

Abstract
While much data exist in the literature about how Neisseria meningitidis adheres to and invades human cells, its behavior inside the host cell is largely unknown. One of the essential meningococcal attributes for pathogenesis is the polysaccharide capsule, which has been shown to be important for bacterial survival in extracellular fluids. To investigate the role of the meningococcal capsule in intracellular survival, we used B1940, a serogroup B strain, and its isogenic derivatives, which lack either the capsule or both the capsule and the lipooligosaccharide outer core, to infect human phagocytic and nonphagocytic cells and monitor invasion and intracellular growth. Our data indicate that the capsule, which negatively affects bacterial adhesion and, consequently, entry, is, in contrast, fundamental for the intracellular survival of this microorganism. The results of in vitro assays suggest that an increased resistance to cationic antimicrobial peptides (CAMPs), important components of the host innate defense system against microbial infections, is a possible mechanism by which the capsule protects the meningococci in the intracellular environment. Indeed, unencapsulated bacteria were more susceptible than encapsulated bacteria to defensins, cathelicidins, protegrins, and polymyxin B, which has long been used as a model compound to define the mechanism of action of CAMPs. We also demonstrate that both the capsular genes (siaD and lipA) and those encoding an efflux pump involved in resistance to CAMPs (mtrCDE) were up-regulated during the intracellular phase of the infectious cycle.
Neisseria meningitidis (meningococcus) is a transitory colonizer of the nasopharynx that occasionally is responsible for life-threatening disease. It is divided into 13 serogroups based on the immunological specificities of its capsular polysaccharides. Of these, five serogroups (A, B, C, Y, and W135) are responsible for most of the diseases worldwide.
However, the meningococcal capsule plays contrasting roles in pathogenesis. On one hand, the antiphagocytic properties of the disease-associated capsules are essential for bacterial growth in the bloodstream, a prerequisite for septicemia and meningitis (13). On the other hand, the expression of the capsular polysaccharide inhibits the colonization and invasion of the nasopharyngeal barrier by masking the meningococcal adhesins/invasins (10).
The presence of the capsule depends on both the possession and expression of the genes for capsule biosynthesis, modification, and transport, which are located in regions A, B, and C of the cps locus, respectively (7), mapping within a putative island of horizontally transferred DNA (IHT-A1) (38). In meningococcus-expressing capsules containing sialic acid (serogroups B, C, Y, and W135), region A comprises the genes siaA, siaB, and siaC, which are required for the synthesis of sialic acid, and the serogroup-specific gene siaD, encoding the polysialyltransferase. Capsular gene expression is regulated both by phase variation via slipped-strand mispairing or reversible insertion of mobile elements (10, 11) and at the transcriptional level. Indeed, there is evidence that capsule biosynthesis and assembly are down-regulated during the early stages of the infectious cycle to facilitate the adhesion to and invasion of the host cells (4).
In this study, the role of the capsular polysaccharide during the later stages of an infection (i.e., the intracellular phase) has been investigated by using a cellular infection model. We found that the capsule is fundamental for the intracellular survival of this microorganism in both human phagocytic and nonphagocytic cells. This finding prompted us to investigate the mechanism(s) by which the capsule protects meningococci in the intracellular environment. In particular, the hypothesis that the capsule may enhance the resistance to cationic antimicrobial peptides (CAMPs) has been tested.
CAMPs are important components of the host innate defense system against microbial infections, constitutively produced by phagocytic cells and inducibly expressed by epithelial cells following exposure to bacterial determinants (12). Based on structural features, CAMPs are classified into different categories: β-sheets stabilized by disulfide bonds, an amphipathic α-helix, loop structures with a single disulfide bond, cyclic peptides, and boat-like extended structures (12). N. meningitidis, as well as other pathogens, utilizes multiple mechanisms to modulate the resistance to CAMPs, including the action of the MtrC-MtrD-MtrE efflux pump, lipid A modification, and the type IV pilin secretion system (41). Although there is evidence that the capsular polysaccharide mediates the resistance of Klebsiella pneumoniae to CAMPs (3), this aspect has never been investigated in N. meningitidis.
