

Abstract
The present study describes a bacteriophage (MSa) active against Staphylococcus aureus, including methicillin-resistant staphylococcal strains. When inoculated into mice simultaneously with S. aureus A170 (108 CFU/mouse), phage (109 PFU) rescued 97% of the mice; when applied to nonlethal (5 × 106 CFU/mouse) 10-day infections, the phage also fully cleared the bacteria. The phage MSa, delivered inside macrophages by S. aureus, kills the intracellular staphylococci in vivo and in vitro. The phage can also prevent abscess formation and reduce the bacterial load and weight of abscesses. These results suggest a potential use of the phage for the control of both local and systemic human S. aureus infections.
Staphylococcus aureus is an extremely flexible organism: it can be a commensal but also a dangerous pathogen, causing skin abscesses, wound infections, endocarditis, osteomyelitis, pneumonia, and toxic shock syndrome (12, 20). S. aureus can also adapt to live inside cells, where it finds protection from host defense mechanisms and antibiotics (9). The number of staphylococcal infections continues to increase—in parallel with the increased use of intravascular devices (20)—while the treatment of these infections becomes ever more difficult because of the emergence of staphylococcal strains resistant to multiple antibiotics, including vancomycin (33, 34). In the United States and the United Kingdom, 40 to 60% of nosocomial S. aureus strains are multidrug resistant (19). The mortality rate due to methicillin-resistant versus methicillin-sensitive S. aureus infections is almost threefold higher (19). This context explains the urgency in developing new antibacterial agents. The present paper describes the isolation of a phage active against local and systemic infections of S. aureus. The phage is also active against intracellular staphylococci and methicillin-resistant staphylococcal strains. The lethal effect of bacteriophages on their bacterial hosts has been known since their discovery (25). However, the advent of antibiotics adversely affected the use of phages as antibacterial agents (25, 32). Rigorous studies conducted with animals (4, 25, 30) and the high incidence of antibiotic-resistant bacteria have resurrected phage therapy (25, 32).
MATERIALS AND METHODS
Bacteria.
The study included 20 S. aureus strains isolated from patients hospitalized at the Medical School of the University of Naples Federico II. Specimens were streaked on Baird-Parker agar base supplemented with egg yolk (Oxoid, Milan, Italy). Single colonies were amplified in Luria-Bertani (LB) broth (Oxoid). Strains were confirmed as S. aureus by the coagulase test, microscopic observation, and PCR assay of the S. aureus-specific gene clfA (21). For in vivo and in vitro experiments, bacteria were grown in LB medium (Difco, Becton Dickinson, Sparks, MD) at 37°C, harvested while in exponential growth phase (optical density at 600 nm, 1.5 to 1.8), centrifuged (8 × 103 × g for 10 min), washed with saline (0.15 M NaCl), and resuspended in saline (106 to 109 CFU/ml). The bacterial genes eta, etb, tst, lukS-PV-lukF-PV, lukE-lukD, lukM, and sea (coding for the exfoliative toxin A, exfoliative toxin B, toxic shock syndrome toxin 1, Panton-Valentine leukocidin components S and F, the leukotoxin LukE-LukD, the leukotoxin LukM, and enterotoxin A, respectively) were detected by PCR (2, 13, 24).
Phage isolation.
Bacteria were grown in LB broth. When cultures reached the exponential growth phase (optical density at 600 nm, 1.5 to 1.8), mitomycin C (final concentration, 1 μg/ml) was added to the cultures. Following incubation with mitomycin C for 30 min, bacteria were washed with LB broth and incubated again for 4 h at 37°C. The supernatants were filtered through a 0.45-μm membrane and screened for the presence of phages by the spot test (1). Supernatants positive in the spot test were tested again by a plaque-forming assay (1). Individual plaques were expanded in LB broth (2 ml) containing the sensitive bacterial host (106 CFU). Phage purification was carried out as described previously (29). Phage DNA was isolated as described previously (3). The phage genes eta, tst, lukSPV-lukFPV, and sea were detected by PCR (2, 13, 24).
Adsorption rate, latent period, and phage burst size.
The adopted procedures were those described previously (1). Briefly, to measure the adsorption rate, 1 ml phage MSa (1.5 × 103 PFU/ml) and 1 ml S. aureus A170 (5 × 108 CFU/ml) were mixed and the number of free phage particles was determined after treatment with chloroform (200 μl). To determine the latent period and burst size, S. aureus A170 bacteria (5 × 108 CFU/ml) were incubated with phage MSa (3 × 103 PFU/ml) for 5 min, washed with cold LB broth to remove free phage particles, and then resuspended in fresh medium. The cell suspension was periodically titrated for newly produced phage on an S. aureus A170 lawn.
Phage selection.
A phage strain able to persist in mouse blood was isolated as described previously (8). Briefly, four mice were injected intraperitoneally with phage WSa (108 PFU/mouse) and, at 12-h intervals, with four doses of the sensitive bacterial host S. aureus A170 (107 CFU/dose). Blood samples were collected periodically from the orbital plexuses of the mice. The phage mutant isolated following this procedure (MSa) persisted in the circulation for 21 days.
Mice.
Experiments were carried out on female BALB/c mice (aged 8 to 10 weeks) at the animal facility of the University of Naples. Phage (106 to 109 PFU in 200 μl saline) and bacteria (106 to 109 CFU in 200 μl saline) were inoculated by the route (intravenous or subcutaneous) indicated for each experiment. To induce abscesses, mice were inoculated subcutaneously on both flanks with 107 CFU S. aureus A170 suspended in 50 μl saline and, concurrently or 4 days later, with phage (109 PFU suspended in 50 μl saline). Organs (hearts, kidneys, and spleens) and abscesses were dissected and weighed. One gram of each sample was homogenized in 1 ml saline and serially diluted in distilled water. CFU were evaluated by plating each dilution on Baird-Parker agar plates, and PFU were evaluated by plating each dilution on a lawn of S. aureus A170. Animal experiments were approved by the Animal Care Committee of the University of Naples.
