113]. In contrast to these animal studies, Tsaknaridis et al demonstrated a decreased ability of purified CD4+, CD25+ from aged human donors to suppress CD4+, CD25− effector cells in vitro, compared to Treg cells from young donors [114]. Functional assays of sorted CD4+, CD25hi T cells by another group demonstrated equivalent regulatory function in young and elderly donors, as measured by suppression of proliferation, expression of intracellular CTLA4 and IFNγ in response to T cell stimulation, and an increase in the number of Tregs in the elderly [111]. Taken together, these data suggest that aged individuals have an increase in peripheral blood regulatory T cells. Whether these cells are naturally derived Tregs from the thymus or induced Tregs from anergized peripheral CD4 T cells, their relevance to immune senescence and disease susceptibility in the aged remains unclear. Characterization of Tregs in aged, thymectomized animals should enhance our understanding of the origination and persistence of these cells throughout life. It is interesting to speculate that if the increase in regulatory T cells is determined to be strongly correlated with age and poor health, this suppression of immune responses would limit the host's ability to mount adequate anti-cancer responses, resolve infections or respond effectively to immunization.
Our understanding of how Tregs contribute to immunological senescence will be aided by the recent development of reagents to definitively characterize and purify these cells for use in functional studies [115]. The use of Foxp3 as a definitive marker of regulatory T cells is problematic, as cells die after fluorescently labelling this protein, due to the fixation and permeablization required to access the antigen. As previously noted, CD25 (IL-2R) is also expressed on recently activated T cells, and some Tregs are Foxp3+ but CD25−. Recently, the observation that the IL-7 receptor (CD127) is not expressed on Foxp3+ cells and a subset of CD4+, CD25+, CD127lo cells are suppressive, but CD127hi cells are not, indicates that this may be a useful staining protocol to identify live Tregs in functional assays [115,116]. Standardized usage of Treg-defining cell markers among researchers would greatly increase our ability to compare the results of different studies and define more efficiently their role in human disease processes and immunosenescence associated with ageing.
Clinical considerations and vaccines
It is well documented that the risk of developing cancer increases dramatically with age [117–120]. Older patients may also exhibit larger and more aggressive tumours [121]. As discussed previously, numerous functions of T and B lymphocytes diminish with age, and it is likely that the increase in cancers with age is directly related to the decline in immune responsiveness to these neoplasms. As chemotherapy and irradiation produce toxic side-effects which can be more pronounced in the elderly, development of alternative targeted therapeutics that are efficacious in all age groups is essential. Unfortunately, only a few studies have investigated anti-tumour responses or the efficacy of immunotherapy in the elderly. These findings and their implications for cancer research and treatment are discussed below.
Using a mouse tumour model, Lustgarten et al investigated primary and memory responses to lymphoma cells in aged and young mice [122]. Young mice have protective immunity to the tumour, which is lethal in aged mice. When the co-stimulatory molecule CD80 was expressed by the tumour cells, aged and young mice rejected the tumours and long-term memory responses were induced in young mice. To elicit memory responses to the tumour in aged mice, additional stimulation, using the agonist monoclonal antibody anti-OX40, was required [122]. OX40 (CD134) is a TNFR family member expressed on T cells. Co-stimulation of OX40 increases T cell effector function, survival, expansion and development into memory cells [123].
Another reported tumour-vaccine strategy utilizes dendritic cells to present tumour antigens to T cells, which in turn elicit a cytotoxic anti-tumour response. A study comparing the efficacy of DC vaccination in young and aged mice demonstrated that the anti-tumour response in aged mice is much lower than that in young mice [124]. As in the study of Haynes et al [89], the addition of recombinant IL-2 enhanced this response in young, but not older, animals. However, administering anti-OX40 or another agonistic monoclonal antibody to the TNFR family member 4-1BB (anti-4-1BB) to provide co-stimulatory signals dramatically increased the ability of the aged animals to inhibit tumour growth [124].
These data indicate that stimulation via co-receptors/co-stimulatory molecules might overcome some of the intrinsic defects of immune responses in the aged, and that age-related changes in immunity can limit the efficacy of anti-tumour therapies. Thus, anti-tumour immunotherapy approaches incorporating co-stimulation may be of value for the elderly. As cancers primarily are a disease of older age, investigations using an aged cohort of animals to determine the efficacy and mechanisms of anti-cancer therapies and prevention are warranted, as the responses of young and aged mice differ dramatically.
Immunosenescence and concurrent health problems involving the lung and heart increase the risk of complications and death from viral infections in the elderly. Influenza is the fifth leading cause of death among people aged 50 and older, and this group is a major target of vaccination campaigns [125]. However, this vaccination strategy is controversial, as it appears to have only a moderate impact on influenza-specific immune responses and infection rates in this age group, a problem compounded when there is a mismatch between the strains included in the vaccine and those circulating in the population [126,127].
Combined data from multiple studies on influenza vaccines show that in the elderly (aged 65+ years), effectiveness (measured by the prevention of influenza-like illness) was 23%, while in healthy children (aged 2+ years) the effectiveness was higher, at 38% [128,129]. Mechanistic studies have demonstrated that in the elderly, humoral and cell-mediated influenza-specific responses are lower following vaccination than in young adults. Deng et al have reported that PBMCs from elderly adults (aged 64+ years) stimulated with an influenza A virus ex vivo had a decrease in IFNγ+ T cells and secretion of IFNγ by individual cells compared to young adults (aged 20–50 years) [130]. Expansion of CD8+ cells was also lower in the aged, and in frailer aged subjects this defect was more pronounced. In addition to the observed decrease in cellular immune responses, the elderly group produced fewer antibodies in response to influenza [130].
Development of vaccination strategies that are effective in all age groups is an important area of research, particularly to protect against new or re-emerging fatal infections, and possible infectious agents weaponized by bioterrorists. There are numerous subtypes and strains of influenza viruses, and vaccination with the strains most likely to be circulating during a given season is required annually for immunity [131,132].
