

Abstract
The population pharmacokinetics of tafenoquine were studied in Australian soldiers taking tafenoquine for malarial prophylaxis. The subjects (476 males and 14 females) received a loading dose of 200 mg tafenoquine base daily for 3 days, followed by a weekly dose of 200 mg tafenoquine for 6 months. Blood samples were collected from each subject after the last loading dose and then at weeks 4, 8, and 16. Plasma tafenoquine concentrations were determined by liquid chromatography-tandem mass spectrometry. Population modeling was performed with NONMEM, using a one-compartment model. Typical values of the first-order absorption rate constant (Ka), clearance (CL/F), and volume of distribution (V/F) were 0.243 h−1, 0.056 liters/h/kg, and 23.7 liters/kg, respectively. The intersubject variability (coefficient of variation) in CL/F and V/F was 18% and 22%, respectively. The interoccasion variability in CL/F was 18%, and the mean elimination half-life was 12.7 days. A positive linear association between weight and both CL/F and V/F was found, but this had insufficient impact to warrant dosage adjustments. Model robustness was assessed by a nonparametric bootstrap (200 samples). A degenerate visual predictive check indicated that the raw data mirrored the postdose concentration-time profiles simulated (n = 1,000) from the final model. Individual pharmacokinetic estimates for tafenoquine did not predict the prophylactic outcome with the drug for four subjects who relapsed with Plasmodium vivax malaria, as they had similar pharmacokinetics to those who were free of malaria infection. No obvious pattern existed between the plasma tafenoquine concentration and the pharmacokinetic parameter values for subjects with and without drug-associated moderate or severe adverse events. This validated population pharmacokinetic model satisfactorily describes the disposition and variability of tafenoquine used for long-term malaria prophylaxis in a large cohort of soldiers on military deployment.
Tafenoquine, a synthetic analog of primaquine, is a new 8-aminoquinoline antimalarial drug being codeveloped by GlaxoSmithKline Pharmaceuticals and the Walter Reed Army Institute of Research (1). Clinical trials have shown tafenoquine to be an effective antimalarial agent that has been generally well tolerated, with transient gastrointestinal discomfort being the most commonly reported adverse event (8, 10, 11, 13, 15). To date, it has been evaluated in more than 2,000 subjects in six phase II clinical studies. Since tafenoquine acts on all malaria stages, it has potential in the chemoprophylaxis of malaria, in radical cure/relapse prevention of Plasmodium vivax infections, and as a transmission-blocking agent (gametocytocidal activity).
The pharmacokinetics of tafenoquine in humans have been derived from studies after oral administration, as no parenteral formulation exists. Tafenoquine is slowly absorbed following oral administration, with maximum plasma concentrations observed at about 12-h postdose in fasted subjects (1). Plasma tafenoquine concentration-time data have been described by a one-compartment model with first-order absorption and elimination (1, 2). The elimination half-life of tafenoquine is about 2 weeks. It is extensively distributed to tissues, with a large volume of distribution and a low clearance, but data on the metabolism of tafenoquine in humans are limited. Although animal studies have shown that absorbed tafenoquine secreted via the bile is found predominantly in the form of metabolites, which accounted for the majority of the drug-related material eliminated in the urine and feces, unchanged tafenoquine was the only drug-related component detected in human plasma by high-performance liquid chromatography-mass spectrometry (HPLC-MS) and HPLC with fluorescence detection (GlaxoSmithKline Pharmaceuticals, unpublished data).
Tafenoquine is highly effective in preventing malaria infections following a weekly dose of either 200 mg or 400 mg for 13 weeks (13) or 400 mg monthly for 6 months (15). In developing the dosage regimen for malaria prophylaxis, a phase III study was conducted to assess the safety, tolerability, and effectiveness of tafenoquine in Australian soldiers deployed for 6 months on peacekeeping duties to an area where malaria is endemic. The full clinical results of that study will be published elsewhere. The soldiers were on a weekly regimen of 200 mg of tafenoquine, and blood samples were collected on four occasions for drug analysis. No malaria infections occurred during the prophylactic phase, but four soldiers were diagnosed with P. vivax infection after returning to Australia.
The primary aim of the present study was to use these data to develop a population pharmacokinetic model for tafenoquine and to estimate the disposition of this drug in the target population of soldiers on military deployment. Secondary aims were to determine whether individual pharmacokinetic estimates for tafenoquine would predict prophylactic outcomes and to investigate if there was any relationship between tafenoquine concentrations and drug-associated adverse events.
MATERIALS AND METHODS
Study design and subjects.
The clinical trial was designed as a prospective, randomized, double-blind comparative study of the safety, tolerability, and effectiveness of tafenoquine and mefloquine in Australian soldiers on weekly malaria prophylaxis. The subjects were deployed on peacekeeping duties to East Timor for 6 months. They all were judged to be healthy by a complete medical history, physical examination, and normal hematological and biochemical values. They had to be glucose-6-phosphate dehydrogenase normal and willing and able to give written informed consent and comply with the study protocol. Females were excluded if they were pregnant, lactating, or unwilling/unable to comply with recognized contraceptive methods. The study protocol received prior written approval by the Australian Defence Human Research Ethics Committee and the U.S. Army Human Subject Research Review Board.