S. aureus A170 transformation.
The plasmid pSK236 was introduced into S. aureus A170 by electroporation. The plasmid contains the construct sarA-gfp-uvr, constitutively expressing green fluorescent protein (GFP) (16). Standard conditions for electroporation were as follows: 150 ng plasmid and 109 CFU bacteria in a 50-μl volume, 2.5 kV/cm, 800 Ω, and 25 μF. Following electroporation, the transformed bacteria (GFP-expressing S. aureus) were incubated for 90 min at 37°C in 1 ml brain heart infusion medium (Oxoid) supplemented with 0.3 M sucrose. Bacteria were then spread on brain heart infusion agar plates supplemented with 15 μg/ml chloramphenicol.
Intracellular killing activity of phage P1.
Peritoneal mouse macrophages were distributed in 24-well plates (105 cells/well), incubated overnight (37°C, 5% CO2) in Dulbecco's modified Eagle medium (DMEM) supplemented with 5% fetal calf serum (Sigma, Milan, Italy), and then infected with S. aureus A170 (104 bacteria/well). The plates were centrifuged (750 × g, 5 min) to facilitate cell contact and then incubated for 1 h at 37°C in 5% CO2. Extracellular bacteria were killed with gentamicin (12.5 μg/well for 1 h). Cells were washed with DMEM and incubated in DMEM containing 5% fetal calf serum at 37°C in 5% CO2. Following 3 h of incubation, cells were infected with phage MSa (5 × 106 PFU/well), incubated for another 45 h, and lysed with Tween 20 (final concentration, 0.03%) at different time points to recover intracellular bacteria. Each lysate was then serially diluted in saline and plated on Baird-Parker agar. Intracellular staphylococci recovered after 48 h of incubation were stained with 30 nM SYTO9 (Molecular Probes, Eugene, OR) and 15 μM propidium iodide (Molecular Probes) for 15 min in the dark and then analyzed by flow cytometry, using a Coulter Epics Elite flow cytometer (Coulter, Miami, FL) and a LIVE-DEAD assay kit (Molecular Probes).
Antibodies.
Antibodies against phage WSa (RαWSa) were raised in Fischer 344 rats. Each rat received two intraperitoneal injections (107 PFU in 500 μl phosphate-buffered saline [PBS] emulsified with an equal volume of complete Freund's adjuvant) at an interval of 2 weeks. The source of antibodies against phage MSa (MαMSa) was the serum of a mouse with MSa circulating in its blood for 20 days. Goat anti-rat immunoglobulin G (IgG) labeled with fluorescein isothiocyanate (GαRIgGFITC) and goat anti-mouse IgG labeled with fluorescein isothiocyanate (GαMIgGFITC) were purchased from Sigma. RαWSa and MαMSa were diluted 1:100. GαRIgGFITC and GαMIgGFITC were diluted 1:600. The volume of antibodies used in each assay was the same (100 μl/tube) for the four reagents.
Phage neutralization test.
Phage MSa (105 PFU suspended in 100 μl PBS) was incubated with MαMSa (diluted 4 × 10−2, 2 × 10−2, and 10−2 in PBS) for 3 h at room temperature. Bacteria (105 CFU in 1 ml tryptic soy broth) were added to the antibody-coated phage particles, and the mixture was incubated for 3 h at 37°C. CFU and PFU numbers were then counted.
Flow cytometry.
Latex particles (4 × 104/tube) with a diameter of 10 μm (Polysciences, Warrington, PA) were incubated overnight with phage MSa or WSa (106 PFU/tube) suspended in 1 ml of 0.1 borate buffer, pH 8.5. Particles were washed with PBS, quenched with 2% milk blocking solution (Kierkegaard & Perry Laboratory, Gaithersburg, MD), washed again with PBS, and incubated for 3 h with MαMSa or RαWSa. The latex particles were then washed and incubated (1 h) with GαMFITC or GαRFITC. Samples were analyzed on a Coulter Epics Elite flow cytometer. In the analysis, a gate was set around the latex particles on the basis of their forward and side scattering characteristics. Standard markers were set by testing a negative control (where the MαMSa or RαWSa reagent was replaced with PBS). For each sample, data for 3,000 events were analyzed.
Real-time reverse transcription-PCR (RT-PCR).
Total RNA was isolated from the mouse kidney by using Trizol reagent (Invitrogen, Milan, Italy) and was reverse transcribed using ImProm-II reverse transcriptase (Promega, Madison, WI) and oligo(dT)18 according to the manufacturer's protocol. Real-time PCR was performed on cDNA, using Brilliant SYBR green master mix (Stratagene, La Jolla, CA) and a Bio-Rad iCycler instrument (Bio-Rad, Hercules, CA). Reactions were performed in 25 μl in triplicate with the following thermal profile: 95°C for 10 min and 45 cycles of 15 s at 95°C and 45 s at 60°C. The PCR primers (500 nM each) were as follows: GAPDH forward, 5′ TTCACCACCATGGAGAAGGC 3′; GAPDH reverse, 5′ GGCATGGACTGTGGTCATGA 3′ (26); IL-6 forward, 5′ AAAGAGTTGTGCAATGGCAATT 3′; IL-6 reverse, 5′ CAGTTTGGTAGCATCCATCAT 3′; TNF-α forward, 5′ TCTCAGCCTCTTCTCATTCCT 3′; and TNF-α reverse, 5′ GTCTGGGCCATAGAACTGATG 3′. Specificity was verified by melting curve analysis and agarose gel electrophoresis. Relative expression levels were calculated by the comparative cycle threshold (CT) method.
Statistical analysis.
Survival rates of mice were analyzed using Fisher's exact test. Bacterial counts, abscess weights, and interleukin-6 (IL-6) levels were analyzed using Student's t test.
RESULTS
Phage isolation.
Following incubation with mitomycin (1 μg/ml for 30 min), the 20 strains of S. aureus included in this study yielded five phages. The phage displaying the largest host range (lysing 7 of the 19 strains tested) was isolated from S. aureus strain A171. This phage was designated WSa (“W” is for wild; “Sa” is for S. aureus) and further characterized with regard to the toxin genes sea, lukSPV-lukFPV (both present), eta, and tst (both absent).
Selection of a phage persisting in the mouse circulation.