The decision of which strains of influenza to include in the inactivated, trivalent (and live, attenuated; LAIV) vaccines produced each year against flu is made by consideration of antigens from recently isolated influenza viruses, epidemiological data and postvaccination serological studies in humans [132]. This complicated process, coupled with viral antigenic drift [133,134] and the previously mentioned decrease in the ability of aged hosts to respond to and clear infections, highlights the need to develop effective vaccine strategies and rapid production techniques to keep pace with the ability of viruses to evolve into strains for which prior exposure (and immunity) does not exist. Recently, the ability of a highly pathogenic avian influenza to infect humans, and possibly be spread from human to human, has highlighted the need for rapid development of vaccines against new subtypes and strains of influenza that will be effective in the elderly.
Summary
A wide spectrum of changes occur in the constitution and function of lymphoid progenitors, tissues and cells across the lifespan. Consideration of how these changes affect susceptibility to cancer and infections is vital to the development of new therapies aimed at enhancing immune responses in the elderly. Understanding thymic atrophy and developing tools to reverse this (increasing the output of new, functional T cells) may overcome many of the defects observed in immunological function with age.
The chronic and steady involution of the thymus gland with increasing age appears to be an active process, with the induction of specific cytokines that cause atrophy of the thymopoietic space. This is in contrast to the perceived view, that the thymus is ‘neglected’ as one ages and therefore withers away due to lack of appropriate cytokine/growth factor stimulation (Figure 4). One hypothesis for this active process is that the thymus is involuting in response to an appropriately aged and full peripheral T cell repertoire that has encountered a wide range of antigens during its years and successfully established a diverse memory pool. In addition, one can speculate that by reducing the output of new T cells throughout ageing, immunological resources will be conserved and the likelihood of exporting a self-reactive T cell will be reduced.
Enhancing thymic export of T cells will theoretically increase the ability to mount successful immune responses to any antigen. In light of the possibility of a highly pathogenic influenza A virus being transmitted directly from birds to humans, and the potential threat of bioterrorism, priority has been placed on developing novel immunization strategies that can be rapidly produced and are efficacious in all age groups. To this end, inclusion of agents to enhance thymopoiesis may prove very useful in promoting a robust, broad T cell response to any antigen (tumour, viral or bacterial) for which a successful vaccine is needed. Critical to this effort will be the ability of multi-disciplinary research teams, composed, for example, of gerontologists, immunologists and stem cell biologists, to build on our current knowledge base and work collaboratively to further dissect and understand the impacts of ageing on immune system ontogeny and function.
Footnotes
No conflicts of interest were declared.
References
- 1.Miller RA. Aging and immune function. Int Rev Cytol. 1991;124:187–215. doi: 10.1016/s0074-7696(08)61527-2. [DOI] [PubMed] [Google Scholar]
- 2.Haynes BF, Markert ML, Sempowski GD, Patel DD, Hale LP. The role of the thymus in immune reconstitution in aging, bone marrow transplantation, and HIV-1 infection. Ann Rev Immunol. 2000;18:529–560. doi: 10.1146/annurev.immunol.18.1.529. [DOI] [PubMed] [Google Scholar]
- 3.Compston JE. Bone marrow and bone: a functional unit. J Endocrinol. 2002;173(3):387–394. doi: 10.1677/joe.0.1730387. [DOI] [PubMed] [Google Scholar]
- 4.Lamberts SW, van den Beld AW, van der Lely AJ. The endocrinology of aging. Science. 1997;278(5337):419–424. doi: 10.1126/science.278.5337.419. [DOI] [PubMed] [Google Scholar]
- 5.French RA, Broussard SR, Meier WA, Minshall C, Arkins S, Zachary JF, et al. Age-associated loss of bone marrow hematopoietic cells is reversed by GH and accompanies thymic reconstitution. Endocrinology. 2002;143(2):690–699. doi: 10.1210/endo.143.2.8612. [DOI] [PubMed] [Google Scholar]
- 6.Wang CQ, Udupa KB, Xiao H, Lipschitz DA. Effect of age on marrow macrophage number and function. Aging (Milano) 1995;7(5):379–384. doi: 10.1007/BF03324349. [DOI] [PubMed] [Google Scholar]
- 7.Goldsby RA, Kindt TJ, Osborne BA, Kuby J. Immunology. 5. New York: W.H. Freeman & Co; 1992. [Google Scholar]
- 8.Tsuboi I, Morimoto K, Hirabayashi Y, Li GX, Aizawa S, Mori KJ, et al. Senescent B lymphopoiesis is balanced in suppressive homeostasis: decrease in interleukin-7 and transforming growth factor-beta levels in stromal cells of senescence-accelerated mice. Exp Biol Med (Maywood) 2004;229(6):494–502. doi: 10.1177/153537020422900607. [DOI] [PubMed] [Google Scholar]