Tafenoquine dosing regimen.
Following a loading dose regimen of 200 mg tafenoquine base daily for three consecutive days, the subjects then received an oral weekly maintenance dose of 200 mg tafenoquine over approximately 6 months. An opaque Swedish Orange size 1 hard gelatin capsule (Capsugel) containing tafenoquine at 200 mg (pure free base) was used as the dosage form. Subjects were directed to take their tafenoquine with food (breakfast or dinner) at the same time each week. Dosage administration was observed and recorded for each subject.
Pharmacokinetic sampling.
The sampling design was guided by the results from a previous smaller study of Thai soldiers (2) and also by logistical issues of the field operations. Blood samples were collected at prerandomized times after the last loading dose and then at prerandomized times at weeks 4, 8, and 16. Samples were collected on predetermined days after dosing on each of the assessment weeks. The predetermined days included day 1 (early postdose; absorption phase), days 3 and 5 (72 to 120 h postdose), and day 7 (predose; trough phase). For example, on week 4, one group of soldiers (about 125 subjects) was bled on day 1, one group was bled on day 3, one group was bled on day 5, and one group was bled on day 7. Thereafter, the groups of soldiers were bled in a cyclical fashion such that at the end of the study each group had been bled on at least one occasion on days 1, 3, 5, and 7. However, the sample for day 2 of the study (1 to 12 h; post-final loading dose) was collected from the study subjects.
Blood (7 ml) was drawn by venipuncture into EDTA tubes and transported on ice bricks to the field laboratory within 3 h of collection. Whole-blood samples were centrifuged at ∼1,200 × g for 15 min (Sigma, Quantum, Australia), and plasmas were separated and stored in liquid nitrogen (<4 weeks) and then air freighted on dry ice to Quintiles Limited (Edinburgh, United Kingdom) for storage at −70°C until analysis. Tafenoquine was stable under these handling and storage conditions.
Measurement of tafenoquine.
Plasma tafenoquine concentrations were determined using a validated HPLC method with a triple-quadrupole mass spectrometer. Briefly, plasma (0.05 ml) was spiked with [2H415N]tafenoquine as a stable-isotope-labeled internal standard, and the protein was precipitated with methanol, followed by centrifugation and then injection of 4 μl of the supernatant fluid onto a reversed-phase HPLC column (4-μm-diameter particles; Genesis C18 column; 30 mm × 2.1-mm internal diameter) held at 40°C. The mobile phase was methanol-1 mM ammonium acetate buffer, pH 2.5 (70:30 [vol/vol]), pumped at 1 ml/min and split approximately 1 to 4 into the TurboIonSpray interface of a PE-Sciex API 3000 LC/MS/MS system (Applied Biosystems) operated in positive-ion multiple-reaction monitoring mode. A chromatographic cycle time of 1.3 min was used, with the peaks being eluted at 0.4 min. The multiple-reaction monitoring transitions monitored were 464 to 379 m/z for tafenoquine and 469 to 379 m/z for stable-isotope-labeled tafenoquine. Linear responses in analyte/internal standard peak area ratios were observed for tafenoquine concentrations ranging from 5 to 500 ng/ml, using a weighted (1/C2) linear regression. Results of a three-run validation gave an intra-assay imprecision (coefficient of variation [CV%]) of <5.8% and an interassay imprecision of <7.3%, with an inaccuracy of 1.5 to 4.4%. The lower limit of quantification of the method was 5 ng/ml.
Population pharmacokinetic modeling.
The population pharmacokinetics of tafenoquine were determined in double precision by using NONMEM (version 5, level 1.1; Globomax LLC, Hanover, MD) in conjunction with a G77 compiler. A one-compartment model with first-order absorption and elimination was fitted to the data, using first-order conditional estimation with interaction. An initial analysis was conducted by permitting NONMEM to estimate the base model parameters (i.e., no covariates). The influence of mean-centered continuous variables, i.e., age, current weight, and estimated creatinine clearance (CLCR [by the Cockcroft-Gault method]), and the categorical variables, i.e., sex or evidence of phospholipidosis, was assessed by adding these to the base model in turn and noting the change in the objective function value (OFV). The inclusion of a covariate improved the fit of the data to the model if there was a decrease in the OFV. The difference between a pair of OFV values when a covariate was included (full model) and then excluded (reduced model) was tested for significance (α = 0.01), using the chi-square statistic with 1 degree of freedom (χ21,0.01 = 6.6).
