

Abstract
Simple, rapid, and reliable fecal indicator tests are needed to better monitor and manage ambient waters and treated waters and wastes. Antibody-coated polymeric bead agglutination assays can fulfill these needs and are inexpensive and portable for nonlaboratory settings, and their reagents can be stored at ambient temperatures for months. The goal of this study was to develop, optimize, and validate a rapid microbial water quality monitoring assay using F+ coliphage culture, latex agglutination, and typing (CLAT) to detect F+ coliphage groups with antibody-coated particles. Rapid (180 min) F+ coliphage culture gave comparable results to those with the 16- to 24-h culture time used in EPA method 1601 and was amenable to CLAT assay detection. CLAT was performed on a cardboard card by mixing a drop of coliphage enrichment culture with a drop of antibody-coated polymeric beads as the detection reagent. Visual agglutination or clumping of positive samples occurred in <60 seconds. The CLAT assay had sensitivities of 96.4% (185/192 samples) and 98.2% (161/164 samples) and specificities of 100% (34/34 samples) and 97.7% (129/132 samples) for F+ RNA and DNA coliphages, respectively. CLAT successfully classified F+ RNA coliphages into serogroups typically obtained from human (groups II and III) and animal (groups I and IV) fecal sources, in similar proportions to those obtained with a nucleic acid hybridization assay. This novel group-specific antibody-based particle agglutination technique for rapid and simple detection and grouping of F+ coliphages provides a new and improved tool for monitoring the microbiological quality of drinking, recreational, shellfishing, and other waters.
Water quality is a global public health concern. In developing countries, there is inadequate access to safe drinking water and its sources. Unsafe water, sanitation, and hygiene cause around 1.7 million deaths each year worldwide, mostly from infectious diarrhea in children in developing countries (55). Microbial pathogens causing gastrointestinal, dermal, and respiratory infections can be spread by drinking, bathing, or cleaning with water polluted with feces (56). In developed countries, waterborne disease outbreaks and discrete disease cases continue to occur despite government regulations on wastewater and drinking water quality, treatment, and monitoring-based warning systems for wastewater effluents, recreational waters, and shellfish-growing waters (11, 27, 36). Fecal indicator microorganisms, such as fecal coliforms, Escherichia coli, and enterococci, are used to measure the efficacy of water and wastewater treatment, drinking water quality, and the sanitary quality of bathing and shellfishing waters (32). However, current microbial indicators are bacteria, and many waterborne pathogens are enteric viruses, for which bacterial indicators are inadequate or unreliable in certain circumstances due to greater virus and bacteriophage resistance to water and wastewater treatment processes (21, 26) and greater virus and bacteriophage persistence in freshwater and seawater (10, 14, 31). Hence, there is a need for quick, easy-to-use, reliable viral indicators and effective methods to detect and assay them.
U.S. ambient water monitoring programs are just one example of the need for improved fecal indicator detection. Bacterial indicator assays used by regulators to monitor ambient water quality require 18 to 96 hours for results, which causes water quality decisions and warnings/advisories to be posted days after contamination events occur (32). Fecal pollution events in water are intermittent and often return to below threshold levels in 24 h (5, 28). The same bacterial indicator assays cannot differentiate human and nonhuman fecal wastes for tracking and controlling their sources without extra and advanced steps, and they have a lack of predictability for enteric virus contamination (12). In 2005, regulators issued around 20,400 days of closures or advisories at U.S. beaches and lakes due to exceedances of bacterial fecal indicators (33). About 75% of those 20,400 exceedances were caused by unknown sources of fecal pollution that could not be tracked, treated, or managed (33).
Coliphages are alternatives to bacterial indicators. Coliphages are bacterial viruses that reside in the guts of animals, sometimes at titers similar to those of bacterial gut flora (1). Coliphages are obligate intracellular parasitic microorganisms that generally do not replicate in environments outside the gut, where host bacterial levels are <104 CFU/ml (50, 54), or in nutrient-poor environments that do not support host growth (54), and coliphage lysis of bacteria only occurs in bacterial cultures undergoing exponential (logarithmic-phase) growth (37). F+ coliphages infect the F pili of coliform bacteria, which stop forming below 25°C (34, 53), further constraining the natural conditions needed for coliphage replication. Coliphages are useful at indicating public health risks for water users and shellfish consumers, as in some studies the presence of coliphages was correlated with the presence of pathogenic human viruses in water and shellfish and with the risk of viral illness (9, 12, 13, 25, 48). F+ coliphages can be divided into two families, namely, the Leviviridae, containing RNA genomes (F+ RNA coliphages), and the Inoviridae, containing DNA genomes (F+ DNA coliphages) (46).
F+ RNA coliphages can be serotyped into distinct groups present in human fecal waste (groups II and III) or animal fecal waste (groups I and IV) (8, 15, 23). Microbial source tracking with F+ RNA coliphages has been used to identify and control human and animal sources of fecal pollution in surface waters (3, 17, 42).
Current coliphage recovery and detection assays are as time-consuming as culture-based bacterial indicator methods, taking 1 to 3 days for coliphage culture and plating methods (44, 45), 1 to 2 days for coliphage serotyping methods (23), and 2 days for molecular coliphage methods, including reverse transcriptase PCR (RT-PCR) and probe hybridization (47). The goals of this study were to develop a same-day microbial water quality monitoring assay using F+ coliphages and, specifically, to (i) develop a straightforward and quick assay to culture and detect F+ coliphages as fecal indicator microbes, (ii) distinguish between F+ RNA and F+ DNA coliphages, and (iii) concurrently subtype F+ RNA coliphages into groups I to IV as microbial source tracking information to distinguish human and animal fecal origins.
This rapid coliphage detection assay is based on an antibody-based immunological approach commonly referred to as “latex agglutination,” which was first performed in the mid-1950s by Singer and Plotz to detect rheumatoid arthritis (39). In this method, particles are coated with antibodies (or antigens) and used to detect by visual means the binding and clumping of target antigens (or antibodies) with adjacent detector particles. Latex agglutination assays are generally rapid, specific, uncomplicated, and inexpensive, which makes them ideal for field or office diagnostic kits, such as those used to detect adenovirus and rotavirus in stool (16, 24) and the parasite Leishmania in urine (6). Agglutination tests are used in doctors’ offices, veterinary offices, clinical diagnostic microbiology laboratories, other medical facilities, and virology laboratories to detect a number of different microbes, including herpes simplex virus (19), tobacco mosaic virus (43), Staphylococcus aureus (40), and Candida dubliniensis (30), as well as antibodies against avian influenza virus subtype H5N1 (57) and human immunodeficiency virus (38). Unlike clinical samples with high titers of antigens, environmental samples usually have low levels of coliphage antigens, which requires that a culture step be used before coliphage detection by particle agglutination. This study describes the development and application of a rapid F+ coliphage enrichment culture and subsequent antibody-mediated particle agglutination test for recovery, detection, and grouping (typing) of F+ coliphages as a tool for monitoring the microbiological quality of drinking, recreational, and shellfishing waters.