- 9.Kohn DB, Bauer G, Rice CR, Rothschild JC, Carbonaro DA, Valdez P, et al. A clinical trial of retroviral-mediated transfer of a rev-responsive element decoy gene into CD34(+) cells from the bone marrow of human immunodeficiency virus-1-infected children. Blood. 1999;94(1):368–371. [PubMed] [Google Scholar]
- 10.de Haan G, Van Zant G. Dynamic changes in mouse hematopoietic stem cell numbers during aging. Blood. 1999;93(10):3294–3301. [PubMed] [Google Scholar]
- 11.Mauch P, Botnick LE, Hannon EC, Obbagy J, Hellman S. Decline in bone marrow proliferative capacity as a function of age. Blood. 1982;60(1):245–252. [PubMed] [Google Scholar]
- 12.Harrison DE, Zhong RK, Jordan CT, Lemischka IR, Astle CM. Relative to adult marrow, fetal liver repopulates nearly five times more effectively long-term than short-term. Exp Hematol. 1997;25(4):293–297. [PubMed] [Google Scholar]
- 13.Rebel VI, Miller CL, Eaves CJ, Lansdorp PM. The repopulation potential of fetal liver hematopoietic stem cells in mice exceeds that of their liver adult bone marrow counterparts. Blood. 1996;87(8):3500–3507. [PubMed] [Google Scholar]
- 14.Liang Y, Van Zant G, Szilvassy SJ. Effects of aging on the homing and engraftment of murine hematopoietic stem and progenitor cells. Blood. 2005;106(4):1479–1487. doi: 10.1182/blood-2004-11-4282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Henckaerts E, Langer JC, Snoeck HW. Quantitative genetic variation in the hematopoietic stem cell and progenitor cell compartment and in lifespan are closely linked at multiple loci in BXD recombinant inbred mice. Blood. 2004;104(2):374–379. doi: 10.1182/blood-2003-12-4304. [DOI] [PubMed] [Google Scholar]
- 16.Yuan R, Astle CM, Chen J, Harrison DE. Genetic regulation of hematopoietic stem cell exhaustion during development and growth. Exp Hematol. 2005;33(2):243–250. doi: 10.1016/j.exphem.2004.10.014. [DOI] [PubMed] [Google Scholar]
- 17.Harrison DE. Long-term erythropoietic repopulating ability of old, young, and fetal stem cells. J Exp Med. 1983;157(5):1496–1504. doi: 10.1084/jem.157.5.1496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chen J, Astle CM, Harrison DE. Development and aging of primitive hematopoietic stem cells in BALB/cBy mice. Exp Hematol. 1999;27(5):928–935. doi: 10.1016/s0301-472x(99)00018-1. [DOI] [PubMed] [Google Scholar]
- 19.Allman D, Sambandam A, Kim S, Miller JP, Pagan A, Well D, et al. Thymopoiesis independent of common lymphoid progenitors. Nat Immunol. 2003;4(2):168–174. doi: 10.1038/ni878. [DOI] [PubMed] [Google Scholar]
- 20.Miller JP, Allman D. Linking age-related defects in B lymphopoiesis to the aging of hematopoietic stem cells. Semin Immunol. 2005;17(5):321–329. doi: 10.1016/j.smim.2005.05.003. [DOI] [PubMed] [Google Scholar]
- 21.Min H, Montecino-Rodriguez E, Dorshkind K. Effects of aging on the common lymphoid progenitor to pro-B cell transition. J Immunol. 2006;176(2):1007–1012. doi: 10.4049/jimmunol.176.2.1007. [DOI] [PubMed] [Google Scholar]
- 22.Stephan RP, Sanders VM, Witte PL. Stage-specific alterations in murine B lymphopoiesis with age. Int Immunol. 1996;8(4):509–518. doi: 10.1093/intimm/8.4.509. [DOI] [PubMed] [Google Scholar]
- 23.Johnson KM, Owen K, Witte PL. Aging and developmental transitions in the B cell lineage. Int Immunol. 2002;14(11):1313–1323. doi: 10.1093/intimm/dxf092. [DOI] [PubMed] [Google Scholar]
- 24.Miller JP, Allman D. The decline in B lymphopoiesis in aged mice reflects loss of very early B-lineage precursors. J Immunol. 2003;171(5):2326–2330. doi: 10.4049/jimmunol.171.5.2326. [DOI] [PubMed] [Google Scholar]
- 25.Sherwood EM, Blomberg BB, Xu W, Warner CA, Riley RL. Senescent BALB/c mice exhibit decreased expression of lambda5 surrogate light chains and reduced development within the pre-B cell compartment. J Immunol. 1998;161(9):4472–4475. [PubMed] [Google Scholar]
- 26.Stephan RP, Lill-Elghanian DA, Witte PL. Development of B cells in aged mice: decline in the ability of pro-B cells to respond to IL-7 but not to other growth factors. J Immunol. 1997;158(4):1598–1609. [PubMed] [Google Scholar]
- 27.Stephan RP, Reilly CR, Witte PL. Impaired ability of bone marrow stromal cells to support B-lymphopoiesis with age. Blood. 1998;91(1):75–88. [PubMed] [Google Scholar]
- 28.Wells JA, de Vos AM. Structure and function of human growth hormone: implications for the hematopoietins. Annu Rev Biophys Biomol Struct. 1993;22:329–351. doi: 10.1146/annurev.bb.22.060193.001553. [DOI] [PubMed] [Google Scholar]
- 29.Hanley MB, Napolitano LA, McCune JM. Growth hormone-induced stimulation of multilineage human hematopoiesis. Stem Cells. 2005;23(8):1170–1179. doi: 10.1634/stemcells.2004-0322. [DOI] [PubMed] [Google Scholar]
- 30.Knyszynski A, Adler-Kunin S, Globerson A. Effects of growth hormone on thymocyte development from progenitor cells in the bone marrow. Brain Behav Immunol. 1992;6(4):327–340. doi: 10.1016/0889-1591(92)90032-j. [DOI] [PubMed] [Google Scholar]