One of the limitations of phage therapy is the rapid clearance of phages by the reticuloendothelial system (14, 25, 35). In vivo amplification of phages in the presence of the bacterial host has been shown to favor the emergence of rare phage mutants able to persist in the mouse circulation (8). Therefore, four mice were injected intraperitoneally with the wild phage WSa (108 PFU/mouse) and, at 12-h intervals, with four doses of the sensitive host S. aureus A170 (individual dose, 107 CFU/mouse). This bacterial strain was chosen for the in vivo experiments since it is positive for only two toxins (exfoliative toxin B and Panton-Valentine leukocidin). Preliminary experiments established that repeated doses of 107 CFU of S. aureus A170 given intraperitoneally did not harm the mice. The procedure allowed the isolation of a phage strain that persisted in the blood for 21 days. This phage strain was designated MSa (“M” is for mutant; “Sa” is for S. aureus) and expanded in vitro from a single plaque. Two mice were given 109 PFU of phage WSa intravenously, and two more mice were given 109 PFU of phage MSa. The phage particles present in the spleen, liver, and blood were counted periodically. While phage MSa remained in the circulation for 20 to 25 days, phage WSa was almost completely trapped inside the spleen and liver within 2 days. The experiment demonstrates that the capacity acquired by phage MSa to persist in the mouse circulation is stable. The mice maintained phage MSa in vivo without displaying visible long-term negative effects. Phage MSa was further characterized with regard to the adsorption rate (1.89 × 10−9 ml/min), latent period (30 min), and burst size (80 PFU).
Phages WSa and MSa are serologically distinct.
The two phages were tested with antibodies against phage WSa prepared in the rat (RαWSa) and with the antibodies present in the serum from one of the mice, which had phage MSa circulating in its blood for about 20 days (MαMSa). The rat antibodies reacted with both phages, but more strongly with phage MSa than with phage WSa, with the latter being the phage used for immunization. The mouse antibodies reacted with phage MSa only (Fig. 1). Presumably, phage MSa, driven by the immune system, lost one or more of the surface antigens present on the wild phage WSa, becoming antigenically distinguishable. Incubation of phage MSa with an excess of mouse anti-MSa antibodies did not interfere with the phage's capacity to lyse S. aureus A170 cells (Table 1). This result supports the conclusion that the antibodies elicited by phage MSa in the mouse are nonneutralizing and explains the coexistence in the mouse circulation of phage MSa and corresponding antibodies. Phages can easily induce neutralizing antibodies (32). The characteristic of phage MSa of not inducing this class of antibodies is therefore biologically relevant and demonstrates how selection can improve the antimicrobial properties of phages.
FIG. 1.
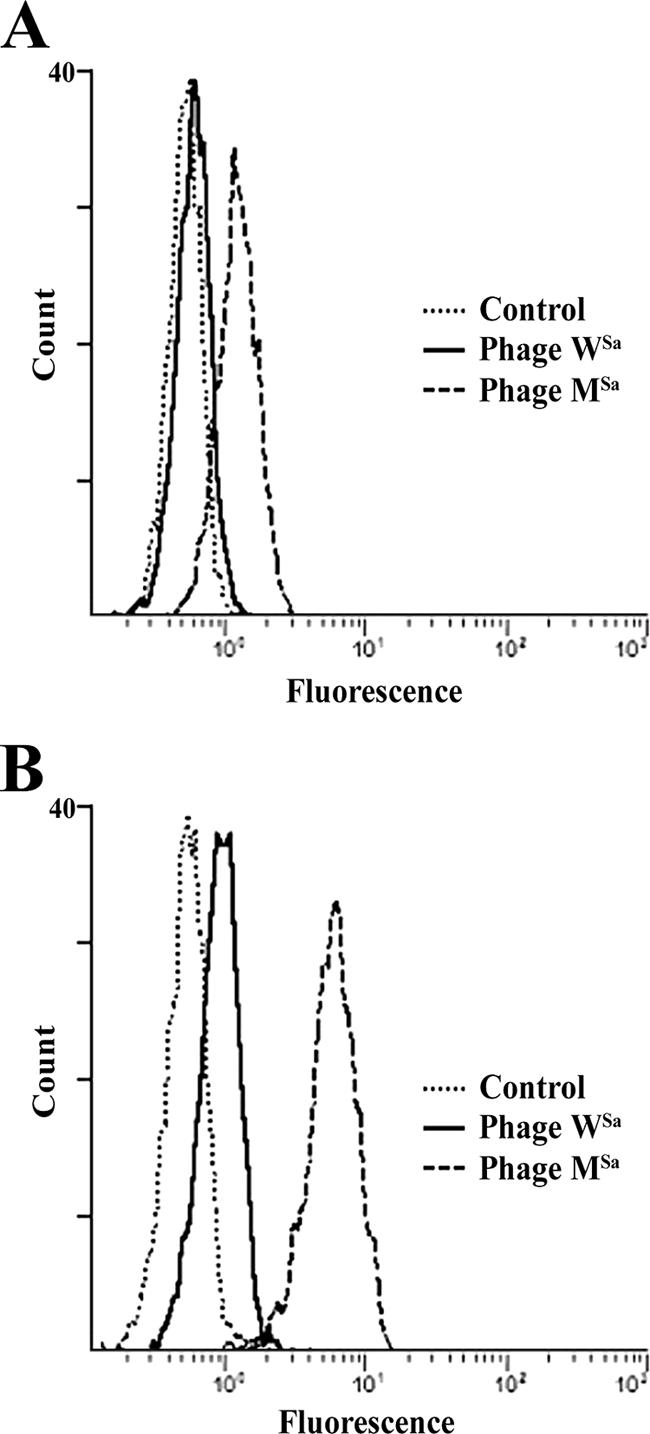
Phages WSa and MSa are serologically distinct. (A) MαMSa antibodies react with the mutant phage MSa but not with the wild phage WSa. The peak of phage WSa coincides with the peak of the negative control. (B) RαWSa antibodies react with the heterologous antigen (phage MSa) more strongly than with the homologous one (phage WSa).
TABLE 1.
Phage MSa is not neutralized by circulating mouse antibodies (MαMSa)a
| MαMSa dilution | Count (log CFU or PFU/ml)
|
|||
|---|---|---|---|---|
| Bacteria
|
Phage
|
|||
| t0 | t3 | t0 | t3 | |
| 4 × 10−2 | 4.95 ± 0.23 | 2.94 ± 0.31 | 5.12 ± 0.20 | 7.70 ± 0.16 |
| 2 × 10−2 | 5.1 ± 0.29 | 2.81 ± 0.19 | 5.23 ± 0.31 | 7.35 ± 0.15 |
| 1 × 10−2 | 4.92 ± 0.19 | 2.75 ± 0.23 | 5.21 ± 0.21 | 7.77 ± 0.23 |
Preincubation with antibodies did not inhibit the killing capacity of phage, as demonstrated by the decreasing number of CFU and the increasing number of PFU at 3 h.
