

Abstract
This research was designed to evaluate surface sampling protocols for use with culture and quantitative PCR (QPCR) amplification assay for detection of the gram-negative bacterial biothreat simulant Erwinia herbicola on a variety of surface materials. Surfaces selected for evaluation were wood laminate, glass and computer monitor screens, metal file cabinets, plastic arena seats, nylon seat cushions, finished concrete flooring, and vinyl tile flooring. Laboratory and test chamber studies were performed to evaluate two sampling methods, a sponge and a macrofoam swab, for detection of E. herbicola on surface materials. In laboratory trials, seven materials were inoculated with a known concentration of E. herbicola cells and samples were collected from the surfaces of the materials to determine sampling efficiencies. Culture analysis was ineffective for assessing E. herbicola collection efficiency because very few culturable cells were obtained from surface samples. QPCR demonstrated that E. herbicola DNA was present in high concentrations on all of the surface samples, and sampling efficiencies ranged from 0.7 to 52.2%, depending on the sampling method and the surface material. The swab was generally more efficient than the sponge for collection of E. herbicola from surfaces. Test chamber trials were also performed in which E. herbicola was aerosolized into the chamber and allowed to settle onto test materials. Surface sampling results supported those obtained in laboratory trials. The results of this study demonstrate the capabilities of QPCR to enhance the detection and enumeration of biocontaminants on surface materials and provide information on the comparability of sampling methods.
Dispersal of biocontaminants in workplaces and residences increases the possibility of occupant exposure and resulting adverse health effects ranging from lost productivity to severe illness. Surface sampling is often used in conjunction with air sampling in the indoor environment to provide information on bioaerosol dispersal and deposition and to locate and identify biocontaminant sources (3, 9, 13). A variety of methods have been used to collect microorganisms from smooth and porous surfaces, including swabs, wipes and sponges, tapes, agar contact plates, and dust and bulk sampling. However, monitoring is hampered by the lack of validated methods that provide precise, accurate, and representative measurements of microbe-contaminated surfaces. The bioterrorism events involving the dispersal of Bacillus anthracis spores that occurred in the United States in 2001 heightened the interest in surface sampling and reemphasized the need for standardized surface sample collection and analysis protocols (7, 12, 13).
While surface sampling methods have been compared for relative effectiveness (1, 4, 12), there is little quantitative information on the collection efficiency of surface sampling methods (2, 8, 10). This research project was designed to evaluate monitoring protocols for use with culture and quantitative PCR (QPCR) amplification assay for detection of Erwinia herbicola, a gram-negative vegetative bacterium that is commonly used as a biothreat simulant, on a variety of surface materials. Surfaces selected for evaluation were wood laminate, glass and computer monitor screens, metal file cabinets, plastic arena seats, nylon seat cushions, finished concrete flooring, and vinyl tile flooring. Laboratory and test chamber studies were performed to evaluate two sampling methods, a sponge (Speci-sponge) and a macrofoam swab (SW Kit), for detection of E. herbicola on surface materials. Laboratory studies were designed to determine the efficiency and sensitivity of the sampling and analysis protocols. Test chamber studies were designed for comparison of methods after aerosol release and settling of E. herbicola cells onto the surface materials.
MATERIALS AND METHODS
Test organism and culture media.
E. herbicola (obtained from Brevard Teaching and Research Laboratories, Palm Bay, FL; USDA PPQ526 Plant Pest Permit) is used as a simulant for Yersinia pestis and Francisella tularensis, both gram-negative bacterial biothreat agents of concern. E. herbicola was cultured on tryptic soy agar amended with 100 μg/ml cycloheximide (TSAC, pH 7.0; Difco Laboratories, Sparks, MD) and incubated at 28°C for 1 to 2 days. For culture analysis, aliquots of the liquid samples were concentrated by filtration and/or serially diluted, depending on the type of sample, followed by inoculation onto TSAC plates and incubation as described above. The CFU were enumerated, and the numbers of CFU per sample and CFU per unit of area sampled were determined. The detection limits of culture analysis were based on the enumeration of 1 CFU/ml and calculated for the two sampling methods by using the equation
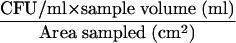 |
Test materials.