- 31.Chen BJ, Cui X, Sempowski GD, Chao NJ. Growth hormone accelerates immune recovery following allogeneic T cell-depleted bone marrow transplantation in mice. Exp Hematol. 2003;31(10):953–958. doi: 10.1016/s0301-472x(03)00196-6. [DOI] [PubMed] [Google Scholar]
- 32.Haynes BF, Denning SM, Singer KH, Kurtzberg J. Ontogeny of T cell precursors: a model for the initial stages of human T cell development. Immunol Today. 1989;10(3):87–91. doi: 10.1016/0167-5699(89)90232-6. [DOI] [PubMed] [Google Scholar]
- 33.Haynes BF, Hale LP. The human thymus. A chimeric organ comprised of central and peripheral lymphoid components. Immunol Res. 1998;18(3):175–192. doi: 10.1007/BF02788778. [DOI] [PubMed] [Google Scholar]
- 34.Kaye J. Regulation of T cell development in the thymus. Immunol Res. 2000;21(2–3):71–81. doi: 10.1385/IR:21:2-3:71. [DOI] [PubMed] [Google Scholar]
- 35.Haynes BF. The role of the thymic microenvironment in promotion of early stages of human T cell maturation. Clin Res. 1986;34(3):422–431. [PubMed] [Google Scholar]
- 36.Haynes BF. Human thymic epithelium and T cell development: current issues and future directions. Thymus. 1990;16(3–4):143–157. [PubMed] [Google Scholar]
- 37.Sempowski GD, Hale LP, Sundy JS, Massey JM, Koup RA, Douek DC, et al. Leukemia inhibitory factor, oncostatin M, IL-6, and stem cell factor mRNA expression in human thymus increases with age and is associated with thymic atrophy. J Immunol. 2000;164(4):2180–2187. doi: 10.4049/jimmunol.164.4.2180. [DOI] [PubMed] [Google Scholar]
- 38.Steinmann GG. Changes in the human thymus during aging. Curr Top Pathol. 1986;75:43–88. doi: 10.1007/978-3-642-82480-7_2. [DOI] [PubMed] [Google Scholar]
- 39.Steinmann GG, Klaus B, Muller-Hermelink HK. The involution of the ageing human thymic epithelium is independent of puberty. A morphometric study. Scand J Immunol. 1985;22(5):563–575. doi: 10.1111/j.1365-3083.1985.tb01916.x. [DOI] [PubMed] [Google Scholar]
- 40.Scollay RG, Butcher EC, Weissman IL. Thymus cell migration. Quantitative aspects of cellular traffic from the thymus to the periphery in mice. Eur J Immunol. 1980;10(3):210–218. doi: 10.1002/eji.1830100310. [DOI] [PubMed] [Google Scholar]
- 41.George AJ, Ritter MA. Thymic involution with ageing: obsolescence or good housekeeping? Immunol Today. 1996;17(6):267–272. doi: 10.1016/0167-5699(96)80543-3. see comments. [DOI] [PubMed] [Google Scholar]
- 42.Kong FK, Chen CL, Six A, Hockett RD, Cooper MD. T cell receptor gene deletion circles identify recent thymic emigrants in the peripheral T cell pool. Proc Natl Acad Sci USA. 1999;96(4):1536–1540. doi: 10.1073/pnas.96.4.1536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Douek DC, McFarland RD, Keiser PH, Gage EA, Massey JM, Haynes BF, et al. Changes in thymic function with age and during the treatment of HIV infection. Nature. 1998;396(6712):690–695. doi: 10.1038/25374. [DOI] [PubMed] [Google Scholar]
- 44.Sempowski GD, Gooding ME, Liao HX, Le PT, Haynes BF. T cell receptor excision circle assessment of thymopoiesis in aging mice. Mol Immunol. 2002;38(11):841–848. doi: 10.1016/s0161-5890(01)00122-5. [DOI] [PubMed] [Google Scholar]
- 45.Sempowski GD, Hale LP, Sundy JS, Massey JM, Koup RA, Douek DC, et al. Leukemia inhibitory factor, oncostatin M, IL-6, and stem cell factor mRNA expression in human thymus increases with age and is associated with thymic atrophy. J Immunol. 2000;164(4):2180–2187. doi: 10.4049/jimmunol.164.4.2180. [DOI] [PubMed] [Google Scholar]
- 46.Hale JS, Boursalian TE, Turk GL, Fink PJ. Thymic output in aged mice. Proc Natl Acad Sci USA. 2006;103(22):8447–8452. doi: 10.1073/pnas.0601040103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jamieson BD, Douek DC, Killian S, Hultin LE, Scripture-Adams DD, Giorgi JV, et al. Generation of functional thymocytes in the human adult. Immunity. 1999;10(5):569–575. doi: 10.1016/s1074-7613(00)80056-4. [DOI] [PubMed] [Google Scholar]
- 48.Sempowski GD, Gooding ME, Liao HX, Le PT, Haynes BF. T cell receptor excision circle assessment of thymopoiesis in aging mice. Mol Immunol. 2002;38:841–848. doi: 10.1016/s0161-5890(01)00122-5. [DOI] [PubMed] [Google Scholar]
- 49.Sempowski G, Thomasch J, Gooding M, Hale L, Edwards L, Ciafaloni E, et al. Effect of thymectomy on human peripheral blood T cell pools in myasthenia gravis. J Immunol. 2001;166(4):2808–2817. doi: 10.4049/jimmunol.166.4.2808. [DOI] [PubMed] [Google Scholar]
- 50.Flores KG, Li J, Sempowski GD, Haynes BF, Hale LP. Analysis of the human thymic perivascular space during aging. J Clin Invest. 1999;104(8):1031–1039. doi: 10.1172/JCI7558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tyan ML. Age-related decrease in mouse T cell progenitors. J Immunol. 1977;118(3):846–851. [PubMed] [Google Scholar]
- 52.Hartwig M, Steinmann G. On a causal mechanism of chronic thymic involution in man. Mech Ageing Dev. 1994;75(2):151–156. doi: 10.1016/0047-6374(94)90083-3. [DOI] [PubMed] [Google Scholar]