MATERIALS AND METHODS
Virus strains, bacterial hosts, and environmental F+ coliphage isolates.
F+ RNA coliphage prototype strains MS2 (serogroup I), GA (serogroup II), Qβ (serogroup III), M11 (serogroup III), SP (serogroup IV), and FI (serogroup IV) and F+ DNA coliphage prototype strains Fd, F1, and M13 were used as positive controls. F+ coliphage field isolates were recovered from samples of shellfish tissue, water, and bird feces at estuaries in Florida, North Carolina, Delaware, New Hampshire, Massachusetts, Rhode Island, and California by previously described methods (41, 44, 45), using a permissive E. coli Famp host (ATCC 700891). F+ coliphage isolates were enriched under conditions described in EPA method 1601, using liquid culture to promote high phage titers. Enriched material was clarified by centrifugation at 1,200 × g for 15 min, and the resulting supernatant was frozen at −80°C in tryptic soy broth (TSB).
Rapid F+ coliphage culture.
A 180-min F+ coliphage culture enrichment was developed as a modified version of the 16- to 24-h culture step of EPA method 1601 (44). Rapid F+ coliphage culture conditions differed from those in EPA method 1601 by the use of an optimized initial log-phase host concentration of 1 × 107 CFU E. coli Famp per ml of culture and lasted 2 to 3 hours in a 35 to 37°C water bath, at which time host bacteria entered stationary-phase growth. Rapid F+ coliphage enrichments were compared for prototype F+ RNA coliphages (MS2, Qβ, SP, and Fi) by inoculating 1 to 3 PFU into 333-ml broth cultures and tracking bacterial and coliphage levels at times throughout the culture period (0, 30, 60, 90, 120, 180, and 360 min). F+ RNA coliphages were quantified on tryptic soy agar (TSA) spot plates containing host E. coli Famp lawns, and E. coli was quantified before and after log-phase growth on TSA plates and during log-phase growth by measuring the optical density at 520 nm in a spectrophotometer (Spectronic 1201; Milton Roy Company).
RNase test for detecting F+ RNA and F+ DNA coliphages.
F+ coliphage field isolates were replated with and without RNase (RNase A; Sigma-Aldrich, St. Louis, MO) to distinguish viral nucleic acid content as DNA or RNA (23). RNase infectivity neutralization tests were performed on spot plates of 0.75% TSA containing the log-phase E. coli Famp host, streptomycin, ampicillin (each at 15 μg/ml), and RNase (100 μg/ml).
F+ coliphage genogrouping.
F+ coliphage isolates were also subjected to molecular typing to distinguish the four groups of F+ RNA coliphages (groups I, II, III, and IV) by RT-PCR of the replicase gene and to F+ DNA coliphage analysis by PCR (47). A reverse line blot (RLB) hybridization assay was performed as previously described (47) to confirm the RT-PCR-amplified products. A new RT-PCR assay was developed to amplify the levivirus capsid region, using the DL10 and DL11 primers at 0.8 μM (Table 1). Capsid region amplification used previously reported reaction conditions (47), with the modification of the RT and annealing steps being increased to a temperature of 50°C. F+ RNA and F+ DNA coliphage field isolates were also genogrouped by nucleic acid sequencing (UNC Nucleic Acids Core Facility, Chapel Hill, NC). Sequences were aligned using free software (Bioedit and Chromas Lite v. 2.0) (18), and phylogenetic trees were created using Jukes and Cantor distance estimation and 100 bootstrap values (TreeCon v. 1.3b).
TABLE 1.
Oligonucleotide primers for amplification of levivirus capsid region
| Oligonucleotide | Sequence (5′-3′)a | Orientation | Tmb (°C) | Location (nt)c |
|---|---|---|---|---|
| DL10 | GTC GAY AAT GGC GGW AC | + | 52 | 1365-1381 |
| DL11 | ATC GCG AGK RHG ATC HAT AC | − | 53.3 | 1795-1814 |
Using IUPAC codes for degenerate positions.
Melting temperature.
Based on strain MS2 sequence (NCBI accession no. NC_001417).
Rabbit antiserum production and collection.
To generate polyclonal antibodies against F+ coliphages, New Zealand White rabbits were given intradermal inoculations with each F+ RNA coliphage group (group I, MS2; group II, GA; group III, Qβ; and group IV, SP and Fi) and with F+ DNA coliphages (Fd, F1, M13, Φ15, Φ16, and Φ18). Initial virus inocula and a 1-month booster had titers of 1010 to 1011 PFU/ml and were partially purified and suspended in Freund's complete adjuvant (7). Antisera were collected from rabbits at 30, 45, 60, and/or 90 days postimmunization to obtain polyclonal rabbit immunoglobulins against coliphage antigens and were stored at −20°C. No purification was performed to separate immunoglobulin G (IgG) or other immunoglobulin classes or other serum constituents. Anti-MS2 serum had a protein concentration of about 3 mg/ml, while other serum protein levels were not measured.
Antiserum labeling of agglutinable particles.
For the F+ coliphage latex agglutination and typing (CLAT) assay, polystyrene particles were first labeled with F+ coliphage antisera. A 1% suspension of 0.29-μm-diameter polystyrene particles (OptiBind particles; Seradyn Inc., Indianapolis, IN) was made from the commercial 10% stock solution of particles by dilution in either phosphate-buffered saline (PBS; 0.136 M sodium chloride, 2.68 mM potassium chloride, 0.88 mM monobasic potassium phosphate, 3.4 mM dibasic sodium phosphate, pH 7.2 and 8.2) or citrate phosphate (CP) buffer (1.36 mM citric acid, 7.28 mM dibasic sodium phosphate, pH 6.2). Rabbit antisera against F+ coliphages or PBS (negative control) was added in an equal volume to that of the polystyrene particles to the 1% polystyrene particle-buffer solution. The antibody-particle-buffer mixtures were agitated by being pipetted up and down for several seconds (not vortexed) and then rocked at 150 rpm on a rotary platform (Orbit shaker; Lab-Line Instruments, Melrose Park, IL) for 1 hour at room temperature to facilitate hydrophobic adsorption of antibodies to particles. Samples were then microcentrifuged at 14,000 rpm (Eppendorf 5415C centrifuge; Brinkman Instruments, Westbury, NY) for 5 minutes, and the unbound antiserum in the supernatant was decanted from the antiserum-labeled particles in the pellet. The pellet was resuspended by pipetting (not vortexing) to give a 1% particle solution in either PBS-0.01% bovine serum albumin (BSA; Sigma-Aldrich, St. Louis, MO) (pH 7.2 or 8.2) or CP-0.01% BSA (pH 6.2) to match the original buffer. BSA was used to block unbound particle binding sites and to create a more stable solution for long-term storage. Labeled particles were stored at 4°C or used directly. Five F+ RNA coliphage antiserum-labeled particle suspensions (anti-MS2, anti-GA, anti-QB, anti-SP, and anti-Fi) and six F+ DNA coliphage antiserum-labeled particle suspensions (anti-Fd, anti-F1, anti-M13, anti-Φ15, anti-Φ16, and anti-Φ18) were prepared.