Phage treatment of systemic infections.
S. aureus is the major cause of septicemia, invasive endocarditis, and septic arthritis in humans (12). To investigate whether phage MSa could protect mice from S. aureus A170 when the pathogen entered the bloodstream, four groups of mice (five mice per group) were injected intravenously with a dose of S. aureus A170 (108 CFU/mouse) which killed 90 to 100% of mice within 4 days. One group served as an untreated control, and the remaining three groups were given the phage MSa (107, 108, or 109 PFU/mouse) intravenously immediately after infection. All mice of the control group and the group treated with the lowest phage dose (107 PFU/mouse) died within 4 days (10/10 mice). The mice treated with the intermediate dose (108 PFU/mouse) were only partially protected (2/5 mice survived). The mice treated with the highest dose (109 PFU/mouse) were all protected from death (5/5 mice). Subsequent experiments were carried out using 108 CFU of S. aureus A170 and 109 PFU of phage MSa per mouse (standard schedule). Cumulative data from six independent experiments carried out according to the standard schedule showed that while 100% (30/30 mice) of the mice of the control group died within 4 days, only 3% (1/30 mice) of the treated mice died (P < 0.0001) (Fig. 2). The clinical advantage of phage treatment was also evident by quantifying the bacterial load 4 days after phage injection. Although all S. aureus A170-infected mice not given phage displayed a high bacterial load, no bacteria were isolated from phage-treated mice (Table 2 and Fig. 3).
FIG. 2.

Phage MSa given concurrently with S. aureus A170 rescues 93% (29/30 mice) of mice artificially infected with a lethal dose of the pathogen. (A) Mice infected with S. aureus A170 (108 CFU/mouse). (B) Mice infected with S. aureus (108 CFU/mouse) and treated immediately after with phage MSa (109 PFU/mouse).
TABLE 2.
Mice infected with S. aureus A170 (108 CFU/mouse) and treated concurrently with phage MSa (109 PFU/mouse) become sterile 4 days after phage treatment
| Mouse group | Bacterial count (mean log CFU/g ± SD)a
|
|||
|---|---|---|---|---|
| Kidney | Heart | Spleen | Lung | |
| Untreated | 7.06 ± 0.34 | 2.99 ± 0.18 | 3.70 ± 0.20 | 3.35 ± 0.19 |
| Treated | 0 | 0 | 0 | 0 |
Organs were homogenized in saline, diluted in distilled water, and plated.
FIG. 3.

In vivo bactericidal activity of phage MSa. (A) Fluorescence microscopy pictures of kidney cells recovered from mice 4 days after intravenous infection with 108 CFU of GFP-expressing S. aureus. (B) Kidney cells from mice infected concurrently with 108 CFU of GFP-expressing S. aureus and 109 PFU of phage MSa. Cells were counterstained with 4′,6′-diamino-2-phenylindole (DAPI). (Left) Cells analyzed with a 340- to 380-nm filter (DAPI). (Center) Cells analyzed with a 450- to 490-nm filter (GFP). (Right) Overlay. Magnification, ×1,000 (oil immersion).
The occurrence of a host threshold for reproduction is common among microbial predators (36). To establish the threshold density of S. aureus A170 required for the replication of phage MSa, mice were infected concurrently with bacteria and phage MSa according to the standard schedule (108 CFU and 109 PFU/mouse). Circulating bacterial and phage titers were measured daily for 20 days. The density of S. aureus A170 at which phage replication began was approximately 104 CFU/ml and was the same for the spleen, heart, and kidneys (Fig. 4). In the blood, S. aureus A170 did not reach this threshold (perhaps it was forced to move out of the bloodstream in search of refuge), and in the absence of adequate bacterial density, phage expansion did not occur (Fig. 4). The threshold host density observed in this study is surprisingly close to that reported for several gram-negative and gram-positive bacteria and lytic as well as lysogenic phages (36), providing further support to the suggestion that the threshold for replication might be similar for most phage-host combinations (36).
FIG. 4.

In vivo phage replication occurs when the bacterial density reaches a threshold of approximately 104 CFU/ml. Mice were treated concurrently with 108 CFU S. aureus A170 and 109 PFU phage MSa. The organs were dissected in saline, diluted in distilled water, and plated. In the bloodstream, bacteria did not reach the threshold for phage replication. The bacterial density thresholds were approximately the same for the various organs.
Phage treatment can drastically reduce inflammation caused by S. aureus infection.
To determine whether phage treatment, by killing bacteria, could modulate inflammation, the levels of the proinflammatory cytokines IL-6 and tumor necrosis factor alpha (TNF-α) were analyzed by real-time RT-PCR 24 h after intravenous infection of mice (3 mice/group) with MSa alone (109 PFU/mouse), S. aureus alone (108 CFU/mouse), or MSa (109 PFU/mouse) and S. aureus (108 CFU/mouse) concurrently. Three control mice provided the average basal levels of the cytokine genes. Analysis was carried out on kidney cells, which are heavily colonized by S. aureus (Table 2). MSa, given concurrently with S. aureus, reduced the IL-6 expression level induced by S. aureus infection about fivefold (P < 0.001) (Fig. 5). Twenty-four hours after infection, the TNF-α expression level did not vary significantly among experimental groups.
FIG. 5.