- 53.Aspinall R. Age-associated thymic atrophy in the mouse is due to a deficiency affecting rearrangement of the TCR during intrathymic T cell development. J Immunol. 1997;158(7):3037–3045. [PubMed] [Google Scholar]
- 54.Plum J, De Smedt M, Leclercq G, Verhasselt B, Vandekerckhove B. Interleukin-7 is a critical growth factor in early human T cell development. Blood. 1996;88(11):4239–4245. [PubMed] [Google Scholar]
- 55.Lacorazza HD, Guevara Patino JA, Weksler ME, Radu D, Nikolic-Zugic J. Failure of rearranged TCR transgenes to prevent age-associated thymic involution. J Immunol. 1999;163(8):4262–4268. [PubMed] [Google Scholar]
- 56.Le PT, Lazorick S, Whichard LP, Yang YC, Clark SC, Haynes BF, et al. Human thymic epithelial cells produce IL-6, granulocyte-monocyte-CSF, and leukemia inhibitory factor. J Immunol. 1990;145(10):3310–3315. [PubMed] [Google Scholar]
- 57.Le PT, Tuck DT, Dinarello CA, Haynes BF, Singer KH. Human thymic epithelial cells produce interleukin 1. J Immunol. 1987;138(8):2520–2526. [PubMed] [Google Scholar]
- 58.Le PT, Kurtzberg J, Brandt SJ, Niedel JE, Haynes BF, Singer KH. Human thymic epithelial cells produce granulocyte and macrophage colony-stimulating factors. J Immunol. 1988;141(4):1211–1217. [PubMed] [Google Scholar]
- 59.Galy AH, de Waal Malefyt R, Barcena A, Peterson SM, Spits H. Untransfected and SV40-transfected fetal and postnatal human thymic stromal cells. Analysis of phenotype, cytokine gene expression and cytokine production. Thymus. 1993;22(1):13–33. [PubMed] [Google Scholar]
- 60.Mackall CL, Fry TJ, Bare C, Morgan P, Galbraith A, Gress RE. IL-7 increases both thymic-dependent and thymic-independent T cell regeneration after bone marrow transplantation. Blood. 2001;97(5):1491–1497. doi: 10.1182/blood.v97.5.1491. [DOI] [PubMed] [Google Scholar]
- 61.Bolotin E, Smogorzewska M, Smith S, Widmer M, Weinberg K. Enhancement of thymopoiesis after bone marrow transplant by in vivo interleukin-7. Blood. 1996;88(5):1887–1894. [PubMed] [Google Scholar]
- 62.von Freeden-Jeffry U, Solvason N, Howard M, Murray R. The earliest T lineage-committed cells depend on IL-7 for Bcl-2 expression and normal cell cycle progression. Immunity. 1997;7(1):147–154. doi: 10.1016/s1074-7613(00)80517-8. [DOI] [PubMed] [Google Scholar]
- 63.Kim K, Lee CK, Sayers TJ, Muegge K, Durum SK. The trophic action of IL-7 on pro-T cells: inhibition of apoptosis of pro-T1, -T2, and -T3 cells correlates with Bcl-2 and Bax levels and is independent of Fas and p53 pathways. J Immunol. 1998;160(12):5735–5741. [PubMed] [Google Scholar]
- 64.Muegge K, Vila MP, Durum SK. Interleukin-7: a co-factor for V(D)J rearrangement of the T cell receptor beta gene. Science. 1993;261(5117):93–95. doi: 10.1126/science.7686307. [DOI] [PubMed] [Google Scholar]
- 65.Andrew D, Aspinall R. Age-associated thymic atrophy is linked to a decline in IL-7 production. Exp Gerontol. 2002;37(2–3):455–463. doi: 10.1016/s0531-5565(01)00213-3. [DOI] [PubMed] [Google Scholar]
- 66.Sempowski GD, Rhein ME, Scearce RM, Haynes BF. Leukemia inhibitory factor is a mediator of Escherichia coli lipopolysaccharide-induced acute thymic atrophy. Eur J Immunol. 2002;32:3066–3070. doi: 10.1002/1521-4141(200211)32:11<3066::AID-IMMU3066>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- 67.Oberholzer C, Oberholzer A, Clare-Salzler M, Moldawer LL. Apoptosis in sepsis: a new target for therapeutic exploration. FASEB J. 2001;15(6):879–892. doi: 10.1096/fj.00-058rev. [DOI] [PubMed] [Google Scholar]
- 68.Oberholzer C, Oberholzer A, Bahjat FR, Minter RM, Tannahill CL, Abouhamaze A, et al. Targeted adenovirus-induced expression of IL-10 decreases thymic apoptosis and improves survival in murine sepsis. Proc Natl Acad Sci USA. 2001;98:11503–11508. doi: 10.1073/pnas.181338198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Howard JK, Lord GM, Matarese G, Vendetti S, Ghatei MA, Ritter MA, et al. Leptin protects mice from starvation-induced lymphoid atrophy and increases thymic cellularity in ob/ob mice. J Clin Invest. 1999;104(8):1051–1059. doi: 10.1172/JCI6762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Madiehe AM, Mitchell TD, Harris RB. Hyperleptinemia and reduced TNF-alpha secretion cause resistance of db/db mice to endotoxin. Am J Physiol Regul Integr Comp Physiol. 2003;284(3):R763–770. doi: 10.1152/ajpregu.00610.2002. [DOI] [PubMed] [Google Scholar]
- 71.Hick RW, Gruver AL, Ventevogel MS, Haynes BF, Sempowski GD. Leptin selectively augments thymopoiesis in leptin deficiency and lipopolysaccharide-induced thymic atrophy. J Immunol. 2006;177(1):169–176. doi: 10.4049/jimmunol.177.1.169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Lord GM, Matarese G, Howard JK, Baker RJ, Bloom SR, Lechler RI. Leptin modulates the T cell immune response and reverses starvation-induced immunosuppression. Nature. 1998;394(6696):897–901. doi: 10.1038/29795. [DOI] [PubMed] [Google Scholar]
- 73.Erickson M, Morkowski S, Lehar S, Gillard G, Beers C, Dooley J, et al. Regulation of thymic epithelium by keratinocyte growth factor. Blood. 2002;100(9):3269–3278. doi: 10.1182/blood-2002-04-1036. [DOI] [PubMed] [Google Scholar]