The interindividual variability (IIV) was modeled, assuming a log-normal distribution, as follows:
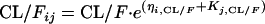 |
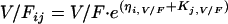 |
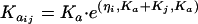 |
where CL/Fjj, V/Fij, and 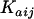 represent the true but unknown values of the parameters for the ith subject on the jth occasion about the typical respective population values CL/F, V/F, and Ka. The parameters ηi,CL/F, ηi,V/F, and
represent the true but unknown values of the parameters for the ith subject on the jth occasion about the typical respective population values CL/F, V/F, and Ka. The parameters ηi,CL/F, ηi,V/F, and 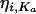 are random variables distributed with means of 0 and respective variances of ω2CL/F, ω2V/F, and
are random variables distributed with means of 0 and respective variances of ω2CL/F, ω2V/F, and 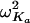 . K (kappa) is a random variable representing the variability of a given pharmacokinetic parameter value on different occasions, with an occasion being defined a priori as a dose or sequential doses followed by at least one observation (in this study, there were typically four occasions). The interoccasion variability (IOV) was assumed to be sampled from a normal distribution having a mean of 0 and a variance of π2. In modeling the IOV, it was assumed that the variances of each parameter were sampled from the same distribution. The residual unexplained variability (RUV) among observed plasma tafenoquine concentrations and those predicted by the final population model were estimated by a combined proportional plus additive error model, as follows: Cij = Cpred,ij(1 + ɛ1,ij) + ɛ2,ij, where Cij is the ith observed concentration in the jth subject, Cpred,ij is the plasma tafenoquine concentration predicted by the pharmacokinetic model, and ɛ1,ij and ɛ2,ij are randomly distributed variables having mean values of 0 and variances of σ12 and σ22, respectively.
. K (kappa) is a random variable representing the variability of a given pharmacokinetic parameter value on different occasions, with an occasion being defined a priori as a dose or sequential doses followed by at least one observation (in this study, there were typically four occasions). The interoccasion variability (IOV) was assumed to be sampled from a normal distribution having a mean of 0 and a variance of π2. In modeling the IOV, it was assumed that the variances of each parameter were sampled from the same distribution. The residual unexplained variability (RUV) among observed plasma tafenoquine concentrations and those predicted by the final population model were estimated by a combined proportional plus additive error model, as follows: Cij = Cpred,ij(1 + ɛ1,ij) + ɛ2,ij, where Cij is the ith observed concentration in the jth subject, Cpred,ij is the plasma tafenoquine concentration predicted by the pharmacokinetic model, and ɛ1,ij and ɛ2,ij are randomly distributed variables having mean values of 0 and variances of σ12 and σ22, respectively.
Model assessment.
The final model was assessed by an inspection of standard diagnostic plots of observed concentration versus population model predicted concentration and separate plots of weighted residual versus model-predicted concentration, elapsed time, subject identification, and screened covariates (3). A degenerate visual predictive check was performed by simulating from the final model 1,000 concentrations at each of 44 sampling times of up to 200 h postdose, at week 1 (after the third loading dose), and then at weeks 4, 8, and 16 during maintenance dosing. The 50th percentile concentration (as an estimator of the population-predicted concentration) and the 5th and 95th percentile concentrations were processed by ActivePerl (v.5.8.4; ActiveState) and then plotted against elapsed time for each of the above four sampling windows. Observed tafenoquine concentrations were superimposed on the plots. Model robustness was assessed by a nonparametric bootstrap, with replacement, of 200 NONMEM runs of the final model, comparing the bootstrapped median parameter values and the percentile bootstrap 90% confidence intervals (4, 5) with the respective values estimated in the final model.
Adverse events, severity rating, and association with drug.
As part of the clinical phase III trial, adverse events were elicited by an investigator asking the subject a nonleading question, such as “Do you feel differently in any way since starting the new treatment?” A physician assessed the level of relationship of any adverse event on the basis of the subject's response and any temporal association and/or known adverse responses to the drug. The physician graded the severity of adverse events as follows: mild, not affecting daily activities; moderate, causing some interference with daily activities; severe, daily duties could not be completed. Attribution or relationship to tafenoquine was judged by the physician to be not related, unlikely to be related, suspected (reasonable probability) to be related, or probably related.
RESULTS
Population characteristics.
The study population consisted of 476 males and 14 females, with a mean (± standard deviation [SD]) age of 25.4 ± 5.3 years (range, 18 to 47 years) and a mean (± SD) weight of 80.9 ± 11.9 kg (range, 50 to 135 kg). All but eight were of Caucasian background. Of the 490 subjects, 2 subjects provided one blood sample, 3 subjects provided two blood samples, 23 subjects provided three blood samples, and the remaining 462 subjects provided four blood samples, giving a total of 1,925 plasma concentration-time points available for the pharmacokinetic analyses.
Population pharmacokinetic modeling.