F+ coliphage agglutination assay and optimization.
Equal 2.5-μl volumes of antibody-labeled particles and coliphage enrichment cultures (or controls) were mixed on a black cardboard card (agglutination cards; Pro-Lab Diagnostics, Austin, TX) with a toothpick and then rocked by hand for 30 seconds. Coliphage-positive samples showed agglutination within 30 to 60 seconds, as visualized by the naked eye, with particles clumping together due to antibodies on different particle-binding coliphages (Fig. 1). Negative samples where no coliphages were detected appeared as a cloudy or “milky” liquid suspension of particles with no visible clumping.
FIG. 1.
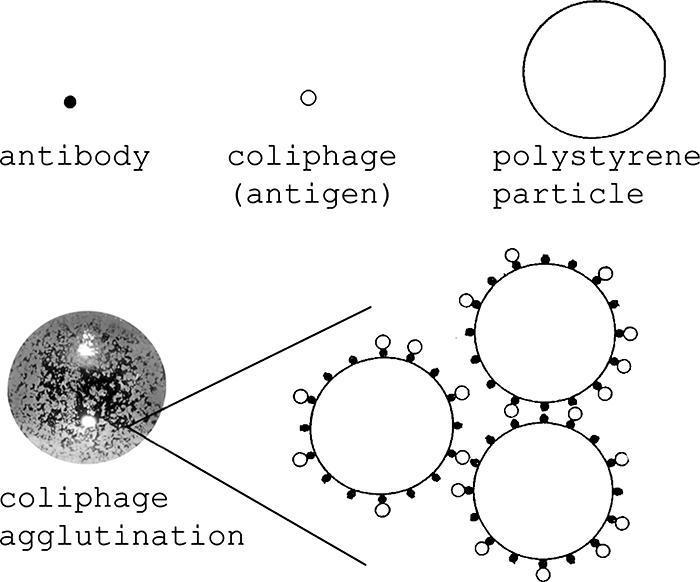
Coliphage agglutination diagram. Coliphage agglutination was visualized after mixing equal volumes of coliphage enrichments with antibody-labeled polystyrene particles for 30 seconds. (Modified from Journal of General Virology [4] with permission of the publisher.)
To determine the appropriate types and concentrations of antisera for F+ coliphage typing and detection, a diverse panel of 32 F+ RNA and F+ DNA coliphage field isolates (confirmed by nucleic acid sequencing of the replicase gene of F+ RNA coliphages and gene IV of F+ DNA coliphages) (47), prototype strains (F+ RNA coliphages MS2, GA, Qβ, SP, and Fi and F+ DNA coliphages F1, Fd, and M13), and negative controls (unlabeled particles and bacterial host cultures in TSB) was tested. Optimization experiments used a “checkerboard” titration system with combinations of various amounts of antigen and antisera (serial twofold dilutions of antisera from 1:4 to 1:128) per sample. The lower detection limit of the CLAT assay was determined using 5-μl volumes of enriched F+ RNA and F+ DNA coliphage prototype strains and fivefold serial dilutions thereof.
Protein assay for antisera.
A protein detection assay (BCA protein assay kit; Pierce, Rockford, IL) was used according to the manufacturer's instructions to determine the levels of antisera adsorbed to polystyrene particles and in stocks of antisera. Briefly, the absorbance at 562 nm (by spectrophotometry) was used to generate an albumin standard curve, which was confirmed to have an r2 value of >99% and then used for comparison to unknown samples. The amount of antiserum labeling particles was taken to be the initial amount of protein added to particles minus the amount of unbound protein in the supernatant after centrifugation.
Statistical methods.
Proportions of coliphages detected by CLAT and RLB hybridization assays were compared using a two-sided Z test with a preset significance level of 0.05, and P values are reported. The Kruskal-Wallis test, a nonparametric analysis of variance, and Dunn's multiple comparison test were used to compare more than two variables, including different buffers for antibody binding efficiency and antibody dilutions. Statistics were calculated in Excel and InStat (version 3.06; GraphPad Software Inc.).
RESULTS
Rapid F+ coliphage culture.
A modified version of EPA method 1601 was used to rapidly enrich F+ RNA coliphage prototype strains in culture broths of host E. coli Famp initially inoculated with 1 to 3 PFU of F+ RNA coliphage (MS2, Qβ, Sp, or Fi) and incubated at 35 to 37°C. Enrichment of these low levels of coliphage produced progeny coliphage at levels of 1.2 × 105 to 5.3 × 106 PFU/ml in 120 min and 4.3 × 106 to 5.5 × 108 PFU/ml in 180 min (Fig. 2). Rapid coliphage culture was achieved by increasing the concentration of the log-phase E. coli Famp host in broth cultures from about 104 CFU/ml in the overnight culture approach to as much as 107 CFU/ml in the new rapid approach. E. coli Famp reached stationary-phase growth in 180 min, with levels of 7.7 × 108 to 4.4 × 109 CFU/ml (Fig. 2).
FIG. 2.

Rapid culture enrichment of F+ RNA coliphage prototype strains (A) MS2, (B) Qβ, (C) Sp, and (D) Fi (squares) with host E. coli Famp (circles). Error bars showing standard deviations for coliphages (n = 3) are obscured by some square data points. The preculture level of E. coli Famp was 1 × 107 CFU/ml. E. coli levels during the experiment were measured by spectrophotometric absorbance at 520 nm.
Efficiency of adsorption of antisera to polystyrene particles.
Because the adsorption of immunoglobulins varies with the isoelectric points of the antibodies in sera, the pH of the adsorption buffer and the electrolyte content were varied by employing three buffer pH levels, 6.2, 7.2, and 8.2, at six antiserum dilutions (1:4 to 1:128) to examine their effects on anti-MS2 serum binding to polystyrene particles. Binding of antiserum to polystyrene particles was measured by a spectrophotometric protein detection assay. The saturation point for polystyrene particles with anti-MS2 serum was the 1:32 dilution, with decreased binding efficiency both above and below this saturation point (Table 2). The highest binding efficiencies were in PBS at pH 7.2, with 106% ± 1% and 100% ± 2% binding of antisera at 1:64 and 1:32 antiserum dilutions, respectively (Table 2). PBS at pH 7.2 was significantly better than CP buffer at pH 6.2 or PBS at pH 8.2 at promoting adsorption of anti-MS2 serum to particles, and significant differences were seen among the three pH buffers at antiserum dilutions of 1:16 (P = 0.0265), 1:32 (P = 0.0036), and 1:64 (P = 0.0379) (Table 2). Subsequent antiserum binding assays used PBS at pH 7.2 as the optimized adsorption buffer.
TABLE 2.