Seven surface materials were selected for evaluation. Wood laminate shelves, painted metal file cabinets and shelves, and computer monitors were obtained from excess office furnishings. Other materials obtained from commercial vendors included finished concrete tiles (untreated; Valentine Construction, Henderson, NV), commercial vinyl composition tile (Armstrong Imperial Texture Standard Excellon, World Industries, Lancaster, PA), nylon seat cushions (Quality Upholstery, Las Vegas, NV), and plastic arena seats (double-wall, high-density polyethylene; Hussey Seating Company, North Berwick, ME). For laboratory trials, glass was used as the test material in place of computer monitor screens.
Surface sampling and sample processing.
Two surface sampling methods were tested, a sponge (Speci-sponge; Nasco, Fort Atkinson, WI) and a macrofoam swab (SW Kit, currently named the Sample Collection and Recovery Device; ASD BioSystems, Danville, VA), by using protocols developed in a previous project (4). The protocol for sponge sampling consisted of moistening the dehydrated sterile sponge in a sterile stomacher bag with 30 ml of sterile 0.01 M potassium phosphate buffer (PB, pH 7.0). The hydrated sponge (4.2 by 8.7 cm) was squeezed to remove the excess buffer and then used to sample surface material sections by wiping a 930-cm2 (1-ft2) area in a horizontal direction. The sponge was turned over to expose the unused side and used to sample the same area in a vertical direction, taking care not to disturb the exposed side of the sponge. The sponge was returned to the stomacher bag, and the sample was hand mixed for 1 min. The liquid sample was eluted from the sponge, the volume was measured, and the sponge was discarded.
The protocol for the swab consisted of removing a macrofoam swab (1.6 by 2.0 cm), premoistened with Tris-buffered saline with 0.1% Tween 20, from a sterile pouch. Surface samples were obtained by swabbing the first half of the surface material area, turning the swab over to expose the unused side, and swabbing the second half of the surface material. A total area of 317 cm2 (49 in2) was sampled. The swab was placed in a 50-ml tube containing 9 ml of Tris-buffered saline with 0.1% Tween 20 and vortexed for 1 min. The liquid sample was eluted from the foam swab, the volume was measured, and the swab was discarded.
DNA extraction.
Following sampling and processing, samples were filter concentrated and E. herbicola DNA was extracted and purified (Fig. 1). Twenty milliliters of the sponge samples and 6 ml of the swab samples were concentrated by filtration for subsequent DNA extraction. The DNA extraction and concentration protocol consisted of filtration of the processed sample through a 0.65-μm-pore-diameter mixed cellulose ester filter membrane (Millipore Corp., Bedford, MA) and resuspending the membrane in 0.5 ml of PB with 0.05% Tween 20 (pH 7.0). The concentrated sample was boiled for 15 min and chilled on ice for 2 min, and bovine serum albumin (0.05%, final concentration) was added to block the binding of DNA to the membrane (Fig. 1). The sample was then incubated for 5 min at 37°C in a rotary shaker at a speed of 230 rpm. After removal of the membrane, the DNA was purified by the Pellet Paint protocol (Novagen, Madison, WI), and the purified DNA pellet was resuspended in 50 μl of Tris-EDTA buffer (pH 8.0).
FIG. 1.

Flow chart summarizing sponge and macrofoam swab sample processing and E. herbicola DNA extraction and purification procedures. Abbreviations: PB, 0.01 M PB; TBS, Tris-buffered saline with 0.1% Tween 20; HA, mixed cellulose esters; PBT, 0.01 M PB with 0.05% Tween 20; BSA, bovine serum albumin; TE, Tris-EDTA buffer.
QPCR amplification.