- 74.Sims JE, Williams DE, Morrissey PJ, Garka K, Foxworthe D, Price V, et al. Molecular cloning and biological characterization of a novel murine lymphoid growth factor. J Exp Med. 2000;192(5):671–680. doi: 10.1084/jem.192.5.671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Min D, Taylor PA, Panoskaltsis-Mortari A, Chung B, Danilenko DM, Farrell C, et al. Protection from thymic epithelial cell injury by keratinocyte growth factor: a new approach to improve thymic and peripheral T-cell reconstitution after bone marrow transplantation. Blood. 2002;99:4592–4600. doi: 10.1182/blood.v99.12.4592. [DOI] [PubMed] [Google Scholar]
- 76.Sokolov VV, Kaplunova OA, Ovseenko TE. Age factors in architectonics of the splenic arterial vessels. Morfologiia. 2003;124(4):57–60. [PubMed] [Google Scholar]
- 77.Cheung HT, Nadakavukaren MJ. Age-dependent changes in the cellularity and ultrastructure of the spleen of Fischer F344 rats. Mech Ageing Dev. 1983;22(1):23–33. doi: 10.1016/0047-6374(83)90004-0. [DOI] [PubMed] [Google Scholar]
- 78.Pahlavani MA, Richardson A, Cheung HT. Age-dependent changes of the mesenteric lymph node of Fischer F344 rats: morphological and histometric analysis. Mech Ageing Dev. 1987;39(2):137–146. doi: 10.1016/0047-6374(87)90005-4. [DOI] [PubMed] [Google Scholar]
- 79.Luscieti P, Hubschmid T, Cottier H, Hess MW, Sobin LH. Human lymph node morphology as a function of age and site. J Clin Pathol. 1980;33(5):454–461. doi: 10.1136/jcp.33.5.454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Connoy AC, Trader M, High KP. Age-related changes in cell surface and senescence markers in the spleen of DBA/2 mice: a flow cytometric analysis. Exp Gerontol. 2006;41(2):225–229. doi: 10.1016/j.exger.2005.11.003. [DOI] [PubMed] [Google Scholar]
- 81.Lazuardi L, Jenewein B, Wolf AM, Pfister G, Tzankov A, Grubeck-Loebenstein B. Age-related loss of naïve T cells and dysregulation of T cell/B cell interactions in human lymph nodes. Immunology. 2005;114(1):37–43. doi: 10.1111/j.1365-2567.2004.02006.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Weksler ME, Szabo P. The effect of age on the B cell repertoire. J Clin Immunol. 2000;20(4):240–249. doi: 10.1023/a:1006659401385. [DOI] [PubMed] [Google Scholar]
- 83.Miller RA. The aging immune system: primer and prospectus. Science. 1996;273(5271):70–74. doi: 10.1126/science.273.5271.70. [DOI] [PubMed] [Google Scholar]
- 84.Yang X, Stedra J, Cerny J. Relative contribution of T and B cells to hypermutation and selection of the antibody repertoire in germinal centers of aged mice. J Exp Med. 1996;183(3):959–970. doi: 10.1084/jem.183.3.959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Whisler RL, Williams JW, Jr, Newhouse YG. Human B cell proliferative responses during aging. Reduced RNA synthesis and DNA replication after signal transduction by surface immunoglobulins compared to B cell antigenic determinants CD20 and CD40. Mech Ageing Dev. 1991;61(2):209–222. doi: 10.1016/0047-6374(91)90018-u. [DOI] [PubMed] [Google Scholar]
- 86.Zheng B, Han S, Takahashi Y, Kelsoe G. Immunosenescence and germinal center reaction. Immunol Rev. 1997;160:63–77. doi: 10.1111/j.1600-065x.1997.tb01028.x. [DOI] [PubMed] [Google Scholar]
- 87.Whisler RL, Grants IS. Age-related alterations in the activation and expression of phosphotyrosine kinases and protein kinase C (PKC) among human B cells. Mech Ageing Dev. 1993;71(1–2):31–46. doi: 10.1016/0047-6374(93)90033-n. [DOI] [PubMed] [Google Scholar]
- 88.Sato H, Dobashi M. The distribution, immune complex trapping ability and morphology of follicular dendritic cells in popliteal lymph nodes of aged rats. Histol Histopathol. 1998;13(1):99–108. doi: 10.14670/HH-13.99. [DOI] [PubMed] [Google Scholar]
- 89.Haynes L, Eaton SM, Burns EM, Randall TD, Swain SL. Newly generated CD4 T cells in aged animals do not exhibit age-related defects in response to antigen. J Exp Med. 2005;201(6):845–851. doi: 10.1084/jem.20041933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Haynes L, Eaton SM, Burns EM, Randall TD, Swain SL. CD4 T cell memory derived from young naïve cells functions well into old age, but memory generated from aged naïve cells functions poorly. Proc Natl Acad Sci USA. 2003;100(25):15053–15058. doi: 10.1073/pnas.2433717100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Eaton SM, Burns EM, Kusser K, Randall TD, Haynes L. Age-related defects in CD4 T cell cognate helper function lead to reductions in humoral responses. J Exp Med. 2004;200(12):1613–1622. doi: 10.1084/jem.20041395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Foy TM, Aruffo A, Bajorath J, Buhlmann JE, Noelle RJ. Immune regulation by CD40 and its ligand GP39. Annu Rev Immunol. 1996;14:591–617. doi: 10.1146/annurev.immunol.14.1.591. [DOI] [PubMed] [Google Scholar]
- 93.Song H, Price PW, Cerny J. Age-related changes in antibody repertoire: contribution from T cells. Immunol Rev. 1997;160:55–62. doi: 10.1111/j.1600-065x.1997.tb01027.x. [DOI] [PubMed] [Google Scholar]