Binding efficiency of diluted MS2 antiserum to polystyrene particles
| Buffer solution | % Binding efficiency for antiserum dilution (mean ± SD)a
|
|||||
|---|---|---|---|---|---|---|
| 1:4 | 1:8 | 1:16* | 1:32* | 1:64* | 1:128 | |
| CP (pH 6.2) | 35 ± 6 | 50 ± 4 | 76 ± 4* | 90 ± 6 | 71 ± 3* | 49 ± 9 |
| PBS (pH 7.2) | 40 ± 2 | 50 ± 3 | 71 ± 0.1 | 100 ± 2* | 106 ± 1* | 39b |
| PBS (pH 8.2) | 38 ± 3 | 46 ± 4 | 54 ± 2* | 74 ± 3* | 78 ± 4 | 50 ± 2 |
Data are means of three replicates. *, statistically significant difference among all three variables in a column (for asterisks in column headings) or between two variables within a column (for asterisks on individual values). Significance was set at an α level of 0.05.
Average of two replicates with no standard deviation.
Optimizing particle agglutination by F+ coliphages.
A series of experiments explored and optimized CLAT with various types and concentrations of antisera in “checkerboard” assays, based on true- and false-positive and true- and false-negative agglutination with a diverse panel of 32 nucleic acid-sequenced F+ coliphage field isolates, F+ coliphage prototype strains, and negative controls. Optimal concentrations of antisera were selected to detect truly positive reactions in the F+ coliphage panel while minimizing nonspecific agglutination and false-positive reactions. As shown in Table 3, CLAT detected and typed F+ RNA coliphage prototype strains into each of four serogroups and gave true negative results for other F+ RNA and F+ DNA coliphage prototype strains, with the exceptions of anti-Fi sera cross-reacting with F+ RNA coliphage strain Sp (Table 3). No agglutination occurred when CLAT was performed with negative controls of TSB alone and stationary-phase E. coli cultures in TSB (Table 3). For F+ DNA coliphage detection, CLAT could detect all F+ DNA coliphage reference strains but could not serotype F+ DNA coliphage field strains. Anti-Fd, anti-F1, anti-M13, and anti-Φ16 sera reacted with the three F+ DNA coliphage prototype strains, while no F+ DNA coliphage antisera reacted with F+ RNA coliphages or negative controls (Table 3). Anti-M13 serum at the 1:8 dilution was the most reactive antiserum and was the only antiserum to detect all 16 F+ DNA coliphage field strains (data not shown). Anti-Φ15 and anti-Φ18 sera gave only weakly positive agglutination at the 1:4 dilution and therefore were not pursued further.
TABLE 3.
Reaction matrix for testing agglutination of antiserum-coated particles with F+ coliphage antigens
| F+ coliphage prototype strain or control | Presence of F+ coliphage antiserum-labeled particlesa
|
Negative control (no antisera) | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| F+ RNA coliphage (antiserum dilution)
|
F+ DNA coliphage (antiserum dilution)
|
|||||||||||
| MS2 (1:16) | GA (1:32) | Qβ (1:8) | SP (1:16) | Fi (1:16) | Fd (1:16) | F1 (1:16) | M13 (1:8) | Φ15 (1:4) | Φ16 (1:16) | Φ18 (1:4) | ||
| F+ RNA coliphages | ||||||||||||
| MS2 | + | − | − | − | − | − | − | − | − | − | − | − |
| GA | − | + | − | − | − | − | − | − | − | − | − | − |
| Qβ | − | − | + | − | − | − | − | − | − | − | − | − |
| SP | − | − | − | + | + | − | − | − | − | − | − | − |
| Fi | − | − | − | − | + | − | − | − | − | − | − | − |
| F+ DNA coliphages | ||||||||||||
| Fd | − | − | − | − | − | + | + | + | + | + | − | − |
| F1 | − | − | − | − | − | + | + | + | − | + | − | − |
| M13 | − | − | − | − | − | + | + | + | + | + | + | − |
| Negative controls | ||||||||||||
| E. coli Famp in TSB | − | − | − | − | − | − | − | − | − | − | − | − |
| TSB | − | − | − | − | − | − | − | − | − | − | − | − |
Checkerboard titration of antiserum-labeled particles (antiserum dilutions of 1:4, 1:8, 1:16, 1:32, 1:64, and 1:128) to empirically determine optimum antibody dilutions for detecting and typing a diverse panel of 32 nucleic acid-sequenced F+ RNA and F+ DNA coliphage field isolates, 8 reference strains, and negative controls. The table gives a summary of agglutination results with F+ coliphage reference strains and controls at optimum antibody dilutions. +, antiserum-labeled particles were detected; −, no particles were detected.
Lower detection limit of CLAT for F+ coliphages.
The detection limit of the CLAT assay was determined using F+ coliphage prototype strains cultured overnight by EPA method 1601. Samples were scored as positive or negative by the presence of agglutination. The detection limit for F+ RNA coliphages ranged from 5 × 103 to 1 × 105 PFU depending upon the antiserum used (Table 4). The detection limit for F+ DNA coliphages was 1 × 106 to 5 × 106 PFU (Table 4).
TABLE 4.
Lower detection limits for F+ coliphage prototype strains, determined using antiserum-labeled polystyrene particles
| F+ coliphage prototype strain count (PFU)a | Detection of F+ coliphage antiserum-labeled particlesb
|
||||||
|---|---|---|---|---|---|---|---|
| F+ RNA coliphage (antiserum dilution)
|
F+ DNA coliphage (antiserum dilution)
|
||||||
| MS2 (1:16) | GA (1:32) | Qβ (1:8) | SP (1:16) | Fi (1:16) | M13 (1:8) | Fd (1:16) | |
| 5 × 107 | ND | ND | ND | ND | ND | + | + |
| 1 × 107 | ND | ND | ND | ND | ND | + | + |
| 5 × 106 | ND | ND | ND | + | + | + | + |
| 1 × 106 | ND | ND | + | + | + | − | + |
| 5 × 105 | + | ND | + | + | + | − | − |
| 1 × 105 | + | ND | + | + | + | − | − |
| 5 × 104 | − | + | − | − | + | − | − |
| 1 × 104 | − | + | − | − | − | − | − |
| 5 × 103 | − | + | − | − | − | − | − |
| 1 × 103 | − | − | − | − | − | − | − |
| 5 × 102 | − | − | − | − | − | − | − |
F+ coliphage prototype strains (MS2, GA, Qβ, SP, Fi, M13, and Fd) were tested against their corresponding antisera.
+, antiserum-labeled particles were detected; −, no particles were detected; ND, not done.
Application of CLAT to serotype F+ RNA coliphage field isolates.