Summary results of the population model-building process are shown in Table 1. The data did not support the inclusion of an absorption lag time in any model. Neither age nor CLCR on CL/F significantly improved the fit, nor did sex or phospholipidosis as indicator variables. Both age and sex effects on V/F produced small but significant decreases in the OFV, of 9 and 12, respectively. Use of an allometric size model scaled to 70 kg for CL/F (power, 0.75) and V/F (power, 1.0) was not supported (OFV = +37). Inclusion of centered linear weight on both CL/F and V/F significantly decreased the OFV, from 22,177 to 22,138. This model predicted that a 1-kg change in weight from the population average value of 80.9 kg would give a commensurate change of 0.0167 liters/h (0.38%) in CL/F and a change of 9.7 liters (0.51%) in V/F. The linear, positive influence of weight on both CL/F and V/F is shown in Fig. 1a and b, respectively.
TABLE 1.
Development of structural model for pharmacokinetics of tafenoquine
| Model | Parameterizationd | ΔOFVa |
|---|---|---|
| 1 | CL/F = θ1; V/F = θ2; Ka = θ3 | |
| 2 | CL/F = θ1·(1 + θ4·age/25.4); V/F = θ2; Ka = θ3 | −2 |
| 3 | CL/F = θ1; V/F = θ2·(1 + θ4·age/25.4); Ka = θ3 | −9b |
| 4 | CL/F = θ1·(1 + θ4·CLCR/121); V/F = θ2; Ka = θ3 | −4 |
| 5 | CL/F = θ1·PHOS + θ4·(1 − PHOS); V/F = θ2; Ka = θ3 | 0 |
| 6 | CL/F = θ1; V/F = θ2·PHOS + θ4·(1 − PHOS); Ka = θ3 | −1b |
| 7 | CL/F = θ1·sex + θ4·(1 − sex); V/F = θ2; Ka = θ3 | −3b |
| 8 | CL/F = θ1; V/F = θ2·sex + θ4·(1 − sex); Ka = θ3 | −12 |
| 9c | CL/F = θ1·(1 + θ4·WT/80.9); V/F = θ2·(1 + θ5·WT/80.9); Ka = θ3 | −39 |
| 10 | CL/F = θ1·(WT/70)0.75; V/F = θ2·(WT/70)1.0; Ka = θ3 | +37b |
ΔOFV, change in OFV from that of model 1 (OFV = 22,177).
Rounding errors occurred during fitting.
Final model.
WT/80.9, body weight (kg) centered on average weight (80.9 kg); age/25.4, age (years) centered on average age (25.4 years); CLCR/121, CLCR (ml/min) centered on average CLCR (121 ml/min); PHOS, phospholipidosis (tested in 77 subjects; 1 = phospholipidosis present, 0 = phospholipidosis not present); sex, male = 0 and female = 1.
FIG. 1.

Relationship of body weight (WT) to individual estimates of (a) CL/F and (b) V/F for tafenoquine.
Modeling the covariance between ω2CL/F and ω2V/F reduced the OFV from 22,265 to 22,248 compared with the corresponding model when ω2CL/F and ω2V/F were assumed to be independent. Inclusion of the IOV for CL/F reduced the OFV further, to 22,177. However, while the addition of IOV to V/F further reduced the OFV, the value for ω2V/F was suspiciously low and the correlation coefficient (r) calculated from the diagonal and off-diagonal elements of the variance matrix [r = ω2CL/F,V/F/(ω2CL/F·ω2CL/F)0.5] was ∼1, indicating an inappropriate variance model. The RUV was best modeled by using a combined proportional and additive model, as seen by an increase in the OFV and by numerical difficulties when the additive and proportional models were used separately.
Parameter values for the final population model and the bootstrap validation are shown in Table 2. The estimated time (Tmax) for peak concentration to occur after a dose was 21.4 ± 8.57 h, calculated from each subject's conditional estimates of Ka and Ke by the standard formula Tmax = ln(Ka/Ke)/(Ka − Ke) for a one-compartment extravascular model. The observed mean (± SD) peak tafenoquine concentration measured in samples drawn within 5% of the time of the estimated mean population Tmax (21.4 h) for 42 subjects at weeks 4, 8, and 16 was 321 ± 63 ng/ml. The observed mean (± SD) trough tafenoquine concentration drawn within 5% of the target 168-h-postdose sampling time for 162 subjects at weeks 4, 8, and 16 was 221 ± 57 ng/ml. The typical population CL/F and V/F values for all subjects, with a mean weight of 80.9 kg, were 4.37 liters/h and 1,901 liters, respectively. The typical value of Ka over all subjects was 0.243 h−1. The IIV about CL/F, V/F, and Ka was 18%, 22%, and 76%, respectively. The IOV for CL/F was 18%. Mean values per kg for CL/F and V/F calculated from conditional estimates for each subject were 0.056 ± 0.013 liters/h/kg and 23.7 ± 4.5 liters/kg, respectively. The elimination half-life (t1/2), derived from the expression t1/2 = (0.693·V/F)/(CL/F) with individual estimates of CL/F and V/F, was 12.7 ± 3.0 days.
TABLE 2.