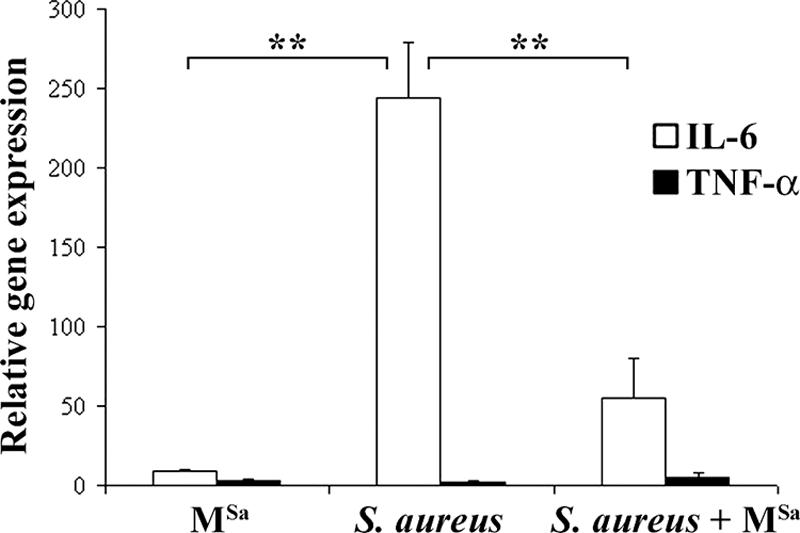
Expression levels of IL-6 and TNF-α genes in the kidneys of mice treated with phage MSa alone, S. aureus alone, or both. Expression levels were measured by real-time RT-PCR 24 h after treatment. Expression levels of the IL-6 and TNF-α genes are given relative to the average levels measured in three control mice. Results refer to three independent experiments, with each one carried out in triplicate. Significant results (at a P value of <0.001) are marked with asterisks.
Phage activity against methicillin-resistant S. aureus strains.
Given the high incidence of methicillin-resistant S. aureus strains (12, 19), it seemed worth investigating whether phage MSa was also active against this class of bacteria. Two groups of mice (10 mice/group) were infected intravenously with the methicillin-resistant A352 strain of S. aureus (108 CFU/mouse). One group of animals served as untreated controls; the second group was given the phage MSa (109 PFU/mouse) intravenously immediately after infection. The survival rate was 20% (2/10) for the control group and 100% (10/10) for the treated group (Fig. 6) (P < 0.0007). Thus, phage MSa represents an antimicrobial potentially effective against human infections with methicillin-resistant S. aureus.
FIG. 6.

Phage MSa is active against the methicillin-resistant strain S. aureus A352. (A) Mice infected with S. aureus A352 (108 CFU/mouse). (B) Mice infected with S. aureus A352 (108 CFU/mouse) and treated immediately after with phage MSa (109 PFU/mouse).
Phage treatment is also effective when phage are administered 10 days after bacterial infection.
To investigate the effects of delaying phage treatment until after S. aureus A170 exposure, mice were infected intravenously with the lowest dose of S. aureus A170 not cleared by innate immunity (5 × 106 CFU/mouse) and then divided into two groups of 10 animals each. One group was given phage MSa (109 PFU/mouse) 10 days after infection. The second group served as an untreated control. The reason for using a low infection dose was twofold, namely, to approximate human S. aureus infection under natural conditions, which is typically started by a small initial inoculum; and to avoid death by an overresponse of the immune system (17, 18) (rather than by the bacteria directly), which would obscure the real contribution of phage. Twenty days after infection with S. aureus (10 days after MSa treatment), the spleens, kidneys, hearts, and blood of the control mice (10/10 mice) were still infected; the same organs of the mice treated with phage (10/10 mice) were sterile (Fig. 7). A repeat experiment with 20 more animals (10 phage-treated mice and 10 control mice) confirmed the above results. The experiment demonstrates clearly that by using the proper infection model, phages can be shown to be highly effective, even when administered 10 days after bacterial infection.
FIG. 7.

Using mouse sterilization as a criterion for measuring phage activity, MSa was shown to be active when administered 10 days after infection. Mice received 5 × 106 CFU S. aureus A170 and, 10 days later, 109 PFU phage MSa. The experiment included 20 phage-treated and 20 untreated animals.
Phage treatment of local infections.
S. aureus accounts for a large proportion of the morbidity and mortality due to surgical wound infections (12, 15). Phage MSa was therefore tested for the capacity to control local infections. The first experiment was designed to test the efficacy of MSa given concurrently with S. aureus. Two groups of mice (5 mice/group) received either S. aureus alone (108 CFU/mouse; untreated group) or S. aureus (108 CFU/mouse) and, immediately after, phage MSa (109 PFU/mouse) (treated group) subcutaneously on both sides of the abdomen.
The second experiment was designed to test the efficacy of phage therapy once the abscesses became well established. Phage MSa (109 PFU/mouse) was therefore administered 4 days after mouse infection with S. aureus (108 CFU/mouse).
The third experiment was designed to test the efficacy of multiple doses of phage MSa. Mice therefore received four daily doses of phage MSa (individual dose, 109 PFU/mouse), starting 4 days after S. aureus infection (109 CFU/mouse).
The parameters used to evaluate the efficacy of phage therapy were the same for the three experiments and included prevention of abscess formation and reductions in the bacterial load (CFU/abscess) and the weight of abscesses. These parameters were evaluated 4 days after phage administration.
Figure 8 illustrates the results of the three experiments. Given concurrently with bacteria, phage MSa inhibited abscess development (P < 0.0001). Furthermore, one single dose or multiple doses of phage MSa given 4 days after bacterial administration did not influence the incidence of abscesses (as expected); however, the phage significantly reduced both the bacterial load and the weight of abscesses (P < 0.001). Multiple doses were more effective than single doses in reducing both the bacterial load and the weight of abscesses (P < 0.001). In view of the adverse conditions present in the abscesses, i.e., low pH, slow bacterial growth, and enzymes protecting bacteria (23, 31), the above results look encouraging.
FIG. 8.

Effect of phage MSa on abscess formation. (A) One single dose of phage MSa, given concurrently with S. aureus, inhibits abscess formation. (B) One single dose of phage MSa, given 4 days after S. aureus, cannot prevent abscess formation but significantly reduces the bacterial load and the weight of abscesses. (C) Multiple doses of phage MSa, given 4 days after S. aureus, are more effective than a single dose in reducing the bacterial load and the weight of abscesses. Significant results (at a P value of <0.001) are marked with asterisks.
Phage activity against intracellular S. aureus.