A diverse panel of F+ RNA and DNA coliphage field isolates were recovered from shellfish and water at 10 estuaries on the East, West, and Gulf Coasts of the United States by lysis zone isolation and overnight reenrichment culture by EPA method 1601. These coliphage isolates were assayed by both CLAT and (for F+ RNA coliphages) RT-PCR amplification with RLB hybridization genotyping (47). Of the 192 F+ RNA coliphage field isolates tested, CLAT correctly serotyped 185 and RLB hybridization correctly genotyped 177. CLAT and RLB hybridization typed the same number of group I isolates, but CLAT typed significantly more group II isolates than did RLB hybridization (P = 0.006) (Table 5). RLB hybridization typed 15 more F+ RNA coliphage group III isolates and 4 more F+ RNA coliphage group IV isolates than did CLAT, which were statistically significant differences (P < 0.0002). The false-negative rates were 4% for CLAT and 8% for RLB hybridization, which is a statistically significant difference (P < 0.0002). Both typing methods gave no false-positive results when challenged with 34 known F+ DNA coliphage field isolates (Table 5). Because CLAT serogrouping and RLB hybridization genogrouping provided different results for a small percentage of isolates, these differences were further explored by nucleic acid sequencing.
TABLE 5.
CLAT detection and serotyping of F+ RNA coliphage field isolatesd
| F+ RNA detection and typing method | No. of sero/genogroup-positive F+ RNA coliphage field isolatesc (n = 192)
|
No. (%) of false-negative results for F+ RNA coliphage field isolatesc | |||
|---|---|---|---|---|---|
| I | II | III | IV | ||
| CLATa | 101 | 90 | 13 | 1 | 7 (3.6) |
| RLB hybridizationb | 101 | 67 | 28 | 5 | 15 (7.8) |
CLAT assay sera were as follows: anti-MS2 serum at 1:16 dilution for group I, anti-GA serum diluted 1:32 for group II, anti-Qβ serum diluted 1:8 for group III, and anti-Sp and anti-Fi sera, both diluted 1:16, for group IV.
See reference 47.
Field isolates were plaque-purified enrichments of coliphages recovered from estuarine water and shellfish.
There were no false-positive results for F+ DNA coliphage field isolates (n = 34).
Capsid nucleic acid sequencing of discordantly typed F+ RNA leviviruses.
The observed inconsistencies between serogrouping and genogrouping results for 24 of 192 F+ RNA coliphage field strains shown in Table 5 were further analyzed by nucleotide sequencing of the capsid genomic region. The capsid regions of the 24 problematic F+ RNA coliphage field strains were RT-PCR amplified, sequenced, and arranged in a phylogenetic tree alongside the CLAT and RLB hybridization grouping results (Fig. 2). Capsid sequence analysis showed 19 isolates clustered with F+ RNA coliphage group I at 90% sequence similarity, 17 of which were classified by CLAT into serogroups I and II (Fig. 3). Five isolates clustered as F+ RNA coliphage group II, with slightly less than 90% sequence similarity, and CLAT serogrouping was in agreement for all five of these isolates (Fig. 3).
FIG. 3.

Phylogenetic tree of F+ RNA coliphage capsid region for 24 field isolates discordantly typed by RLB hybridization genogrouping and CLAT serogrouping. The phylogenetic tree is based on a 344-nucleotide region of the 392-nucleotide levivirus capsid gene, using Jukes and Cantor distance estimations and 100 bootstrap values, as indicated at tree nodes. na, no typing data available.
Application of CLAT to detect and type F+ DNA coliphage field isolates.
A diverse panel of 164 F+ DNA coliphage field isolates and 132 F+ RNA coliphage field isolates were recovered from shellfish and water at 10 estuaries on the East, West, and Gulf Coasts of the United States by lysis zone isolation and overnight reenrichment culture by EPA method 1601. Subsequently, these coliphage isolates were assayed by both the CLAT assay and an RNase infectivity neutralization assay for F+ DNA coliphages. The RNase infectivity neutralization assay scored coliphages as having either RNA or DNA nucleic acid and was used as a standard for comparing CLAT results. The CLAT assay detected 161 of 164 F+ DNA coliphage field isolates (98%), which was not statistically different from the 164 detections (100%) of the RNase assay (P = 0.82) (Table 6). The CLAT assay failed to detect 3 of 164 F+ DNA coliphage isolates and gave false-positive detection of 3 of 132 F+ RNA coliphage field isolates (2%) (Table 6). The detection rate for F+ DNA coliphages with M13 antiserum-coated particles was 83% (data not shown), which was improved to 98% detection by including a second level of screening of all negative samples with Fd antiserum particles.
TABLE 6.
CLAT detection of F+ DNA coliphage field isolates
| F+ DNA coliphage detection method | No. (%) of F+ DNA coliphage field isolatesc (n = 164)
|
No. (%) of false-positive results for F+ RNA coliphage field isolatesc (n = 132) | |
|---|---|---|---|
| True positive | False negative | ||
| CLATa | 161 (97.7)b | 3 (2.3) | 3 (1.8) |
| RNase neutralization assay | 164 (100) | 0 | 0 |
CLAT for F+ DNA coliphages used both M13 antiserum (1:8 antiserum dilution) and Fd antiserum (1:16 antiserum dilution).
No significant difference was detected between the proportions of truly positive F+ coliphage DNAs detected by the two methods (P value = 0.82).
Field isolates were plaque-purified enrichments of coliphages recovered from estuarine water and shellfish.
DISCUSSION
The simple, rapid, and inexpensive F+ coliphage CLAT method is a novel tool for monitoring the microbiological quality of water and other environmental media in both the developing and the developed world and for identifying and tracking human and animal fecal waste sources. The CLAT assay is a novel application of the agglutination immunoassay originally developed for use in clinical medicinal diagnostics. While clinical diagnostic samples typically have high titers of antigens and do not require a culture step before agglutination assays, water and other environmental samples have low levels of antigens (in our case, coliphages) and require a culture step or other antigen enrichment step before detection by particle agglutination.
Other investigators have determined the lowest host concentration needed for bacteriophage attachment and replication (50, 54), while in this study the host concentration was increased to determine the highest level of host useful for rapid coliphage culture. This approach employed a 120- to 180-min culture step, modifying EPA method 1601, to rapidly enrich both F+ RNA and DNA coliphages to levels amenable to particle agglutination. Preliminary trials with water and mussels from San Diego Bay, CA, and Tijuana River, CA, showed that rapid F+ coliphage cultures gave equivalent results to those obtained by EPA method 1601 with overnight enrichment (data not shown). The rapid F+ coliphage culture enrichment can be assayed directly by CLAT, with no plaque purification or centrifugation.
CLAT was developed with the long-term goal of field-portable application, which necessitates the use of simple methods, robust but nonsterile techniques, and inexpensive and stable detection materials. A possible impediment to future field adaptability of the CLAT method is the culturing of organisms in a field-portable water bath. Exploratory work on the field adaptability of culturing used an insulated cooler, an aquarium heater, and a deep-cycle marine battery to create an inexpensive 35°C water bath that runs for more than 5 h (data not shown). More research is needed to develop and evaluate field-portable culturing methods for coliphages and fecal indicator bacteria for use in both the developing and the developed world.