MATERIALS AND METHODS
Bacterial strains and growth conditions.
N. meningitidis strains have been reported previously (2, 6, 16, 25, 31, 32). In particular, the origin, genotype, and phenotype of B1940 and its derivatives, the B1940 cps mutant, the B1940 siaD(−C) mutant, and the B1940 siaD(+C) mutant (where −C and +C indicate the deletion and insertion of a cytosine in a polycytidine repeat in the coding region of siaD, respectively), have been described previously (6, 10). The B1940 cps mutant lacks both the capsule and the lipooligosaccharide (LOS) outer core. The B1940 siaD(−C) and B1940 siaD(+C) mutants lack the capsule but retain the LOS outer core due to frameshift mutations, a cytosine deletion and a cytosine insertion in a polycytidine repeat located in the coding region of the polysialyltransferase gene (siaD), respectively. MC58, H44/76, NGP165, BF65, 1000, NGE31, NGF26, and NGH15 are serogroup B strains. All meningococci were cultured in gonococcus (GC) broth or agar with 1% Polyvitox at 37°C in a 5% CO2 incubator.
Adherence and invasion assays.
N. meningitidis adherence and invasion assays were performed as described previously (1, 44).
For standard invasion and intracellular viability assays, HeLa cells (ATCC CCL-2) were used. Several experiments were also repeated with HEp-2 cells (ATCC CCL-23) and Chang conjunctiva cells (ATCC CCL-20.2). It should be noted, however, that as a result of a well-known contamination event, HEp-2 (thought to be derived from an epidermoid carcinoma of the larynx) and Chang conjunctiva (thought to be derived from normal conjunctiva) cells present HeLa markers and have been established via HeLa cell contamination, as stated by the ATCC (http://www.atcc.com). These cells were grown in Dulbecco's modified Eagle medium (DMEM) with 10% fetal bovine serum and 2 mM l-glutamine. Invasion and intracellular viability assays were also done using THP-1 (ATCC TIB-202) and U-937 (ATCC CRL-1593.2) cell lines. These human monocyte cell lines were grown in RPMI 1640 medium supplemented with 2 mM l-glutamine and 10% fetal bovine serum and were differentiated to macrophages using 0.8 nM (for U937) or 8 nM (for THP-1) phorbol myristic acid for 3 to 5 days.
HeLa (or HEp-2 or Chang conjunctiva) cells were infected at a multiplicity of infection (MOI) of 50, while THP-1 and U-937 cells were infected at an MOI of 5 for 1 h, washed twice with phosphate-buffered saline to eliminate the majority of extracellular bacteria, exposed to gentamicin to kill remaining extracellular bacteria, and subsequently disrupted using saponin to release intracellular bacteria. Bacteria were plated, and CFU were counted the day after. Adherence was determined by omitting gentamicin treatment. When required, cells were reincubated in culture medium for various times after gentamicin treatment. Gentamicin treatment was performed at 100 μg/ml, a concentration 10-fold above the MIC for 30 min. Cells were then washed extensively with phosphate-buffered saline to remove gentamicin and dead extracellular bacteria and then lysed or reincubated in medium. In some experiments, during reincubation, cells were washed and medium was changed every hour to avoid reinfection. All strains used in this study had comparable MICs of gentamicin and were equally insensitive to saponin. In most experiments with HeLa, HEp-2, or Chang conjunctiva cells, bacteria were centrifuged (60 × g) onto cells to start the infection.
For dual infection/competition experiments, HeLa cells were coinfected with equal amounts of B1940 and B1940 siaD(−C) mutant bacteria at an MOI of 50. After 2, 4, or 6 h, HeLa cells were disrupted with saponin to release and count the number of intracellular bacteria by CFU method. The proportions of encapsulated and unencapsulated bacteria were determined by Wellcogen latex agglutination test for the direct detection of capsular antigen to N. meningitidis group B and Escherichia coli K1 (Abbott Murex, United Kingdom) on 100 randomly selected colonies.