The data reported in Table 2 prove that 4 days after phage administration, mice are free of the bacteria used to infect them. In view of the evidence that S. aureus can adopt an intracellular lifestyle (9, 28), the complete eradication of infecting staphylococci supposes that phage MSa might kill both extracellular and intracellular staphylococci. To test this hypothesis, mouse peritoneal macrophages (105/well) were incubated in vitro with S. aureus A170 (104 CFU/well), and infection was allowed to occur for 1 h. The monolayer was then incubated with gentamicin for 1 h (to kill the extracellular bacteria), washed with DMEM (to remove the excess antibiotic), and further incubated for 3 h. Phage MSa (106 PFU/well), alone or adsorbed to S. aureus (104 CFU), was then added to the individual wells. The control wells were left without phage or phage-infected bacteria. Following incubation (24 to 48 h), macrophages were lysed and the number of surviving intracellular bacteria was determined by plating the lysate on Baird-Parker agar. The results (Fig. 9A) demonstrate that phage MSa alone cannot penetrate inside cells. Instead, when delivered inside cells by S. aureus, MSa efficiently killed intracellular bacteria at 24 and 48 h (Fig. 9A). Flow cytometry, by directly measuring the proportions of dead and live bacteria, confirmed the killing activity of phage MSa against intracellular bacteria. About 70% of the intracellular bacteria isolated from macrophages treated in vitro with phage-infected bacteria for 48 h were dead (Fig. 9B), while the intracellular bacteria isolated from macrophages treated with phage MSa alone were almost all alive (Fig. 9C). Thus, in vitro (Fig. 9) and in vivo (Table 2) results concur in demonstrating that phage MSa, once shuttled inside the host cells by S. aureus, also kills intracellular staphylococci.
FIG. 9.

MSa kills intracellular S. aureus A170. (A) Numbers of live bacteria recovered in the 0- to 48-h time interval from mouse peritoneal macrophages (105/well) infected with S. aureus A170 (104 CFU/well) and subsequently treated with phage MSa alone (▪), infected with S. aureus A170 preincubated with phage MSa (⧫), or left untreated (▴). (B) Flow cytometric analysis of dead and live intracellular staphylococci recovered from mouse peritoneal macrophages infected with S. aureus A170 and subsequently treated for 48 h with S. aureus A170 preincubated with phage MSa. (C) Flow cytometric analysis of dead and live intracellular staphylococci recovered from mouse peritoneal macrophages infected with S. aureus A170 and subsequently treated for 48 h with phage MSa. When used concurrently, propidium iodide stains dead bacteria and SYTO9 stains the bacteria which are alive.
Evolution of phage resistance.
A concern often expressed about phage therapy is that bacteria can become refractory to phages. This study provides data relevant to this point. Different organs (spleen, kidneys, and heart) and the blood of more than 50 mice infected with S. aureus A170 and treated with phage MSa concurrently (Fig. 4) or 10 days after infection (Fig. 7) were all found to be sterile when the animals were sacrificed at the end of the experiment. In vivo, phage-resistant staphylococci might be cleared rapidly by the immune system. The frequency of phage-resistant bacteria was therefore also measured in vitro. Five large bacterial cultures (108 CFU/ml) in rich (LB) medium were infected with phage WSa (109 PFU/ml) and incubated at 37°C overnight. The observed frequency of phage-resistant bacteria was 1.3 × 10−8 ± 4.16 × 10−9. The above results confirm previous studies (8, 11, 32) indicating that phage resistance is a rare event, perhaps rarer than antibiotic resistance (11, 32).
DISCUSSION
Treatment of staphylococcal infections with antibiotics is becoming increasingly difficult in view of the widespread presence of S. aureus strains resistant to multiple antibiotics. The present study highlights phage therapy as a possible solution. Given concurrently with a lethal dose of S. aureus A170, phage MSa rescued 97% of mice and completely eradicated bacteria in vivo within 4 days of phage treatment (Table 2 and Fig. 3). The phage was active against systemic (Fig. 2), local (Fig. 8), and intracellular (Fig. 9) bacterial infections. Most importantly, the phage lysed methicillin-resistant staphylococci (Fig. 6). Mortality is certainly an important criterion to judge the efficacy of a treatment, yet the present study also provides additional evidence, as follows: phage MSa was well tolerated by the animals, it drastically reduced inflammation (Fig. 5), and it did not stimulate the production of neutralizing antibodies. In all experiments, bacterial rebound or adverse effects due to rapid bacterial lysis (10) were not observed. Phage-treated mice, in fact, remained healthy 20 days after infection (Fig. 2), when the experiment was ended. The above results become more convincing when examined in the context of numerous reports documenting phage efficacy in vivo against several bacterial species (4, 28), including S. aureus (22, 37).
We also attempted to test the efficacy of phage MSa when bacterial infection was already established. To demonstrate that phage can be effective several days after experimental infection, we shifted the criterion for measuring phage efficacy from the survival rate for animals treated with a lethal bacterial dose to the sterilization rate for animals infected with a low bacterial dose. In this regard, our study differs significantly from previous ones on phage therapy. We infected mice with the smallest dose of S. aureus A170 not cleared by the innate immunity of the mouse (5 × 106 CFU/mouse) and then determined how many days after bacterial exposure phage therapy was successful. By these means, we mimicked human staphylococcal infection more closely and, at the same time, avoided sepsis, which could mask phage therapy effectiveness. Given 10 days after artificial infection, phage MSa sterilized the totality (20/20 mice) of the phage-treated mice, while the control mice (20/20 mice) were still infected (Fig. 7). The above experiment clearly demonstrates that phages are not temporally constrained in their ability to kill bacteria. This is perhaps the most significant contribution of this article to the application of phage therapy in a clinical context. Also, it supports earlier observations on the limits of some experimental infection designs and the opportunity to exploit long-term infections for treatment with phages (6).
In the case of systemic infections, just one single phage injection was sufficient for effective recovery. In the case of abscesses, clearance of bacteria required repeated doses. Both outcomes can be interpreted as direct consequences of the phage property of replicating only when the bacterial density is above a threshold. This threshold was reached in the course of systemic infections (Fig. 4), but presumably not in abscesses, which contain large numbers of slowly growing or static bacteria (31). Thus, systemic infections offer an example of active phage therapy (with in vivo phage multiplication), and abscesses give an example of passive therapy (without in vivo phage multiplication) (27). Clear cases of both active and passive therapy have been found (27).