Five microliters of sample DNA was amplified by QPCR as described below. The ABI Prism 7700 Sequence Detection System (Applied Biosystems, Foster City, CA) was used for QPCR analysis. Amplification conditions, according to the protocol specified by the Naval Medical Research Center (Silver Spring, MD) and with Applied Biosystems reagents, included E. herbicola template DNA, 1× TaqMan buffer A, 5 mM MgCl2, 0.1 mM dATP, 0.1 mM dCTP, 0.1 mM dGTP, 0.2 mM dUTP, 2.5 U of AmpliTaq Gold, 0.5 U of AmpErase Uracyl N-Glycosylase, each primer at 0.3 μM, and 0.2 μM probe, for a total reaction volume of 50 μl. The TaqMan cycling conditions were 2 min at 50°C, 10 min at 95°C, and 40 cycles of 15 s at 95°C followed by 1 min at 60°C. Amplification of E. herbicola DNA was carried out with a Naval Medical Research Center-specified primer pair and a fluorescently labeled probe that targeted the chorismate mutase (aroQ) gene. The sequences of the forward and reverse primers used were GCTGCAAAACGCACAACA (EH492F) and CGTGAACAAACGGCTCCA (EH550R). The TaqMan probe (EH512T) sequence was 6-carboxyfluorescein-5′-CCGGGCTTGAACCCCACTCC-3′-6-carboxytetramethyl rhodamine. Primers were obtained from Operon Biotechnologies (Huntsville, AL), and the probe was obtained from Applied Biosystems. PCR quantitation standards were prepared by using serial dilutions of E. herbicola cell suspensions of known concentrations enumerated electronically with a Coulter Multisizer II particle counter (Beckman Coulter, Inc., Miami, FL). Standards were prepared by using the same extraction and purification methods used to process samples as described above, and the cell number of each suspension extracted was used as the template concentration for that standard. The PCR standards (100 to 105 E. herbicola templates per reaction) were amplified in duplicate at the same time and under the same conditions as the replicate unknown samples. Once amplification was completed, the data were analyzed with the software provided with the 7700 Sequence Detection System. Concentrations for the unknown samples were extrapolated from the standard curve by the software and reported as the mean of two replicates. The detection limits of QPCR analysis were determined for the two sampling methods based on the sensitivity of the assay (≤1 template per reaction) and the enumeration of 1 template per reaction. The detection limits were calculated with the equation
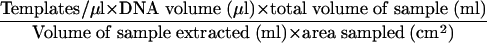 |
Laboratory trials.
Laboratory trials were conducted with the swab and sponge to determine the efficiency and sensitivity of the surface sampling methods for detection of E. herbicola cells from surfaces. A log-phase culture (optical density at 600 nm, ca. 0.6) of E. herbicola was harvested by centrifugation, washed twice, and resuspended in PB. Cell concentration (CFU per milliliter) was determined by serial dilution, spread plating onto TSAC, and incubation as described above. One-milliliter volumes of the culture, containing a known concentration of cells (108 CFU/ml), were used to inoculate triplicate sections of each test material in a biosafety cabinet, and the materials were covered and allowed to dry. Test materials consisted of wood laminate, glass (to represent computer monitor screens), metal shelves (to represent file cabinets), plastic arena seats, nylon seat cushions, finished concrete flooring, and vinyl tile flooring. Three samples were collected from each of the seven materials by each sampling method. Sponge and swab surface samples were analyzed by culture and QPCR. For QPCR, 20 ml of sponge samples and 6 ml of swab samples were concentrated by filtration prior to DNA extraction. DNA extraction and purification were performed as described above.
Test chamber trials.
E. herbicola aerosolization trials were conducted in a room-sized test chamber. As previously reported, the room measures 4.0 m by 4.0 m by 2.2 m high and has a sheet vinyl tile floor (5). The interior walls, exterior walls, and ceiling are sheetrocked and coated with interior latex paint. The chamber is equipped with a heating, ventilation, and air conditioning system that is sized to simulate a residential system with rectangular bare metal ductwork. During aerosolization experiments, the heating, ventilation, and air conditioning system was operated with an airflow of 4.2 m3/min, resulting in approximately five room air volume exchanges per h. An anteroom equipped with a HEPA-filtered air shower attached to the room entrance reduces mixing of air resulting from entering and exiting the chamber during experiments. During all activities in the test chamber, technicians wore full-face respirators and nonwoven protective clothing. Upon completion of each series of test chamber trials, contaminated surface materials were either decontaminated or removed and discarded and the interior surfaces of the chamber were decontaminated with a 0.5% sodium hypochlorite solution. For test surface materials, decontamination was followed by rinsing with sterile water.