Immunofluorescence analysis.
HeLa cells were seeded onto glass coverslips, infected, and processed for immunofluorescence to distinguish extracellular and intracellular bacteria as described previously (10, 17). Rabbit polyclonal anti-N. meningitidis antibody was obtained from ViroStat, while monoclonal H4A3 anti-Lamp1 was obtained from the Developmental Studies Hybridoma Bank at the University of Iowa. Primary and secondary antibodies diluted 1:500 were used. Specimens were viewed with a Zeiss LSM510 confocal microscope.
RNA procedures.
Total RNAs were extracted from intracellular bacteria during the infection of HeLa cells or control bacteria grown in culture medium as described previously (22).
The procedure for limited transcriptional analysis has been detailed previously (22, 25). Briefly, a partial Sau3AI-restricted genomic library from serogroup B strain H44/76 was constructed. Individual clones were digested, and Southern blot analysis was performed using cDNA probes derived from intracellular bacteria after 8 h of infection of HeLa cells or control bacteria grown for 8 h in culture medium. This procedure detects mRNA species present in the initial population at a frequency of at least 1 in 200.
Semiquantitative analysis of the siaD, lipA, mtrD, and mtrE transcripts, normalized to 16S rRNA or rho mRNA, was performed by real-time reverse transcriptase (RT) PCR (RT-PCR). Total RNAs (1 μg) from either intracellular bacteria or control bacteria grown without HeLa cells were reverse transcribed by using random hexamer (2.5 μM) with Superscript RT (Invitrogen). About 0.1 to 1% of each RT reaction was used to run real-time PCR on a SmartCycler system (Cepheid) with SYBR green JumpStart Taq ReadyMix (Sigma-Aldrich) and the primer pairs 16Suniv-1/16S-r (specific for 16S rRNA; amplicon length, 185 bp), rho-f/rho-r (specific for rho; amplicon length, 110 bp), siaD-f/siaD-r (specific for siaD; amplicon length, 144 bp), lipA-f/lipA-r (specific for lipA; amplicon length, 196 bp), mtrC-f/mtrC-r (specific for mtrC; amplicon length, 120 bp), mtrD-f/mtrD-r (specific for mtrD; amplicon length, 114 bp), and mtrE-f/mtrE-r (specific for mtrE; amplicon length, 124 bp) (Table 1). Real-time PCR samples were run in triplicate. The real-time PCR conditions were 30 s at 94°C, 30 s at 55°C, and 30 s at 72°C for 35 cycles; detection of PCR products was performed at 83°C.
TABLE 1.
Oligonucleotide primers used in this study
| Primer | Target gene | Sequence | Positionsa |
|---|---|---|---|
| 16Suniv-1 | 16S rRNA | 5′-CAGCAGCCGCGGTAATAC-3′ | 491-508 |
| 16S-r | 16S rRNA | 5′-CTACGCATTTCACTGCTACACG-3′ | 654-675 |
| rho-f | rho | 5′-CAAAACATTGCCCACGCCGTTACCG-3′ | 565-589 |
| rho-r | rho | 5′-CCACGGACGGAGCGGCTCATTTCGG-3′ | 650-674 |
| siaD-f | siaD | 5′-CGACTCATTTAATTGATGAAGG-3′ | 440-461 |
| siaD-r | siaD | 5′-CATGCAATATGTCAAAATGATC-3′ | 562-583 |
| lipA-f | lipA | 5′-CACATCAGTAAATACAACGTCGG-3′ | 1462-1484 |
| lipA-r | lipA | 5′-GATGCGGTTTGTAGATGATATAG-3′ | 1635-1657 |
| mtrC-f | mtrC | 5′-CAAAGTTTCCGAAGGTACGTTGC-3′ | 582-604 |
| mtrC-r | mtrC | 5′-CGCAATTTCATCACTTCGGATGC-3′ | 679-701 |
| mtrD-f | mtrD | 5′-GATTCTGGAGTTGGGCAACGGTTC-3′ | 1998-2021 |
| mtrD-r | mtrD | 5′-GCACGCATTTTCTGAATCAACTCG-3′ | 2088-2111 |
| mtrE-f | mtrE | 5′-CTGTATCAGATCGACAGTTCCAC-3′ | 298-320 |
| mtrE-r | mtrE | 5′-CAACCAAAGGCTTGTATCGCGCC-3′ | 399-421 |
Positions are relative to the ATG start codon for those genes that code for proteins.