Comparison of parameter estimates for the population model with the results of 200 bootstrapped runs
| Parameter and model | Final model value | Bootstrap value (n = 200) (median [90% CI])b |
|---|---|---|
| Structural modela | ||
| CL/F (θ1; liters/h) | 3.02 | 3.01 (2.42-3.52) |
| V/F (θ2; liters) | 1,110 | 1,110 (874-1,382) |
| Ka (θ3; h−1) | 0.243 | 0.245 (0.212-0.280) |
| Weight centered on CL/Fc | 0.448 | 0.447 (0.249-0.816) |
| Weight centered on V/Fd | 0.713 | 0.713 (0.371-1.20) |
| Variance model | ||
| IIVCL/F (CV%) | 18 | 18 (16-20) |
| IIVV/F (CV%) | 22 | 22 (20-25) |
| IIVKa (CV%) | 76 | 75 (64-85) |
| IOVCL/F (CV%) | 18 | 18 (16-20) |
| RUV (CV%) | 5.9 | 5.9 (4.7-7.4) |
| RUV (ng/ml) | 22.9 | 23.1 (18.7-26.3) |
CL/F = θ1·(1 + θ4·WT/80.9); V/F = θ2·(1 + θ5·WT/80.9); Ka = θ3.
Percentile bootstrap 90% confidence interval (5th to 95th percentiles).
Linear coefficient (θ4) for weight centered on CL/F.
Linear coefficient (θ5) for weight centered on V/F.
Routine diagnostic weighted residuals versus population model-predicted values (data not shown) were symmetrically distributed and were mostly within about 3 units of the null ordinate, indicating a good fit of the model to the data. Plots of weighted residuals versus both subject identification and time (data not shown) were distributed symmetrically in a band with no obvious trend and were mostly within approximately 3 units of the null ordinate, indicating that no time-related factor affected the data and that no subject's data contributed to any marked deviation from the model. The bootstrapped median parameter values very closely agreed with the respective values from the final population model (Table 2). The degenerate visual predictive check showed the observed data to be symmetrically distributed about the 50th percentile profile, with approximately 10% of the data distributed outside the 5th- to 95th-percentile boundaries (Fig. 2a, b, c, and d).
FIG. 2.

Degenerate visual predictive check of the final population model for tafenoquine. Plots are shown for plasma tafenoquine concentration versus postdose time in sampling windows of (a) week 1 (post-loading dose), (b) week 4, (c) week 8, and (d) week 16. The population-predicted profile (50th percentile) is shown by the solid line, and the 90% prediction intervals estimated from 1,000 simulated concentrations over 200 h (postdose) are encompassed by the broken lines in each plot.
Individual pharmacokinetics of tafenoquine in subjects with malaria and with drug-associated adverse events.
The four subjects who had a relapse after returning to Australia had a mean (± SD) CL/F of 0.060 ± 0.014 liters/h/kg, a V/F of 23.2 ± 8.0 liters/kg, and a t1/2 of 11.1 ± 2.3 days, calculated from conditional parameter estimates for each individual.
One or more adverse events with a suspected/probable relationship to tafenoquine were reported by 73 subjects. These were ranked as mild in 67 subjects (91.8%), moderate in 5 subjects (6.8%), and severe in 1 subject (1.4%) and encompassed the following: nausea, abdominal pain, flatulence, vomiting, vertigo, agitation, amnesia, headache, eye abnormality, reflux, dreaming abnormality, insomnia, somnolence, diarrhea, hyperesthesia, tremor, paranoia, headache, anorexia, depression, coordination abnormality, appetite increase, and thirst. Tafenoquine was not withdrawn in any of the 67 mild cases, but it was withdrawn for three subjects who reported either moderate hyperesthesia, abdominal pain, or depression. Assessment for phospholipidosis was carried out in a subgroup of 77 subjects because tafenoquine has cationic amphiphilic characteristics and, therefore, the potential to cause phospholipid accumulation. Table 3 shows adverse events reported in the five moderate cases and one severe case where tafenoquine was suspected to cause the discomfort, together with individual estimates of the pharmacokinetic responses for these subjects. All moderate adverse events were experienced 1 to 24 days after the initiation of tafenoquine, while the single subject with severe effects reported diarrhea and abdominal pain 2 days after commencing tafenoquine treatment.
TABLE 3.
Tafenoquine pharmacokinetic data for six subjects reporting at least one adverse effect classified as severe (n = 1) or moderate (n = 5)
| Adverse event | Treatment duration (days)a | Cumulative dose (mg)b | Dosing stopped | Clast (ng/ml)c | CL/F (liters/h/kg) | V/F (liters/kg) | t½ (days) |
|---|---|---|---|---|---|---|---|
| Severe event | |||||||
| Diarrhea and/or abdominal pain | 2 | 400 | No | * | 0.059 | 24.4 | 12.0 |
| Moderate events | |||||||
| Insomnia | 1 | 200 | No | * | 0.059 | 23.2 | 11.3 |
| Hyperesthesia | 12 | 800 | Yes | 283 | 0.046 | 20.7 | 13.1 |
| Abdominal pain | 20 | 1,000 | Yes | 253 | 0.053 | 27.8 | 15.1 |
| Depression | 24 | 1,000 | Yes | 275 | 0.061 | 25.1 | 12.0 |
| Vomiting and/or nausea | 3 | 600 | No | 315 | 0.077 | 26.1 | 9.8 |
Number of days from starting dosing until adverse event reported.