- 94.Miller RA, Garcia G, Kirk CJ, Witkowski JM. Early activation defects in T lymphocytes from aged mice. Immunol Rev. 1997;160:79–90. doi: 10.1111/j.1600-065x.1997.tb01029.x. [DOI] [PubMed] [Google Scholar]
- 95.Nel AE, Slaughter N. T cell activation through the antigen receptor. Part 2: role of signaling cascades in T cell differentiation, anergy, immune senescence, and development of immunotherapy. J Allergy Clin Immunol. 2002;109(6):901–915. doi: 10.1067/mai.2002.124965. [DOI] [PubMed] [Google Scholar]
- 96.Effros RB, Walford RL. The immune response of aged mice to influenza: diminished T cell proliferation, interleukin 2 production and cytotoxicity. Cell Immunol. 1983;81(2):298–305. doi: 10.1016/0008-8749(83)90237-x. [DOI] [PubMed] [Google Scholar]
- 97.Powers DC. Influenza A virus-specific cytotoxic T lymphocyte activity declines with advancing age. J Am Geriatr Soc. 1993;41(1):1–5. doi: 10.1111/j.1532-5415.1993.tb05938.x. see comment. [DOI] [PubMed] [Google Scholar]
- 98.Po JL, Gardner EM, Anaraki F, Katsikis PD, Murasko DM. Age-associated decrease in virus-specific CD8+ T lymphocytes during primary influenza infection. Mech Ageing Dev. 2002;123(8):1167–1181. doi: 10.1016/s0047-6374(02)00010-6. [DOI] [PubMed] [Google Scholar]
- 99.el Refaei M, Blank KJ, Murasko DM. Prolonged E55+ retrovirus expression in aged mice is associated with a decline in the antivirus immune response. Virology. 2001;290(2):281–289. doi: 10.1006/viro.2001.1128. [DOI] [PubMed] [Google Scholar]
- 100.Wing K, Suri-Payer E, Rudin A. CD4+ CD25+-regulatory T cells from mouse to man. Scand J Immunol. 2005;62(1):1–15. doi: 10.1111/j.1365-3083.2005.01634.x. [DOI] [PubMed] [Google Scholar]
- 101.Rouse BT, Sarangi PP, Suvas S. Regulatory T cells in virus infections. Immunol Rev. 2006;212:272–286. doi: 10.1111/j.0105-2896.2006.00412.x. [DOI] [PubMed] [Google Scholar]
- 102.Belkaid Y, Rouse BT. Natural regulatory T cells in infectious disease. Nat Immunol. 2005;6(4):353–360. doi: 10.1038/ni1181. [DOI] [PubMed] [Google Scholar]
- 103.Chatila TA. Role of regulatory T cells in human diseases. J Allergy Clin Immunol. 2005;116(5):949–959. doi: 10.1016/j.jaci.2005.08.047. quiz, 960. [DOI] [PubMed] [Google Scholar]
- 104.Toda A, Piccirillo CA. Development and function of naturally occurring CD4+ CD25+ regulatory T cells. J Leukoc Biol. 2006 doi: 10.1189/jlb.0206095. in press. [DOI] [PubMed] [Google Scholar]
- 105.Bennett CL, Christie J, Ramsdell F, Brunkow ME, Ferguson PJ, Whitesell L, et al. The immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome (IPEX) is caused by mutations of FOXP3. Nat Genet. 2001;27(1):20–21. doi: 10.1038/83713. [DOI] [PubMed] [Google Scholar]
- 106.Kim JM, Rudensky A. The role of the transcription factor Foxp3 in the development of regulatory T cells. Immunol Rev. 2006;212:86–98. doi: 10.1111/j.0105-2896.2006.00426.x. [DOI] [PubMed] [Google Scholar]
- 107.Wing K, Ekmark A, Karlsson H, Rudin A, Suri-Payer E. Characterization of human CD25+ CD4+ T cells in thymus, cord and adult blood. Immunology. 2002;106(2):190–199. doi: 10.1046/j.1365-2567.2002.01412.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Dejaco C, Duftner C, Schirmer M. Are regulatory T cells linked with aging? Exp Gerontol. 2006;41(4):339–345. doi: 10.1016/j.exger.2006.01.008. [DOI] [PubMed] [Google Scholar]
- 109.van Amelsfort JM, Jacobs KM, Bijlsma JW, Lafeber FP, Taams LS. CD4(+) CD25(+) regulatory T cells in rheumatoid arthritis: differences in the presence, phenotype, and function between peripheral blood and synovial fluid. Arthritis Rheum. 2004;50(9):2775–2785. doi: 10.1002/art.20499. [DOI] [PubMed] [Google Scholar]
- 110.Gottenberg JE, Lavie F, Abbed K, Gasnault J, Le Nevot E, Delfraissy JF, et al. CD4 CD25high regulatory T cells are not impaired in patients with primary Sjogren's syndrome. J Autoimmun. 2005;24(3):235–242. doi: 10.1016/j.jaut.2005.01.015. [DOI] [PubMed] [Google Scholar]
- 111.Gregg R, Smith CM, Clark FJ, Dunnion D, Khan N, Chakraverty R, et al. The number of human peripheral blood CD4+ CD25high regulatory T cells increases with age. Clin Exp Immunol. 2005;140(3):540–546. doi: 10.1111/j.1365-2249.2005.02798.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Trzonkowski P, Szmit E, Mysliwska J, Mysliwski A. CD4+ CD25+ T regulatory cells inhibit cytotoxic activity of CTL and NK cells in humans — impact of immunosenescence. Clin Immunol. 2006;119(3):307–316. doi: 10.1016/j.clim.2006.02.002. [DOI] [PubMed] [Google Scholar]
- 113.Nishioka T, Shimizu J, Iida R, Yamazaki S, Sakaguchi S. CD4+ CD25+ Foxp3+ T cells and CD4+ CD25− Foxp3+ T cells in aged mice. J Immunol. 2006;176(11):6586–6593. doi: 10.4049/jimmunol.176.11.6586. [DOI] [PubMed] [Google Scholar]
- 114.Tsaknaridis L, Spencer L, Culbertson N, Hicks K, LaTocha D, Chou YK, et al. Functional assay for human CD4+ CD25+ Treg cells reveals an age-dependent loss of suppressive activity. J Neurosci Res. 2003;74(2):296–308. doi: 10.1002/jnr.10766. [DOI] [PubMed] [Google Scholar]