CAMP susceptibility assays.
Polymyxin B, β-defensin 1, β-defensin 2, defensin HNP-1, and defensin HNP-2 were purchased from Sigma. Human cathelicidin LL-37 and murine cathelicidin-related peptides CRAMP and CRAMP-18 were purchased from Vinci-Biochem (Italy), and protegrin PG-1 was synthesized by PRIMM (Italy). To determine the MICs of CAMPs, meningococci (106 CFU ml−1) were inoculated in GC broth with 1% Polyvitox in the presence of CAMPs (0.125 to 1,024 μg ml−1). Cultures were grown at 37°C with shaking for 18 h, and the optical density at 550 nm was then measured. Susceptibility assays testing the bactericidal effects of CAMPs were performed essentially as described previously (33), with minor modifications. Briefly, meningococci were grown to mid-logarithmic phase (optical density at 550 nm of 0.6). The cultures were then diluted 1:100 in GC broth containing 1% Polyvitox, and 90 μl of the diluted cultures was added to 96-well polypropylene microtiter wells containing 10 μl of twofold serial CAMP dilutions (0.125 to 1,024 μg ml−1). Incubations were carried out for 45 min at 37°C in a 5% CO2 incubator. Finally, dilutions of bacteria from each well were plated onto GC medium with 1% Polyvitox to determine CFU. All assays were performed with triplicate samples. Statistical significance was determined by the Student t test.
RESULTS
Adherence to and invasion of nonphagocytic human cells.
We measured N. meningitidis adherence to and invasion of HeLa cells as previously described (1, 44). Consistent with previous findings (28), encapsulated strain B1940 was about a hundredfold less adherent to cells than the isogenic B1940 cps mutant (Fig. 1A) lacking both the capsule and the LOS outer core (see Fig. S1 in the supplemental material). The B1940 cps mutant was also more efficiently internalized, especially at early time points (Fig. 1A). However, if bacteria were centrifuged onto cells to start infection, as previously described for Neisseria gonorrhoeae (1, 44), we observed, at maximum, a twofold difference between B1940 and the B1940 cps mutant (Fig. 1B). Thus, the absence of a capsule gene complex region favored adherence and, indirectly, internalization. Similarly to what was shown for N. gonorrhoeae, invasion efficiency with centrifugation was similar to that without centrifugation, but it was obtained in a shorter time period (1, 44). Therefore, all subsequent experiments on HeLa cells were done by initiating infection by centrifugation.
FIG. 1.