Total amount of drug taken before adverse event reported.
Last plasma tafenoquine concentration before adverse event reported. *, adverse event was reported before first plasma sample was drawn.
DISCUSSION
This study of the population pharmacokinetics of tafenoquine in 490 Australian soldiers is the largest undertaken by far with this promising new oral antimalarial agent. Previously, a two-stage dose-ranging pharmacokinetic study was performed with 48 healthy adult males (Caucasian [n = 20], African-American [n = 12], and Hispanic [n = 16]) (1), while a subsequent population pharmacokinetic study was reported for 104 Thai soldiers on a monthly prophylactic regimen of tafenoquine (2). The present findings confirm the knowledge of tafenoquine disposition in humans and considerably extend the pharmacokinetic data to a large population of healthy, Caucasian military personnel deployed in field operations.
The apparent V/F was similar to that reported by Edstein et al. (2), but the systemic CL/F was greater (4.37 liters/h versus 3.20 liters/h). The derived typical elimination t1/2 of 12.7 days was slightly shorter than the 14 to 16 days reported previously, which may partly reflect the fact that the last samples were drawn at only up to 1 week postdose and therefore the presumed “terminal” phase may have included some components of a distribution phase, but not substantial enough to be supported by a two-compartment model. The mean values for CL/F and V/F obtained by Brueckner et al. (1) for fasted subjects of similar average weight to that from this study were 5.7 liters/h and 2,558 liters, respectively, which are 30% to 35% higher than the present typical values. However, in the current study, the subjects took tafenoquine with food, which reportedly can increase the bioavailability (F) by up to one-third (R. P. Brueckner, personal communication), which brings the respective CL/F and V/F values into closer agreement when corrected for F. While the extent of tafenoquine absorption may be greater, food could also slow the rate of drug absorption, as evidenced by the typical Ka of 0.243 h−1, compared with 0.391 h−1 and 0.694 h−1 reported by Brueckner et al. (1) and Edstein et al. (2), respectively. As a result, the average Tmax of 21.4 h was greater than the 8.6 h to 13.8 h reported previously (1, 2), which as well as the influence of food, may reflect continuous absorption along the intestinal tract, perhaps due in part to microprecipitation and redissolution of tafenoquine, which is only slightly water soluble (1). Unpublished data on file (GlaxoSmithKline) for healthy volunteers showed mean (CV%) Tmax values of 18.6 h (84%) and 26.3 h (126%) under fasted conditions and when administered with a standard high-fat meal, respectively, indicating that the Tmax and its variability were increased by food. Nonetheless, it should be remembered that Tmax is a model-dependent parameter in that the true value is likely to be overestimated when a one-compartment model is used compared with that for a two-compartment model. In agreement with previous reports (1, 2), there was marked IIV in the Tmax, reflecting the considerable variability in both Ka and Ke, with the latter being estimated from conditional estimates of V/F and CL/F for each subject.
The variability in CL/F and V/F was not excessive, at 18% to 22%, most likely reflecting the uniformity of the military subjects. The variance model supported estimation of the IOV in CL/F but not that in V/F or Ka. While Edstein et al. (2) used a proportional (exponential) model for RUV, presently a combined additive-proportional RUV model was supported, which is the preferred model wherever possible, especially where the range of concentration data is as wide as in this study. There was a positive linear association between weight and both CL/F and V/F, but attempts to model these parameters using an allometric size model scaled to 70 kg were not supported by the data, most likely because of the reasonably narrow range of body weights. Although heavier subjects tended to have a slightly greater CL/F and V/F, this would not have any major implications for changes in the way that tafenoquine would be prescribed, at least on the basis of the pharmacokinetic data alone. Using the present steady-state plasma tafenoquine concentrations as the appropriate clinical target, a 20-kg change in weight would require changes in the loading dose and maintenance dose of only about 10% and 7.5%, respectively. Unpublished data (GlaxoSmithKline) indicated that a considerable fraction of a tafenoquine dose may be excreted unchanged, while the clinical data from the trial of which the present study was a part showed that mean serum creatinine concentrations increased 12.1 mmol/liter from baseline until the end of the prophylaxis. However, estimated creatinine clearance explained an insignificant amount of the variability about CL/F. Age explained a small yet significant amount of the variability in both V/F and CL/F but was positively correlated with weight and thus was not considered further.