- 115.Liu W, Putnam AL, Xu-Yu Z, Szot GL, Lee MR, Zhu S, et al. CD127 expression inversely correlates with FoxP3 and suppressive function of human CD4+ T reg cells. J Exp Med. 2006;203(7):1701–1711. doi: 10.1084/jem.20060772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Seddiki N, Santner-Nanan B, Martinson J, Zaunders J, Sasson S, Landay A, et al. Expression of interleukin (IL)-2 and IL-7 receptors discriminates between human regulatory and activated T cells. J Exp Med. 2006;203(7):1693–700. doi: 10.1084/jem.20060468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Gann PH. Risk factors for prostate cancer. Rev Urol. 2002;4 suppl 5:S3–10. [PMC free article] [PubMed] [Google Scholar]
- 118.Lowenfels AB, Maisonneuve P. Epidemiology and risk factors for pancreatic cancer. Best Pract Res Clin Gastroenterol. 2006;20(2):197–209. doi: 10.1016/j.bpg.2005.10.001. [DOI] [PubMed] [Google Scholar]
- 119.Rosenkranz KM, Bedrosian I, Feng L, Hunt KK, Hartman K, Lucci A, et al. Breast cancer in the very elderly: treatment patterns and complications in a tertiary cancer center. Am J Surg. 2006;192(4):541–544. doi: 10.1016/j.amjsurg.2006.06.019. [DOI] [PubMed] [Google Scholar]
- 120.Yancik R. Cancer burden in the aged: an epidemiologic and demographic overview. Cancer. 1997;80(7):1273–1283. [PubMed] [Google Scholar]
- 121.Caraco C, Marone U, Botti G, Celentano E, Lastoria S, Mozzillo N. Age as predictor in patients with cutaneous melanoma submitted to sentinel lymph node biopsy. Eur J Surg Oncol. 2006 doi: 10.1016/j.ejso.2006.07.007. in press. [DOI] [PubMed] [Google Scholar]
- 122.Lustgarten J, Dominguez AL, Thoman M. Aged mice develop protective antitumor immune responses with appropriate co-stimulation. J Immunol. 2004;173(7):4510–5451. doi: 10.4049/jimmunol.173.7.4510. [DOI] [PubMed] [Google Scholar]
- 123.Murata S, Ladle BH, Kim PS, Lutz ER, Wolpoe ME, Ivie SE, et al. OX40 co-stimulation synergizes with GM-CSF whole-cell vaccination to overcome established CD8+ T cell tolerance to an endogenous tumor antigen. J Immunol. 2006;176(2):974–983. doi: 10.4049/jimmunol.176.2.974. [DOI] [PubMed] [Google Scholar]
- 124.Sharma S, Dominguez AL, Lustgarten J. Aging affect the antitumor potential of dendritic cell vaccination, but it can be overcome by co-stimulation with anti-OX40 or anti-4–1BB. Exp Gerontol. 2006;41(1):78–84. doi: 10.1016/j.exger.2005.10.002. [DOI] [PubMed] [Google Scholar]
- 125.Bridges CB, Winquist AG, Fukuda K, Cox NJ, Singleton JA, Strikas RA. Prevention and control of influenza: recommendations of the Advisory Committee on Immunization Practices (ACIP) Morb Mortal Wkly Rep Recomm Rep. 2000;49(RR3):1–38. [PubMed] [Google Scholar]
- 126.Rivetti D, Jefferson T, Thomas R, Rudin M, Rivetti A, Di Pietrantonj C, et al. Vaccines for preventing influenza in the elderly. Cochrane Database Syst Rev. 2006;3:CD004876. doi: 10.1002/14651858.CD004876.pub2. [DOI] [PubMed] [Google Scholar]
- 127.de Jong JC, Beyer WE, Palache AM, Rimmelzwaan GF, Osterhaus AD. Mismatch between the 1997/1998 influenza vaccine and the major epidemic A(H3N2) virus strain as the cause of an inadequate vaccine-induced antibody response to this strain in the elderly. J Med Virol. 2000;61(1):94–99. [PubMed] [Google Scholar]
- 128.Jefferson T, Rivetti D, Rivetti A, Rudin M, Di Pietrantonj C, Demicheli V. Efficacy and effectiveness of influenza vaccines in elderly people: a systematic review. Lancet. 2005;366(9492):1165–1174. doi: 10.1016/S0140-6736(05)67339-4. [DOI] [PubMed] [Google Scholar]
- 129.Jefferson T, Smith S, Demicheli V, Harnden A, Rivetti A, Di Pietrantonj C. Assessment of the efficacy and effectiveness of influenza vaccines in healthy children: systematic review. Lancet. 2005;365(9461):773–780. doi: 10.1016/S0140-6736(05)17984-7. [DOI] [PubMed] [Google Scholar]
- 130.Deng Y, Jing Y, Campbell AE, Gravenstein S. Age-related impaired type 1 T cell responses to influenza: reduced activation ex vivo, decreased expansion in CTL culture in vitro, and blunted response to influenza vaccination in vivo in the elderly. J Immunol. 2004;172(6):3437–3446. doi: 10.4049/jimmunol.172.6.3437. [DOI] [PubMed] [Google Scholar]
- 131.Swain GR, Ransom J. Universal influenza vaccination recommendations: local health department perspectives. J Publ Health Manag Pract. 2006;12(4):317–320. doi: 10.1097/00124784-200607000-00003. [DOI] [PubMed] [Google Scholar]
- 132.Update: influenza activity — United States. Morb Mortal Wkly Rep. 2005 2004–05 Season;54(13):328–331. [PubMed] [Google Scholar]
- 133.Schwartz B, Wortley P. Mass vaccination for annual and pandemic influenza. Curr Top Microbiol Immunol. 2006;304:131–152. doi: 10.1007/3-540-36583-4_8. [DOI] [PubMed] [Google Scholar]
- 134.Shoham D. Review: Molecular evolution and the feasibility of an avian influenza virus becoming a pandemic strain — a conceptual shift. Virus Genes. 2006;33(2):127–132. doi: 10.1007/s11262-005-0047-3. [DOI] [PubMed] [Google Scholar]