Three E. herbicola releases were conducted in the test chamber. For each release, seven test materials were placed in the test chamber and sampling areas on each material were identified. Test materials were the same as described above for the laboratory trials, except that an actual file cabinet and computer monitors (power on) were substituted for the metal shelves and glass, respectively. Test materials were oriented horizontally, with the exception of the computer monitor screens, which were oriented vertically. A log-phase culture of E. herbicola was harvested, washed, and resuspended in PB as described above. Ten milliliters of the washed culture was added to a Collison nebulizer (BGI Inc., Waltham, MA), and the nebulizer was placed in the experimental room. With the chamber air-handling system on, the Collison nebulizer was operated at a pressure of 20 lb/in2 for 10 min to aerosolize E. herbicola cells in the room. The bioaerosol concentration was monitored during the release with a BioSampler (SKC, Inc., Eighty-Four, PA) supplied with 20 ml of PB and operated at 12.5 liters/min. The chamber air-handling system was then turned off to allow the bioaerosol to settle onto the test materials. After overnight settling of the bioaerosol, biocontamination levels were measured on surfaces in the chamber over a 2-day period. Surface sampling was conducted on all test surfaces by the sponge and swab sampling methods. The first set of samples was collected on day 1, and duplicate samples were collected on day 2 for each material. Samples were processed as described above for the laboratory trials and analyzed by culture and QPCR.
RESULTS
Laboratory trials.
The concentrations of the cell suspensions used to inoculate the seven materials were measured by both culture, reflecting the presence of culturable organisms, and QPCR, reflecting the presence of total cells, i.e., amplifiable templates (Table 1). With one exception, QPCR measurements were slightly greater than culture measurements. Sampling efficiency was measured with the equation
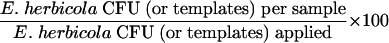 |
Data from laboratory trials with triplicate samples of each test material showed that very few culturable E. herbicola cells were obtained from surface samples (culture detection limits: sponge, 30 CFU/sample = 0.03 CFU/cm2; swab, 9 CFU/sample = 0.03 CFU/cm2). Therefore, sampling efficiencies measured by culture analysis were very low and the greatest efficiency measurement was only 0.006% (Table 2). Conversely, QPCR analysis showed that total E. herbicola cells, both living and dead, were present in high concentrations on all of the samples (QPCR detection limits: sponge, 15 templates/sample = 0.02 templates/cm2; swab, 15 templates/sample = 0.05 templates/cm2). Sampling efficiencies measured by QPCR ranged from 0.7 to 52.2%, depending on the sampling method and the surface material. The swab was generally more efficient than the sponge for collection of E. herbicola from surfaces. For both sampling methods, collection efficiency was highest for glass surfaces and lowest for finished concrete. When data were expressed as templates collected per sample, the sponge data were comparable to the swab data due to the larger area sampled by the sponge (Table 2).
TABLE 1.
Concentrations of E. herbicola cell suspensions used to inoculate triplicate sections of test materials evaluated in laboratory studies, as determined by culture and QPCR analysis
| Material | Inoculum concn
|
|
|---|---|---|
| No. of CFU/ml | No. of templates/ml | |
| Metal file cabinet | 6.00 × 108 | 4.34 × 108 |
| Glass | 6.35 × 108 | 6.80 × 108 |
| Wood laminate | 4.15 × 108 | 4.38 × 108 |
| Vinyl tile | 3.30 × 108 | 3.68 × 108 |
| Plastic seat | 3.90 × 108 | 5.04 × 108 |
| Finished concrete | 4.50 × 108 | 4.74 × 108 |
| Nylon cushion | 4.10 × 108 | 4.30 × 108 |
TABLE 2.