In assessing performance, model robustness was evaluated via a nonparametric bootstrap, which indicated that randomly selected combinations of data gave very similar results to those obtained with the original data set. In addition, a degenerate visual predictive check showed that the raw data obtained after the third split loading dose and at week 4, 8, and 16 during maintenance dosing mirrored the corresponding profiles obtained from simulations using point estimates of the final model parameter values. This convenient approach has been shown elsewhere (16) to give a good approximation of the full posterior predictive check, in which the simulations are performed using posterior distributions of the parameter values (6), which are difficult to calculate from the NONMEM output. The predictive check showed, firstly, that the structural model was satisfactory by the symmetrical distribution of the raw data about the 50th percentile profile and, secondly, that the variance model was appropriate, with about 10% of the raw data lying outside the 5th and 95th percentiles.
The prophylactic efficacy of tafenoquine is determined by its ability to prevent parasitemia from developing, which is associated with the susceptibility of malaria parasites to tafenoquine concentrations achieved in the target population. Tafenoquine has both causal prophylactic activity against the hepatic stages of the parasite and suppressive activity, which eradicates the erythrocytic stages of the parasite (1). In the present study, no subject developed parasitemia during the 6 months of prophylaxis, but four had a relapse of P. vivax infection after returning to Australia. In contrast, one subject in a population of 104 Thai soldiers on 400 mg tafenoquine monthly for 6 months developed vivax malaria during prophylaxis (15). At the time of diagnosis, the Thai soldier had a plasma tafenoquine concentration of 40 ng/ml, which was >5-fold lower than the mean steady-state trough tafenoquine concentration of 221 ng/ml presently recorded. Six Australian soldiers had tafenoquine concentrations of <100 ng/ml at either week 4, 8, or 16. Of those, only one subject had consistently lower tafenoquine concentrations (<120 ng/ml) on the three occasions sampled and therefore may have had a reduced margin of suppressive protection against malaria infection. The Thai soldier who developed parasitemia also had consistently lower tafenoquine concentrations during the prophylactic phase (15). Unlike the Thai soldier, the four Australian soldiers who relapsed had comparable tafenoquine concentrations to subjects who did not have a recurrence of malaria. Although the number of subjects who relapsed was small, the individual estimates of the pharmacokinetic responses for these subjects did not provide a prediction or correlation with tafenoquine's prophylactic efficacy.
There was no apparent correlation between either the pharmacokinetic parameter values predicted for individual subjects or the last tafenoquine concentration measured in subjects reporting moderate or severe adverse events. These findings suggested that plasma tafenoquine concentrations are not the primary predictor of tafenoquine tolerability. This lack of an association between plasma drug concentrations and adverse events has also been seen with another antimalarial agent, mefloquine, which shares similar pharmacokinetic properties with tafenoquine (12) in that both are lipophilic, are slowly absorbed from the gastrointestinal tract, are extensively bound to tissues, and have elimination t1/2 values of about 2 weeks (1, 2, 9, 14).
In conclusion, the pharmacokinetic properties of tafenoquine determined in this study support a weekly dosing regimen for prolonged periods. Although body weight influenced CL/F and V/F, it was not considered to have sufficient impact to warrant changing the maintenance or loading dose for any individual from such a population. Nonetheless, dose changes may be warranted for other patients who are markedly overweight or underweight compared with this homogenous group of soldiers. Any dosing requirements for markedly overweight subjects may need special consideration, as reviewed recently (7). Tafenoquine was generally well tolerated. Individual pharmacokinetic parameter estimates for subjects with malaria did not predict prophylactic outcomes, and plasma concentrations at steady state did not appear to be related to the occurrence of adverse events. Since this population was a homogenous group of healthy Australian soldiers of predominantly Caucasian background, additional pharmacokinetic studies may be required for other populations.
Acknowledgments
We thank all the Australian soldiers who participated in the phase III study for providing blood samples, the Australian Army Malaria Institute personnel who conducted the clinical trial, and Kym Ward for giving coordination and monitoring support. We are grateful to William Prescott (U.S. Army Medical Materiel Development Activity [USAMMDA]) and Philip Pickford (GlaxoSmithKline) for being the project managers. We acknowledge the support of Thomas Travers in data assessment. We also thank Jürgen Bulitta (IBMP, Nürnberg, Germany) for assistance with the ActivePerl script used for the visual predictive checks.
Financial support for the study was provided by USAMMDA, GlaxoSmithKline, and the Australian Defence Force.
The opinions expressed are those of the authors and do not necessarily reflect those of the Australian Defence Health Service or any extant Australian Defence Force policy.
Footnotes
Published ahead of print on 21 May 2007.