Although traditionally considered an extracellular pathogen, S. aureus can internalize and survive inside mouse macrophages infected in vivo (Fig. 9). Phage MSa added alone to the infected macrophages did not reduce the number of intracellular bacteria (Fig. 9A) (very likely because phage particles could not penetrate inside the cells). However, S. aureus previously infected with MSa particles significantly reduced the number of intracellular bacteria (Fig. 9A). Flow cytometry confirmed this result (Fig. 9B and C). The simplest explanation for these results is that MSa is delivered to bacteria residing within macrophages by phage-infected staphylococci. If we assume that the same phenomenon also takes place in vivo (if we assume that MSa infects the extracellular bacteria and that these, in turn, carry phage particles inside the cells), we can understand how 4 days of phage treatment of mice resulted in complete sterility (Table 2). We are not the first to describe a phage active against an intracellular pathogen. Broxmeyer et al. (5) showed that phage TM4 can kill intracellular bacilli (Mycobacterium tuberculosis or Mycobacterium avium) in vitro. They proposed the use of attenuated strains of mycobacteria as delivery systems. Perhaps an animal model of infection might have shown these authors that TM4, like MSa, works equally well if given alone, shuttled inside the cells by extracellular mycobacteria.
One of the limitations of phages as antimicrobials is their rapid uptake by the reticuloendothelial system (14, 25, 35). This problem, originally solved by Merril et al. (25), was addressed again recently, with the devising of a protocol which permitted isolation of a phage mutant (specific for Escherichia coli O157:H7) persisting in the mouse circulation for over 1 month (8). The protocol also proved successful in the present study and very likely is of general applicability. The opportunity to easily select long-lasting phages expands phage use to the prophylaxis of bacterial infections (27, 30); more importantly, it demonstrates how phages can be manipulated by selection to suit a specific purpose. At the same time, given the complex phage pharmacodynamics (7, 27) and the rapidity with which the persisting phage evolved in this study, we cannot exclude that the wild phage WSa might have performed equally well in treating infections.
Rapid clearance in the spleen, an inability to kill intracellular bacteria, and stimulation of neutralizing antibodies are the recurrent objections to the use of phages against bacteria (14, 32, 35). As discussed above, MSa is free of these shortcomings and, in addition, lyses methicillin-resistant strains of S. aureus. The evidence that MSa can lyse only 7 of the 19 S. aureus strains tested does not seriously limit its potential use as an antimicrobial agent. In addition to MSa, we have isolated four more phages, which lyse the remaining 12 strains. More generally, the number of phage species is astonishingly high. Therefore, if a bacterial strain develops resistance or is naturally resistant to a bacteriophage, a new one can be isolated in a few days. In conclusion, given the cost in terms of mortality and morbidity imposed by the widespread presence of multidrug-resistant S. aureus strains (19) and the present lack of an effective solution to the problem, investigating the potential of MSa in the management of human diseases associated with S. aureus infections seems a reasonable suggestion.
Footnotes
Published ahead of print on 21 May 2007.
Contribution no. DISSPAPA 146.
REFERENCES
- 1.Adams, M. H. 1959. Methods of study of bacterial viruses, p. 447-448. In Bacteriophages. Interscience Publishers, London, United Kingdom.
- 2.Becker, K., R. Roth, and G. Peters. 1998. Rapid and specific detection of toxigenic Staphylococcus aureus: use of two multiplex PCR enzyme immunoassays for amplification and hybridization of staphylococcal enterotoxin genes, exfoliative toxin genes, and toxic shock syndrome toxin 1 gene. J. Clin. Microbiol. 36:2548-2553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Binetti, A. G., B. Del Río, M. Cruz Martìn, and M. A. Álvarez. 2005. Detection and characterization of Streptococcus thermophilus bacteriophages by use of the antireceptor gene sequence. Appl. Environ. Microbiol. 71:6069-6103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Biswas, B., S. Adhya, P. Washart, B. Paul, A. N. Trostel, B. Powell, R. Carlton, and C. R. Merril. 2002. Bacteriophage therapy rescues mice bacteremic from a clinical isolate of vancomycin-resistant Enterococcus faecium. Infect. Immun. 70:204-210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Broxmeyer, L., D. Sosnowska, E. Miltner, O. Chacon, D. Wagner, J. McGarvey, R. G. Barletta, and L. E. Bermudez. 2002. Killing of Mycobacterium avium and Mycobacterium tuberculosis by a mycobacteriophage delivered by a nonvirulent mycobacterium: a model for phage therapy of intracellular bacterial pathogens. J. Infect. Dis. 186:1155-1160. [DOI] [PubMed] [Google Scholar]
- 6.Bull, J. J., B. R. Levin, T. DeRouin, N. Walker, and C. A. Bloch. 2002. Dynamics of success and failure in phage and antibiotic therapy in experimental infections. BMC Microbiol. 2:35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bull, J. J., and R. R. Regoes. 2006. Pharmacodynamics of non-replicating viruses, bacteriocins and lysins. Proc. Biol. Sci. 273:2703-2712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Capparelli, R., I. Ventimiglia, S. Roperto, D. Fenizia, and D. Iannelli. 2006. Selection of an Escherichia coli O157:H7 bacteriophage for persistence in the circulatory system of mice infected experimentally. Clin. Microbiol. Infect. 12:248-253. [DOI] [PubMed] [Google Scholar]
- 9.Clement, S., P. Vaudaux, P. Francois, J. Schrenzel, E. Huggler, S. Kampf, C. Chaponnier, D. Lew, and J. S. Lacroix. 2005. Evidence of an intracellular reservoir in the nasal mucosa of patients with recurrent Staphylococcus aureus rhinosinusitis. J. Infect. Dis. 192:1023-1028. [DOI] [PubMed] [Google Scholar]
- 10.Dixon, B. 2004. New dawn for phage therapy. Lancet Infect. Dis. 4:186. [DOI] [PubMed] [Google Scholar]