Efficiency of surface sampling methods for detection of E. herbicola in laboratory studiesa
| Sampling method and surface material | Culture analysis
|
QPCR analysis
|
||
|---|---|---|---|---|
| Mean no. of CFU/ sample ± 1 SE | Sampling efficiency ± 1 SE (%) | Mean no. of templates/ sample ± 1 SE | Sampling efficiency ± 1 SE (%) | |
| Sponge | ||||
| Metal file cabinet | 5.90 × 102 ± 6.57 × 101 | <0.005 | 1.94 × 108 ± 2.79 × 106 | 44.8 ± 0.6 |
| Glass | 3.64 × 104 ± 7.61 × 103 | 0.006 ± 0.001 | 3.22 × 108 ± 7.06 × 106 | 47.3 ± 1.0 |
| Wood laminate | <25 | <0.005 | 5.01 × 107 ± 2.88 × 106 | 11.4 ± 0.7 |
| Vinyl tile | <29 | <0.005 | 9.48 × 107 ± 4.22 × 106 | 25.8 ± 1.1 |
| Plastic seat | 4.83 × 102 ± 2.27 × 102 | <0.005 | 9.15 × 107 ± 2.12 × 107 | 18.1 ± 4.2 |
| Finished concrete | 6.66 × 101 ± 6.66 × 101 | <0.005 | 3.17 × 106 ± 8.23 × 105 | 0.7 ± 0.2 |
| Nylon cushion | 1.42 × 102 ± 5.59 × 101 | <0.005 | 4.87 × 107 ± 7.55 × 106 | 11.3 ± 1.8 |
| Swab | ||||
| Metal file cabinet | 3.68 × 103 ± 6.63 × 102 | <0.005 | 1.72 × 108 ± 2.29 × 107 | 39.5 ± 5.3 |
| Glass | 3.20 × 104 ± 1.15 × 104 | 0.005 ± 0.002 | 3.55 × 108 ± 5.64 × 107 | 52.2 ± 8.3 |
| Wood laminate | 2.97 × 101 ± 2.97 × 101 | <0.005 | 1.25 × 108 ± 2.91 × 107 | 28.6 ± 6.6 |
| Vinyl tile | 2.06 × 101 ± 1.28 × 101 | <0.005 | 1.51 × 108 ± 1.28 × 107 | 41.1 ± 3.5 |
| Plastic seat | 7.09 × 102 ± 9.39 × 101 | <0.005 | 1.57 × 108 ± 7.55 × 106 | 31.2 ± 1.5 |
| Finished concrete | <9 | <0.005 | 3.78 × 106 ± 7.10 × 105 | 0.8 ± 0.1 |
| Nylon cushion | 1.71 × 101 ± 4.99 × 100 | <0.005 | 1.67 × 107 ± 6.05 × 105 | 3.9 ± 0.1 |
The sponge and macrofoam swab methods were used to sample triplicate areas of 930 cm2 and 317 cm2, respectively, of surface materials inoculated with 108 E. herbicola cells.
Test chamber trials.
Three E. herbicola releases were conducted in the test chamber. The airborne cell concentrations for the three releases, determined by SKC air sampling and QPCR analysis, were 3.4 × 106, 3.0 × 106, and 2.6 × 106 templates/m3. The corresponding measurements of culturable airborne E. herbicola were 2.5 × 104, 7.0 × 104, and 4.5 × 104 CFU/m3. Culture and QPCR analyses were performed on all surface samples. The results supported those obtained in laboratory trials. Very few culturable cells were obtained from surface samples (<0.04 CFU/cm2), presumably due to losses of viability from aerosolization and desiccation stresses (6). QPCR analysis was necessary for detection of E. herbicola. QPCR results indicated that the swab was generally more efficient than the sponge (Fig. 2). For both sampling methods, the greatest collection efficiency was obtained with the metal file cabinet, vinyl tile, and wood laminate surfaces. The lowest retrieval of E. herbicola was obtained from the computer monitor screens and finished concrete.
FIG. 2.

Summary of QPCR results obtained from surface samples in the experimental room. Materials were contaminated by aerosolization of E. herbicola into the room, followed by settling of the bioaerosol onto surfaces in the room. Duplicate surface samples were collected by sponge (930 cm2 = 1 ft2) and macrofoam swab (317 cm2 = 49 in2) for each of three releases. Bar heights represent the mean of six samples ± 1 standard error.
DISCUSSION
The collection efficiency of a surface sampling method is determined by both the efficiency of removal of microorganisms from the surface and the efficiency of their removal from the collection material (8). Numerous factors can influence the results of surface sampling, including the sampling method, the surface material, the sample processing protocol, the properties of the target microorganisms, and the analysis method (10). Results from this study showed that culture analysis, while useful for measuring the presence of microorganisms such as B. anthracis that form stress-resistant endospores (7, 10-12), was ineffective for assessing the efficiency of E. herbicola collection from surface samples. In contrast, QPCR, which measures both living and dead cells, demonstrated that E. herbicola DNA was present in high concentrations on all of the samples, with the exception of sponge samples from concrete. The ability of QPCR to detect nonculturable cells is an important consideration because some nonculturable microorganisms may still be viable and certain bacteria, fungi, and viruses are able to repair damage due to aerosolization and remain infective (6). Therefore, while both culture and QPCR could be used effectively to detect the presence of B. anthracis spores in surface samples, efficient detection of vegetative bacterial cells that may be nonculturable requires an alternative approach such as QPCR analysis. These data also point out one of the limitations of using simulants instead of actual biothreat agents. Previous research on the persistence of Y. pestis demonstrated that this microorganism maintains culturability for several days on environmental surfaces (11), in contrast with the results obtained in this research with E. herbicola.