REFERENCES
- 1.Brueckner, R. P., K. C. Lasseter, E. T. Lin, and B. G. Schuster. 1998. First-time-in-humans safety and pharmacokinetics of WR 238605, a new antimalarial. Am. J. Trop. Med. Hyg. 58:645-649. [DOI] [PubMed] [Google Scholar]
- 2.Edstein, M. D., D. A. Kocisko, T. G. Brewer, D. S. Walsh, C. Eamsila, and B. G. Charles. 2001. Population pharmacokinetics of the new antimalarial agent tafenoquine in Thai soldiers. Br. J. Clin. Pharmacol. 52:663-670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ette, E. I., and T. M. Ludden. 1995. Population pharmacokinetic modelling: the importance of informative graphics. Pharm. Res. 12:1845-1855. [DOI] [PubMed] [Google Scholar]
- 4.Ette, E. I. 1997. Population modelling stability and performance. J. Clin. Pharmacol. 37:486-495. [DOI] [PubMed] [Google Scholar]
- 5.Ette, E. I., P. J. Williams, and J. R. Lane. 2004. Population pharmacokinetics. III. Design, analysis, and application of population pharmacokinetic studies. Ann. Pharmacother. 38:2136-2144. [DOI] [PubMed] [Google Scholar]
- 6.Gelman, A., X.-L. Meng, and H. Stern. 1996. Posterior predictive assessment of model fitness via realized discrepancies. Stat. Sinica 6:733-807. [Google Scholar]
- 7.Green, B., and S. B. Duffull. 2004. What is the best size descriptor to use for pharmacokinetic studies in the obese? Br. J. Clin. Pharmacol. 58:117-118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hale, B. R., S. Owusu-Agyei, D. J. Fryauff, K. A. Koram, M. Adjuik, A. R. Oduro, W. R. Prescott, J. K. Baird, F. Nkrumah, T. L. Ritchie, E. D. Franke, F. N. Binka, J. Horton, and S. L. Hoffman. 2003. A randomized, double-blind, placebo-controlled, dose-ranging trial of tafenoquine for weekly prophylaxis against Plasmodium falciparum. Clin. Infect. Dis. 36:541-549. [DOI] [PubMed] [Google Scholar]
- 9.Karbwang, J., and N. J. White. 1990. Clinical pharmacokinetics of mefloquine. Clin. Pharmacokinet. 19:264-279. [DOI] [PubMed] [Google Scholar]
- 10.Lell, B., J. F. Faucher, M. A. Missinou, S. Borrmann, O. Dangelmaier, J. Horton, and P. G. Kremsner. 2000. Malaria chemoprophylaxis with tafenoquine: a randomised study. Lancet 355:2041-2045. [DOI] [PubMed] [Google Scholar]
- 11.Nasveld, P. E., S. Kitchener, M. D. Edstein, and K. H. Rieckmann. 2002. Comparison of tafenoquine (WR238605) and primaquine in the post exposure prophylaxis of vivax malaria in Australian Defence Force personnel. Trans. R. Soc. Trop. Med. Hyg. 96:683-684. [DOI] [PubMed] [Google Scholar]
- 12.Schlagenhauf, P., R. Steffen, H. Lobel, R. Johnson, R. Letz, A. Tschopp, N. Vranjes, Y. Bergqvist, O. Ericsson, U. Hellgren, L. Rombo, S. Mannino, J. Handschin, and D. Sturchler. 1996. Mefloquine tolerability during chemoprophylaxis: focus on adverse event assessments, stereochemistry and compliance. Trop. Med. Int. Health 1:485-494. [DOI] [PubMed] [Google Scholar]
- 13.Shanks, G. D., A. J. Oloo, G. M. Aleman, C. Ohrt, F. W. Klotz, D. Braitman, J. Horton, and R. Brueckner. 2001. A new primaquine analogue, tafenoquine (WR 238605), for prophylaxis against Plasmodium falciparum malaria. Clin. Infect. Dis. 33:1968-1974. [DOI] [PubMed] [Google Scholar]
- 14.Simpson, J. A., R. Price, F. ter Kuile, P. Teja-Isavatharm, F. Nosten, T. Chongsuphajaisiddhi, S. Looareesuwan, L. Aarons, and N. J. White. 1999. Population pharmacokinetics of mefloquine in patients with acute falciparum malaria. Clin. Pharmacol. Ther. 66:472-484. [DOI] [PubMed] [Google Scholar]
- 15.Walsh, S. D., C. Eamsila, T. Sasiprapha, S. Sangkharomya, P. Khaewsathien, P. Supakalin, D. B. Tang, P. Jarasrumgsichol, C. Cherdchu, M. D. Edstein, K. H. Rieckmann, and T. G. Brewer. 2004. Efficacy of monthly tafenoquine for prophylaxis of Plasmodium vivax and multidrug-resistant P. falciparum malaria. J. Infect. Dis. 190:1456-1463. [DOI] [PubMed] [Google Scholar]
- 16.Yano, Y., S. L. Beal, and L. B. Sheiner. 2001. Evaluating pharmacokinetic/pharmacodynamic models using the posterior predictive check. J. Pharmacokinet. Pharmacodyn. 28:171-192. [DOI] [PubMed] [Google Scholar]