In this study, CLAT results were scored as positive or negative, while quantification is possible by most-probable-number culture enrichment where replicate volumes in dilution are scored as positive or negative. The presence of host bacteria in enrichment cultures did not adversely affect the detectability of F+ coliphages by CLAT. Coliphage-enriched water samples were analyzed by CLAT, which accurately detected and subtyped prototype F+ RNA coliphage strains into serogroups I, II, III, and IV and did not react with F+ DNA coliphage prototype strains or controls. Subgrouping of F+ RNA coliphages is useful for microbial source tracking (15, 17, 23, 35) but is not used routinely because it is time-consuming and more expensive than bacteriological analysis and requires scientific knowledge and technical skill. This study improves access to F+ RNA coliphage detection and source tracking by making it easier, as affordable as bacteriological analysis, and rapid, which invites future work on a field-portable coliphage method.
In validation studies of the use of the F+ RNA coliphage CLAT assay for serotyping a large panel of F+ coliphage field strains, CLAT sensitivity was 96.4%, and its specificity was 100%. These findings are similar to those of previous F+ RNA coliphage characterization studies, where serotyping classified 99.5% of isolates (23), genotyping by probe hybridization classified 96.6% of isolates (23), and RT-PCR followed by probe hybridization correctly classified 97.8% of isolates (47). The CLAT assay had a similar performance and typing ability as an RT-PCR-probe hybridization assay (47), compared using the same panel of F+ coliphage field strains. Hsu et al. (23) also compared genotyping and serotyping outcomes by using a common isolate panel and arrived at a similar grouping outcome performance to that reported in this study.
The few inconsistencies found between serogrouping and genogrouping results were further investigated in this study by examining virus capsid genes to better interpret the CLAT results. It was hypothesized that studying the virus capsid genes of these problematic F+ RNA coliphage strains would provide a robust genetic approach consistent with antiserum binding to distinct capsid (antigen) epitopes and might reconcile inconsistencies between genogrouping and serogrouping (2). A new RT-PCR assay targeting the levivirus capsid gene was created for this purpose, which itself may be a stand-alone method for F+ RNA coliphage genogroup I and II source tracking. When amplified levivirus capsid genes were sequenced and grouped phylogenetically, the findings did not agree 100% with either RLB hybridization genogrouping or CLAT serogrouping findings, which indicates that there may be multiple reasons for typing differences. Previous F+ RNA coliphage serotyping and genotyping inconsistencies were shown to result from a change in three amino acids of the coat protein—causing an F+ RNA coliphage group II strain to be serotyped as group I, although it was 95% genetically similar to group II (2, 20). In our study, environmental F+ RNA coliphage isolates may be serological intermediaries between groups I and II by sharing surface proteins for antibody binding as a result of prior genetic crossover by recombination events (antigenic shifts) or by progressive mutations common to single-stranded RNA viruses that occur at rates of 10−3 to 10−4 per incorporated nucleotide (genetic drifts) (22). Also, the group-specific antiserum generated against a single prototype strain may not be representative of the diversity of strains in the environment. Further analysis of levivirus epitopes by nucleic acid and protein microarrays and by monoclonal antibody screening may give better insights into the reasons for these discrepancies and the robustness of serogroup predictions by CLAT.
The developed F+ DNA coliphage CLAT assay provides a simple, robust, rapid, and affordable means to facilitate detection of all F+ coliphages, regardless of whether or not F+ RNA coliphages are present. F+ DNA coliphages as fecal indicator viruses have been isolated from wastewater treatment plants and from swine, gull, and cattle waste (8). They have been found in higher proportions than F+ RNA coliphages in surface waters impacted by humans and animals during storm events than during background flows, in warmer waters (8), and in epidemiological-microbiological studies of illness risks from recreational use of water contaminated by nonpoint fecal sources (9). Efforts to subtype F+ DNA coliphages have not been as successful as those for F+ RNA coliphages. In the CLAT assay, six F+ DNA coliphage polyclonal antisera were cross-reactive among the F+ DNA coliphage strains tested and thus could not be used for subtyping, as previously observed (29). Attempts at F+ coliphage genotyping have shown three genetic clusters, called M13, Fd, and CH, based on >5% nucleotide sequence diversity in a 190-nucleotide region of inovirus gene IV (47). However, analyses of other inovirus genes are needed to confirm these distinct gene clusters and to determine if a confirmed grouping method can be established for reliable and practical F+ DNA coliphage fecal source tracking (47).
The CLAT detection limit was sufficiently low for both F+ RNA and DNA coliphages that they can be detected readily after enrichment of water samples for 2 to 3 h. While the lower detection limit was lower for F+ RNA coliphages than for F+ DNA coliphages, this difference does not pose a problem for F+ DNA coliphage CLAT detection, because these coliphages enrich to 2- to 3-log10 higher levels than do F+ RNA coliphages. In this study, all but 3 of 164 F+ DNA coliphage field isolates were detected by the CLAT assay. No difference was observed in the speed or strength of agglutination for the small, icosahedral RNA coliphages compared to the long, rod-shaped DNA coliphages tested (data not shown), suggesting that virus morphology has little influence on CLAT detection. Antiserum concentration (dilution) had an important role in the sensitivity and specificity of the CLAT assay, but the concentrations of the specific immunoglobulin types responsible for coliphage agglutination were not determined. Characterization of the anti-coliphage immunoglobulin types and their concentrations and agglutination reactivities would be informative for the development of standard reagents for the CLAT assay. Further efforts to create and test a coliphage monoclonal antibody library may also improve the sensitivity, specificity, and availability of the CLAT assay, if the monoclonal antibodies are as amenable to and as effective in agglutination assays as were the polyclonal sera tested.
While agglutination assays are available for viruses of plants and animals (4, 24, 38), we were unable to find evidence of their existence for bacteriophages or, specifically, coliphages. Other fecal indicator viruses, such as Bacteroides fragilis phages, Salmonella phages, and somatic coliphages as well as phages in terrestrial and marine environments, could possibly be detected by agglutination assays. Phages in aquatic and terrestrial environments are not well characterized because often <1% of their natural hosts are culturable, resulting in the “great plaque count anomaly” (49, 51). Agglutination assays can potentially detect bacteriophage strains that infect bacterial hosts but do not form plaques, thereby obviating or circumventing the need for conventional serotyping methods based on neutralization of virus infectivity. Marine bacteriophages grow to titers as high as 108 PFU/ml of seawater (52), a titer that may be compatible with direct agglutination detection for further characterization and a better understanding of their occurrence and ecology. The success of this newly developed CLAT assay for F+ coliphages suggests that additional applications of this assay to other bacteriophages may also be possible and provide useful information about coliphage occurrence, ecology, properties, and public health risks.
Acknowledgments
This work was supported by graduate research fellowships from the National Water Research Institute and by National Estuarine Research Reserve grant NA06NOS4200057.
We thank Douglas Wait and Greg Lovelace for their technical assistance.
Footnotes
Published ahead of print on 4 May 2007.
REFERENCES
- 1.Abedon, S. T. 1990. The ecology of bacteriophage T4. Ph.D. thesis. The University of Arizona, Tucson.