The type of surface material sampled also affected the efficiency of recovery of E. herbicola from surfaces. Some differences were observed between laboratory trials using a liquid inoculum and test chamber trials with aerosolization and deposition of cells onto the test materials. In general, the greatest concentrations of E. herbicola were measured on smooth, nonporous materials such as glass and metal. In laboratory trials, the greatest concentrations were obtained in surface samples from glass. In chamber trials, the results obtained from the vertically oriented surface of the computer monitor screens (power on) were lower than all of the other materials except concrete. In contrast, previous experiments with aerosolized dry endospores of the simulant Bacillus atrophaeus showed that surface samples of computer monitor screens had greater concentrations of spores than any of the other materials tested (unpublished data), presumably due to electrostatic attraction of the spores to the surface of the monitor. Because microbial contamination may be present on all types of surfaces, it is important to develop effective sampling methods for both porous and nonporous surfaces and be able to reliably determine the contamination levels on these materials, especially for assessing the efficacy of decontamination efforts.
E. herbicola sampling efficiencies measured by QPCR ranged from 0.7 to 52.2%, depending on the sampling method and the surface material. The swab was generally more efficient than the sponge in laboratory studies for collection of E. herbicola from surfaces, as determined by QPCR analysis. Surface sampling results from test chamber aerosolization trials supported those obtained in laboratory trials. The SW Kit consists of a premoistened macrofoam swab. In a previous study, the collection efficiencies of four types of swabs, cotton, macrofoam, polyester, and rayon, were compared by inoculating steel coupons with known concentrations of B. anthracis spores and surface sampling with both wet and dry swabs (10). Results indicated that premoistened macrofoam and cotton were the swab materials that had the greatest collection efficiency. In this study, collection efficiencies measured for the swab with metal surfaces were comparable to those obtained previously with steel coupons, although the test organisms and analysis methods differed. It is possible that one of the reasons for the lower collection efficiencies obtained with sponge samples was the difference in the collection buffers used. The swab was obtained premoistened with a buffer containing the surfactant Tween 20, while the sponge was wetted with buffer without a surfactant added. Rose et al. suggest that the use of a surfactant in the collection buffer may enhance the collection of spores (10), but thorough buffer evaluation studies have not been conducted. Another possible explanation for the lower collection efficiencies observed with the sponge is that a component of the sponge material had an inhibitory effect on QPCR. In our previous sponge sampling experiments with B. atrophaeus using an internal positive control in the QPCR, inhibition was not observed for any of the materials except concrete. Results indicated that sponge samples from concrete were completely inhibited and swab samples were slightly inhibited (unpublished data). Therefore, the presence of inhibitory compounds associated with concrete likely accounted for the low measurements of E. herbicola obtained with sponge samples. For swabs, less area was sampled and the concentration of inhibitory compounds in the sample was probably lower. In this study, inhibition of the QPCR assay was not determined.
Although the sponge method was less efficient than the swab for surface sampling, their detection sensitivities are similar. The larger surface area sampled by the sponge (930 cm2) resulted in comparable concentrations of E. herbicola in samples for both the swab and the sponge (Table 2). In previous research that measured the collection efficiency and sensitivity of the Biological Sampling Kit, a device designed for large-area surface sampling, results showed that the large surface area (1 m2) sampled by the Biological Sampling Kit resulted in higher sensitivity than the swab sampling methods tested (2). Another potential advantage of large-area sampling in the field is greater coverage that results in fewer samples collected per site. Potential uses for the sponge include intermediate-to-large-area surface sampling, particularly on smooth, nonporous surfaces, whereas swab sampling is most applicable for sampling small areas, nonsmooth surfaces, and locations where access is difficult. Based on the results obtained in this research, the selection of the sample analysis method and the type of surface to sample were more important considerations for maximizing the detection of E. herbicola than the type of sampling method chosen.