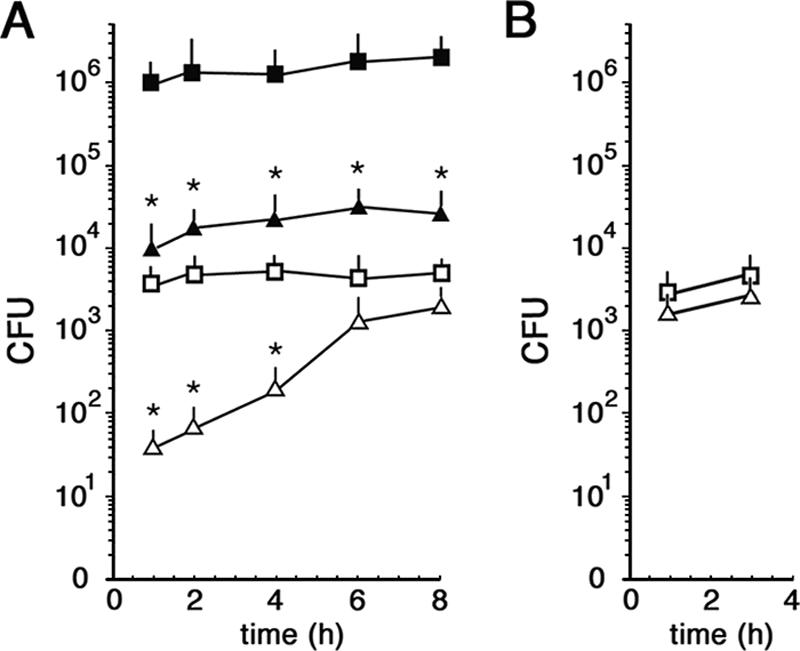
Adherence to and invasion of HeLa cells by N. meningitidis strains. (A) Adherence and invasion assays without centrifugation. HeLa cells were infected with bacteria for the indicated times. The amounts of internalized B1940 (open triangles) and B1940 cps mutant (open squares) bacteria were quantified after saponin treatment. The amounts of adherent B1940 (closed triangles) and B1940 cps mutant (closed squares) bacteria were quantified by omitting the gentamicin step. (B) Invasion assays with centrifugation. The numbers of internalized B1940 (open triangles) and B1940 cps mutant (open squares) bacteria after starting the infection by centrifugation are shown. In A and B, values are means of at least 10 independent experiments made with triplicate samples with standard errors. Asterisks mark statistically significant differences in adherence and invasion values between B1940 and the B1940 cps mutant (P value of <0.05).
These data were confirmed by immunofluorescence analysis using anti-N. meningitidis antibody before and after permeabilization to distinguish between adherent and intracellular bacteria (10, 17) (Fig. 2). Indeed, immediately after gentamicin treatment (time zero), the B1940 cps mutant showed high adherence to cells, and only few bacteria were detected inside (Fig. 2A), while B1940 showed low adherence (Fig. 2B). At longer time points (8 h), it became more difficult to find intracellular bacteria for the B1940 cps mutant (data not shown), while most B1940 bacteria were actually intracellular (Fig. 2C).
FIG. 2.

Immunofluorescence analysis of cells infected with the B1940 cps mutant or B1940. HeLa cells were infected with the B1940 cps mutant or B1940 as shown. Images were taken after gentamicin treatment for the B1940 cps mutant (A, A′, and A′′) or after gentamicin treatment (B, B′, and B′′) or 8 h after gentamicin treatment (C, C′, C′′) for B1940. To distinguish between extracellular and intracellular bacteria, the anti-N. meningitidis antibody (α-N. meningitidis) and its secondary antibody were used before (OUT) (A, B, and C) or after (IN + OUT) (A′, B′, and C′) permeabilization of cells with saponin. The secondary antibody used before permeabilization was Cy5 conjugated, while the one used after permeabilization was fluorescein isothiocyanate conjugated. To detect a cellular marker, we used anti-Lamp1 followed by a tetramethyl rhodamine isothiocyanate-conjugated secondary antibody. Merged images of different channels are shown in A′′, B′′, and C′′. Bars, 10 μm.
The ability of several encapsulated strains (MC58, H44/76, NGP165, BF65, 1000, NGE31, NGF26, and NGH15) to invade HeLa cells was also determined. All tested strains had very similar invasion efficiencies under these experimental conditions. All these experiments were repeated by using alternative cell lines, such as HEp-2 or Chang conjunctiva, with very similar results (data not shown).
Influence of capsulation on meningococcal survival in human cells.
The capsule negatively affects meningococcal adhesion to epithelial cells and subsequent internalization by masking surface-exposed adhesins, and capsule biosynthesis and assembly are down-regulated during the early stages of the infectious cycle (4).