- 2.Adhin, M. R., A. Hirashima, and J. Van Duin. 1989. Nucleotide sequences from the ssRNA bacteriophages JP34 resolves the discrepancy between serological and biophysical classification. Virology 170:238-242. [DOI] [PubMed] [Google Scholar]
- 3.Alderisio, K. A., D. A. Wait, and M. D. Sobsey. 1996. Detection and characterization of male-specific RNA coliphages in a New York City reservoir, p. 133-142. In J. J. McDonnell, D. L. Leopold, J. B. Stribling, and L. R. Neville (ed.), Watershed restoration management New York City water supply studies. American Water Resources Association, Herndon, VA.
- 4.Bercks, R., and G. Querfurth. 1971. The use of the latex test for the detection of distant serological relationships among plant viruses. J. Gen. Virol. 12:25-32. [DOI] [PubMed] [Google Scholar]
- 5.Boehm, A. B., S. B. Grant, J. H. Kim, S. L. Mowbray, C. D. McGee, C. D. Clark, D. M. Foley, and D. E. Wellman. 2002. Decadal and shorter period variability of surf zone water quality at Huntington Beach, California. Environ. Sci. Technol. 36:3885-3892. [DOI] [PubMed] [Google Scholar]
- 6.Chappuis, F., S. Rijal, U. K. Jha, P. Desjeux, B. M. S. Karki, S. Koirala, L. Loutan, and M. Boelaert. 2006. Field validity, reproducibility and feasibility of diagnostic tests for visceral leishmaniasis in rural Nepal. Trop. Med. Int. Health 11:31-40. [DOI] [PubMed] [Google Scholar]
- 7.Chung, H.-M. 1993. F-specific coliphages and their serogroups, and Bacteroides fragilis phages as indicators of estuarine water and shellfish quality. Ph.D. thesis. The University of North Carolina, Chapel Hill.
- 8.Cole, D., S. D. Long, and M. D. Sobsey. 2003. Evaluation of F+ RNA and DNA coliphages as source-specific indicators of fecal contamination in surface waters. Appl. Environ. Microbiol. 69:6507-6514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Colford, J. M., Jr., T. J. Wade, K. C. Schiff, C. C. Wright, J. G. Griffith, S. K. Sandhu, S. Burns, J. Hayes, M. Sobsey, G. Lovelace, and S. B. Weisberg. 2007. Water quality indicators and the risk of illness in non-point source impacted recreational waters. Epidemiology 18:27-35. [DOI] [PubMed] [Google Scholar]
- 10.Contreras-Coll, N., F. Lucena, K. Mooijman, A. Havelaar, V. Pierz, M. Boque, A. Gawler, C. Holler, M. Lambiri, G. Mirolo, B. Moreno, M. Niemi, R. Sommer, B. Valentin, A. Wiedenmann, V. Young, and J. Jofre. 2002. Occurrence and levels of indicator bacteriophages in bathing waters throughout Europe. Water Res. 36:4963-4974. [DOI] [PubMed] [Google Scholar]
- 11.Craun, G. F., R. L. Calderon, and M. F. Craun. 2005. Outbreaks associated with recreational water in the United States. Int. J. Environ. Health Res. 15:243-262. [DOI] [PubMed] [Google Scholar]
- 12.Dore, W. J., K. Henshilwood, and D. N. Lees. 2000. Evaluation of F-specific RNA bacteriophage as a candidate human enteric virus indicator for bivalve molluscan shellfish. Appl. Environ. Microbiol. 66:1280-1285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dufour, A. P. 1984. Health effects criteria for fresh recreational waters. EPA-600-1-84-004. U.S. EPA, Cincinnati, OH.
- 14.Duran, A. E., M. Muniesa, X. Mendez, F. Valero, F. Lucena, and J. Jofre. 2002. Removal and inactivation of indicator bacteriophages in fresh waters. J. Appl. Microbiol. 92:338-347. [DOI] [PubMed] [Google Scholar]
- 15.Furuse, K., T. Sakurai, A. Hirashima, M. Katsuki, A. Ando, and I. Watanabe. 1978. Distribution of ribonucleic acid coliphages in south and east Asia. Appl. Environ. Microbiol. 35:995-1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Grandien, M., C.-A. Pettersson, L. Svensson, and I. Uhnoo. 1987. Latex agglutination test for adenovirus diagnosis in diarrheal disease. J. Med. Virol. 23:311-316. [DOI] [PubMed] [Google Scholar]
- 17.Griffin, D. W., R. Stokes, J. B. Rose, and J. H. Paul III. 2000. Bacterial indicator occurrence and the use of an F+ specific RNA coliphage assay to identify fecal sources in Homosassa Springs, Florida. Microb. Ecol. 39:56-64. [DOI] [PubMed] [Google Scholar]
- 18.Hall, T. A. 1999. BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp. Ser. 41:95-98. [Google Scholar]
- 19.Halstead, D. C., D. G. Beckwith, R. L. Sautter, L. Plosika, and K. A. Schneck. 1987. Evaluation of a rapid latex slide agglutination test for herpes simplex virus as a specimen screen and culture identification method. J. Clin. Microbiol. 25:936-937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Harigai, H., K. Furuse, Y. Inokuchi, and A. Hirashima. 1986. Characterization of an intergroup serological mutant from group II RNA phage GA. Microbiol. Immunol. 30:1247-1257. [DOI] [PubMed] [Google Scholar]
- 21.Harwood, V. J., A. D. Levine, T. M. Scott, V. Chivukula, J. Lukasik, S. R. Farrah, and J. B. Rose. 2005. Validity of the indicator organism paradigm for pathogen reduction in reclaimed water and public health protection. Appl. Environ. Microbiol. 71:3163-3170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Holland, J. J. 2006. Transitions in understanding of RNA viruses: a historical perspective. Curr. Top. Microbiol. Immunol. 299:371-401. [DOI] [PubMed] [Google Scholar]
- 23.Hsu, F. C., Y. S. Shieh, J. van Duin, M. J. Beekwilder, and M. D. Sobsey. 1995. Genotyping male-specific RNA coliphages by hybridization with oligonucleotide probes. Appl. Environ. Microbiol. 61:3960-3966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hughes, J. H., A. V. Tuomari, D. R. Mann, and V. V. Hamparian. 1984. Latex immunoassay for rapid detection of rotavirus. J. Clin. Microbiol. 20:441-447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jiang, S., R. Noble, and W. Chu. 2001. Human adenoviruses and coliphages in urban runoff-impacted coastal waters of southern California. Appl. Environ. Microbiol. 67:179-184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jofre, J., E. Olle, F. Ribas, A. Vidal, and F. Lucena. 1995. Potential usefulness of bacteriophages that infect Bacteroides fragilis as model organisms for monitoring virus removal in drinking water treatment plants. Appl. Environ. Microbiol. 61:3227-3231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Koopmans, M. 2005. Outbreaks of viral gastroenteritis: what's new in 2004? Curr. Opin. Infect. Dis. 18:295-299. [DOI] [PubMed] [Google Scholar]
- 28.Leecaster, M. K., and S. B. Weisberg. 2001. Effect of sampling frequency on shoreline microbiology assessments. Mar. Pollut. Bull. 42:1150-1154. [DOI] [PubMed] [Google Scholar]
- 29.Long, S. C., S. S. El-Khoury, S. Oudejans, M. D. Sobsey, and J. Vinjé. 2005. Assessment of sources and diversity of male-specific coliphages for source tracking. Environ. Eng. Sci. 22:367-377. [Google Scholar]
- 30.Marot-Leblond, A., B. Beucher, S. David, S. Nail-Billaud, and R. Robert. 2006. Development and evaluation of a rapid latex agglutination test using a monoclonal antibody to identify Candida dubliniensis colonies. J. Clin. Microbiol. 44:138-142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Moce-Llivina, L., F. Lucena, and J. Jofre. 2005. Enteroviruses and bacteriophages in bathing waters. Appl. Environ. Microbiol. 71:6838-6844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.National Research Council of the National Academies. 2004. Indicators of waterborne pathogens. The National Academies Press, Washington, DC.