The aftermath of the bioterrorism events of 2001 involving B. anthracis spores has emphasized the importance of surface sampling for detection of biological contaminants and the need for additional research to determine the collection efficiencies and sensitivities of surface sampling methods (7, 12, 13). The results of this research provided quantitative data on the collection efficiencies of two surface sampling methods and demonstrated the abilities of QPCR to enhance the detection and enumeration of biocontaminants on surface materials. Additional studies designed to minimize environmental interference and optimize sampling and processing methods are ongoing to improve the field applicability of surface sampling for detection of biocontaminants in indoor environments. The protocols developed could be adapted for use in other environmental microbiological applications to enhance the detection and measurement of surface-associated microorganisms.
Acknowledgments
The research described in this article was funded by the Technical Support Working Group, Arlington, VA.
Footnotes
Published ahead of print on 6 April 2007.
REFERENCES
- 1.Buttner, M. P., P. Cruz, L. D. Stetzenbach, A. K. Klima-Comba, V. L. Stevens, and T. D. Cronin. 2004. Determination of the efficacy of two building decontamination strategies by surface sampling with culture and quantitative PCR analysis. Appl. Environ. Microbiol. 70:4740-4747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Buttner, M. P., P. Cruz, L. D. Stetzenbach, A. K. Klima-Comba, V. L. Stevens, and P. A. Emanuel. 2004. Evaluation of the Biological Sampling Kit (BiSKit) for large-area surface sampling. Appl. Environ. Microbiol. 70:7040-7045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Buttner, M. P., P. Cruz-Perez, L. D. Stetzenbach, P. J. Garrett, and A. E. Luedtke. 2002. Measurement of airborne fungal spore dispersal from three types of flooring materials. Aerobiologia 18:1-11. [Google Scholar]
- 4.Buttner, M. P., P. Cruz-Perez, and L. D. Stetzenbach. 2001. Enhanced detection of surface-associated bacteria in indoor environments by quantitative PCR. Appl. Environ. Microbiol. 67:2564-2570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Buttner, M. P., and L. D. Stetzenbach. 1993. Monitoring of fungal spores in an experimental indoor environment to evaluate sampling methods and the effects of human activity on air sampling. Appl. Environ. Microbiol. 59:219-226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cox, C. S. 1989. Airborne bacteria and viruses. Sci. Prog. 73:469-500. [PubMed] [Google Scholar]
- 7.Higgins, J. A., M. Cooper, L. Schroeder-Tucker, S. Black, D. Miller, J. S. Karns, E. Manthey, R. Breeze, and M. L. Perdue. 2003. A field investigation of Bacillus anthracis contamination of U.S. Department of Agriculture and other Washington, D.C., buildings during the anthrax attack of October 2001. Appl. Environ. Microbiol. 69:593-599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kirschner, L. E., and J. R. Puleo. 1979. Wipe-rinse technique for quantitating microbial contamination on large surfaces. Appl. Environ. Microbiol. 38:466-470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Martyny, J. W., K. F. Martinez, and P. R. Morey. 1999. Source sampling, p. 12-1-12-8. In J. Macher (ed.), Bioaerosols: assessment and control. American Conference of Governmental Industrial Hygienists, Cincinnati, OH.
- 10.Rose, L., B. Jensen, A. Peterson, S. N. Banerjee, and M. J. Arduino. 2004. Swab materials and Bacillus anthracis spore recovery from nonporous surfaces. Emerg. Infect. Dis. 10:1023-1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rose, L. J., R. Donlan, S. N. Banerjee, and M. J. Arduino. 2003. Survival of Yersinia pestis on environmental surfaces. Appl. Environ. Microbiol. 69:2166-2171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sanderson, W. T., M. J. Hein, L. Taylor, B. D. Curwin, G. M. Kinnes, T. A. Seitz, T. Popovic, H. T. Holmes, M. E. Kellum, S. K. McAllister, D. N. Whaley, E. A. Tupin, T. Walker, J. A. Freed, D. S. Small, B. Klusaritz, and J. H. Bridges. 2002. Surface sampling methods for Bacillus anthracis spore contamination. Emerg. Infect. Dis. 8:1145-1151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Weis, C. P., A. J. Intrepido, A. K. Miller, P. G. Cowin, M. A. Durno, J. S. Gebhardt, and R. Bull. 2002. Secondary aerosolization of viable Bacillus anthracis spores in a contaminated U.S. Senate office. JAMA 288:2853-2858. [DOI] [PubMed] [Google Scholar]