- 11.Fischetti, V. 2004. Vincent Fischetti—following phages for life. Lancet Infect. Dis. 4:246-249. [DOI] [PubMed] [Google Scholar]
- 12.Foster, J. F. 2005. Immune evasion by staphylococci. Nat. Rev. Microbiol. 3:948-958. [DOI] [PubMed] [Google Scholar]
- 13.Jarraud, S., C. Mougel, J. Thioulouse, G. Lina, H. Meugnier, F. Forey, X. Nesme, J. Etienne, and F. Vandenesch. 2002. Relationships between Staphylococcus aureus genetic background, virulence factors, agr groups (alleles), and human disease. Infect. Immun. 70:631-641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lederberg, J. 1996. Smaller fleas … ad infinitum: therapeutic bacteriophage redux. Proc. Natl. Acad. Sci. USA 93:3167-3168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Leeb, M. 2004. Antibiotics: a shot in the arm. Nature 431:892-893. [DOI] [PubMed] [Google Scholar]
- 16.Leid, J. G., M. E. Shirtliff, J. W. Costerton, and P. Stoodley. 2002. Human leukocytes adhere to, penetrate, and respond to Staphylococcus aureus biofilms. Infect. Immun. 70:6339-6345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Levin, B. R., and D. E. Rozen. 2006. Non-inherited antibiotic resistance. Nat. Rev. Microbiol. 4:556-561. [DOI] [PubMed] [Google Scholar]
- 18.Levin, B. R., and R. Antia. 2001. Why we do not get sick: the within host dynamics of bacterial infections. Science 292:1112-1115. [DOI] [PubMed] [Google Scholar]
- 19.Levy, S. B., and B. Marshall. 2004. Antibacterial resistance worldwide: causes, challenges and responses. Nat. Med. 10:122-129. [DOI] [PubMed] [Google Scholar]
- 20.Lowy, F. D. 1998. Staphylococcus aureus infections. N. Engl. J. Med. 339:520-532. [DOI] [PubMed] [Google Scholar]
- 21.Mason, W. J., J. S. Blevins, K. Beenken, N. Wibowo, N. Ojma, and M. S. Smeltzer. 2001. Multiplex PCR protocol for the diagnosis of staphylococcal infections. J. Clin. Microbiol. 39:3332-3338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Matsuzaki, S., M. Yasuda, H. Nishikawa, M. Kuroda, T. Ujihara, T. Shuin, Y. Shen, Z. Jin, S. Fujmoto, M. D. Nasimuzzan, H. Wakiguchi, S. Suguhara, T. Sugiura, S. Koda, A. Muraoka, and S. Imai. 2003. Experimental protection of mice against lethal Staphylococcus aureus infection by novel bacteriophage φMR11. J. Infect. Dis. 187:613-624. [DOI] [PubMed] [Google Scholar]
- 23.McClean, K. L., G. J. Sheehan, and K. M. Harding. 1994. Intraabdominal infection: a review. Clin. Infect. Dis. 19:100-116. [DOI] [PubMed] [Google Scholar]
- 24.Mehrotra, M., G. Wang, and W. M. Johnson. 2000. Multiplex PCR for detection of genes for Staphylococcus aureus enterotoxins, exfoliative toxins, toxic shock syndrome toxin 1, and methicillin resistance. J. Clin. Microbiol. 38:1032-1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Merril, C. R., B. Biswas, R. Carlton, N. C. Jensen, G. J. Creed, S. Zullo, and S. Adhya. 1996. Long circulating bacteriophage as antibacterial agents. Proc. Natl. Acad. Sci. USA 93:3188-3192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Palliser, D., D. Chowdhury, Q. Y. Wang, S. J. Lee, R. T. Bronson, D. M. Knipe, and J. Lieberman. 2006. An siRNA-based microbicide protects mice from lethal herpes simplex virus 2 infection. Nature 439:89-94. [DOI] [PubMed] [Google Scholar]
- 27.Payne, J. H., and V. A. Jansen. 2000. Phage therapy: the peculiar kinetics of self-replicating pharmaceutical. Clin. Pharmacol. Ther. 68:225-229. [DOI] [PubMed] [Google Scholar]
- 28.Qazi, S. N., S. E. Harrison, T. Self, P. Williams, and J. Hill. 2004. Real-time monitoring of intracellular Staphylococcus aureus replication. J. Bacteriol. 186:1065-1077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sambrook, J., E. F. Fritsch, and T. Maniatis. 1989. Molecular cloning: a laboratory manual, 2nd ed. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- 30.Smith, H. W., M. B. Huggins, and K. M. Shaw. 1987. The control of experimental Escherichia coli diarrhoea in calves by means of bacteriophages. J. Gen. Microbiol. 133:1111-1126. [DOI] [PubMed] [Google Scholar]
- 31.Stearne, L. E., I. C. Gyssens, W. H. Goessens, J. W. Mouton, W. J. Oyen, J. W. Van der Meer, and H. A. Verbrugh. 2001. In vivo efficacy of trovafloxacin against Bacteroides fragilis in mixed infection with either Escherichia coli or a vancomycin-resistant strain of Enterococcus faecium in an established-abscess murine model. Antimicrob. Agents Chemother. 45:1394-1401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sulakvelidze, A., Z. Alavidze, and J. G. Morris, Jr. 2001. Bacteriophage therapy. Antimicrob. Agents Chemother. 45:649-659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tenover, F. C., L. M. Weigel, P. C. Appelbaum, L. K. McDougal, J. Chaitram, S. McAllistar, N. Clark, G. Killgore, C. M. O'Hara, L. Jevitt, J. B. Patel, and B. Bozdogan. 2004. Vancomycin-resistant isolate from a patient in Pennsylvania. Antimicrob. Agents Chemother. 48:275-280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Weigel, L. M., D. B. Clewell, S. R. Gill, N. C. Clark, L. K. McDougal, S. E. Flannagan, J. F. Kolonay, J. Shetty, G. E. Killgore, and F. C. Tenover. 2003. Genetic analysis of a high-level vancomycin-resistant isolate of Staphylococcus aureus. Science 302:1569-1571. [DOI] [PubMed] [Google Scholar]
- 35.Westwater, C., L. M. Kasman, A. Schofield, P. A. Werner, J. W. Dolan, M. G. Schmidt, and J. S. Norris. 2003. Use of genetically engineered phage to deliver antimicrobial agents to bacteria: an alternative therapy for treatment of bacterial infections. Antimicrob. Agents Chemother. 47:1301-1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wiggins, B. A., and M. Alexander. 1985. Minimum bacterial density for bacteriophage replication: implications for significance of bacteriophages in natural ecosystems. Appl. Environ. Microbiol. 49:19-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wills, Q. F., C. Kerrigan, and J. S. Soothill. 2005. Experimental bacteriophage protection against Staphylococcus aureus abscesses in a rabbit model. Antimicrob. Agents Chemother. 49:1220-1221. [DOI] [PMC free article] [PubMed] [Google Scholar]