- 33.National Resources Defense Council. 2006. Testing the waters 2006: a guide to water quality at vacation beaches. National Resources Defense Council, New York, NY. http://www.nrdc.org/water/oceans/ttw/titinx.asp.
- 34.Novotny, C. P., and K. Lavin. 1971. Some effects of temperature on the growth of F pili. J. Bacteriol. 107:671-682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Osawa, S., K. Furuse, and I. Watanabe. 1981. Distribution of ribonucleic acid coliphages in animals. Appl. Environ. Microbiol. 41:164-168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Poullis, D. A., R. W. Attwell, and S. C. Powell. 2005. The characterization of waterborne-disease outbreaks. Rev. Environ. Health 20:141-149. [PubMed] [Google Scholar]
- 37.Ricciuti, C. P. 1972. Host-virus interactions in Escherichia coli: effects of stationary phase on viral release from MS2-infected bacteria. J. Virol. 10:162-165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Riggin, C. H., G. A. Beltz, C. H. Hung, R. M. Thorn, and D. J. Marciani. 1987. Detection of antibodies to human immunodeficiency virus by latex agglutination with recombinant antigen. J. Clin. Microbiol. 25:1772-1773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Singer, J. M., and C. M. Plotz. 1956. The latex fixation test. I. Application to the serologic diagnosis of rheumatoid arthritis. Am. J. Med. 21:888-892. [PubMed] [Google Scholar]
- 40.Smole, S. C., E. Aronson, A. Durbin, S. M. Brecher, and R. D. Arbeit. 1998. Sensitivity and specificity of an improved rapid latex agglutination test for identification of methicillin-sensitive and -resistant Staphylococcus aureus isolates. J. Clin. Microbiol. 36:1109-1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sobsey, M. D., K. J. Schwab, and T. R. Hanzel. 1990. A simple membrane filter method to concentrate and enumerate male-specific RNA coliphages. J. Am. Water Works Assoc. 82:52-59. [Google Scholar]
- 42.Stewart-Pullaro, J., J. W. Daugomah, D. E. Chestnut, D. A. Graves, M. D. Sobsey, and G. I. Scott. 2006. FRNA coliphage typing for microbial source tracking in surface waters. J. Appl. Microbiol. 101:1015-1026. [DOI] [PubMed] [Google Scholar]
- 43.Tsuda, S., M. Lameya-Iwaki, K. Hanada, Y. Kouda, M. Nikata, and K. Tomaru. 1992. A novel detection and identification technique for plant viruses: rapid immunofilter paper assay (RIPA). Plant Dis. 76:466-469. [Google Scholar]
- 44.U.S. EPA. 2001. Method 1601. Male-specific (F+) and somatic coliphage in water by two-step enrichment procedure. EPA-821-R-01-030. U.S. EPA, Washington, DC.
- 45.U.S. EPA. 2001. Method 1602. Male-specific (F+) and somatic coliphage in water by single agar layer (SAL) procedure. EPA-821-R-01-029. U.S. EPA, Washington, DC.
- 46.Van Regenmortel, M. H. V., C. M. Fauquet, D. H. L. Bishop, E. B. Carstens, M. K. Estes, S. M. Lemon, D. J. McGeoch, J. Maniloff, M. A. Mayo, C. R. Pringle, and R. B. Wickner. 2000. Virus taxonomy: classification and nomenclature of viruses. Seventh ICTV report. Academic Press, San Diego, CA.
- 47.Vinjé, J., S. J. Oudejans, J. R. Stewart, M. D. Sobsey, and S. C. Long. 2004. Molecular detection and genotyping of male-specific coliphages by reverse transcription-PCR and reverse line blot hybridization. Appl. Environ. Microbiol. 70:5996-6004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wade, T. J., N. Pai, J. N. S. Eisenberg, and J. M. Colford, Jr. 2003. Do U.S. Environmental Protection Agency water quality guidelines for recreational waters prevent gastrointestinal illness? A systematic review and meta-analysis. Environ. Health Perspect. 111:1102-1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Weinbauer, M. G. 2004. Ecology of prokaryotic viruses. FEMS Microbiol. Rev. 28:127-181. [DOI] [PubMed] [Google Scholar]
- 50.Wiggins, B. A., and M. Alexander. 1985. Minimum bacterial density for bacteriophage replication: implications for significance of bacteriophages in natural ecosystems. Appl. Environ. Microbiol. 49:19-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Williamson, K. E., M. Radosevich, and K. E. Wommack. 2005. Abundance and diversity of viruses in six Delaware soils. Appl. Environ. Microbiol. 71:3119-3125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wommack, K. E., and R. R. Colwell. 2000. Virioplankton: viruses in aquatic ecosystems. Microbiol. Mol. Biol. Rev. 64:69-114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Woody, M. A., and D. O. Cliver. 1995. Effects of temperature and host cell growth phase on replication of F-specific RNA coliphage Qβ. Appl. Environ. Microbiol. 61:1520-1526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Woody, M. A., and D. O. Cliver. 1997. Replication of coliphage Qβ as affected by host cell number, nutrition, competition from insusceptible cells and non-FRNA coliphages. J. Appl. Microbiol. 82:431-440. [DOI] [PubMed] [Google Scholar]
- 55.World Health Organization. 2002. World Health Report 2002: reducing risks, promoting healthy life. World Health Organization, Geneva, Switzerland. http://www.who.int/whr/2002/en/whr02_en.pdf.
- 56.World Health Organization. 2004. Guidelines for drinking-water quality, 3rd ed., vol. 1., p. 121-144. World Health Organization, Geneva, Switzerland. http://www.who.int/water_sanitation_health/dwq/gdwq3/en/. [Google Scholar]
- 57.Xu, X., M. Jin, Z. Yu, H. Li, D. Qiu, Y. Tan, and H. Chen. 2005. Latex agglutination tests for monitoring antibodies to avian influenza virus subtype H5N1. J. Clin. Microbiol. 43:1953-1955. [DOI] [PMC free article] [PubMed] [Google Scholar]