Here, we monitored the entry, survival, and spread of H44/76, B1940, the B1940 cps mutant, and the B1940 siaD(−C) mutant (lacking the capsule but possessing the LOS outer core). After infection (started by centrifugation) and gentamicin treatment, HeLa cells were reincubated for various times at 37°C (Fig. 3A and B). The different strains behaved similarly during the first 2 h after gentamicin treatment (Fig. 3A and B). Indeed, the number of intracellular CFU decreased dramatically. The decrease was actually more pronounced for the B1940 siaD(−C) mutant, and more so for the B1940 cps mutant, suggesting that the absence of the capsule and/or the LOS outer core rendered these strains less resistant to intracellular degradation. At later time points, a remarkable difference in the behavior of the different strains was observed. Indeed, while the B1940 siaD(−C) and B1940 cps mutants were more degraded with time (Fig. 3B), the number of CFU of the other two strains (H44/76 and B1940) increased up to 50-fold 8 h after gentamicin treatment (Fig. 3A and B). Thus, the presence of an intact capsule seems to confer resistance to degradation to internalized bacteria. This finding is consistent with the results of a recent paper addressing the role of capsule in meningococcal survival/growth within HEp-2 and human brain microvascular endothelial cells (24). We observed similar kinetics of infection (a drop in the number of intracellular bacteria 2 h after infection of HEp-2 cells) and rates of intracellular multiplication of the encapsulated strain. As in our study, in that study, unencapsulated bacteria failed to multiply within infected cells, although they were internalized much more efficiently than encapsulated bacteria, and, at variance with our results, they were not killed significantly more than the encapsulated bacteria during the 2 h after gentamicin treatment. These discrepancies might be partially due to the fact that in that study, infection was not initiated by centrifugation.
FIG. 3.

Survival/growth of meningococcal strains in HeLa and THP-1 cells. (A and B) HeLa cells (105 cells /well) were infected with different N. meningitidis strains (H44/76 [open circles], B1940 [open triangles], the B1940 cps mutant [open squares], and the B1940 siaD(−C) mutant [closed squares]) at an MOI of 50, treated with gentamicin, and reincubated in DMEM for the indicated times. Bacteria were centrifuged onto cells to start the infection. After saponin lysis, CFU from intracellular bacteria were scored. Values are means of at least 10 independent experiments made in triplicate with standard errors. Asterisks mark statistically significant differences in the numbers of CFU between H44/76 and B1940 (A) and between B1940 and the B1940 cps/B1940 siaD(−C) mutant (P value of <0.05). (C and D) THP-1 cells (105 cells /well) differentiated with 8 nM phorbol myristic acid for 4 days were infected with different N. meningitidis strains (H44/76 [open circles], B1940 [open triangles], the B1940 cps mutant [open squares], and the B1940 siaD(−C) mutant [closed squares]) at an MOI of 5 and treated as described above. Values are means of at least four independent experiments made in triplicate with standard errors. Asterisks mark statistically significant differences in the numbers of CFU between H44/76 and B1940 (C) and between B1940 and the B1940 cps/B1940 siaD(−C) mutant (P value of <0.05).
Noticeably, in our assays, the increase in the number of intracellular bacteria was accompanied by a proportional increase of infected HeLa cells as scored by immunofluorescence microscopy (Fig. 2C). To avoid reinfection, cells were washed once every hour, and new medium was added. Even under these conditions, infected cells were increasing with time proportionally to intracellular bacteria. These data strongly suggest the spreading of the bacteria between HeLa cells.
Experiments of invasion and intracellular viability were also performed using differentiated THP-1 or U937 human monocyte cell lines (Fig. 3C and D), with similar results. Indeed, in the first 3 h after gentamicin treatment, a decrease in the number of intracellular bacteria was observed for all strains challenging THP-1 cells (Fig. 3C and D). At later time points (6 to 9 h), B1940 showed a marked increase in the number of intracellular bacteria compared to the value measured at time zero, namely, after gentamicin treatment. Such an increase was barely detectable for the B1940 siaD(−C) mutant, while the B1940 cps mutant was efficiently killed intracellularly (Fig. 3C and D).
These data also demonstrate that in different human cell lines, including monocyte cell lines differentiated to macrophages, capsule and LOS outer core are important for intracellular survival. This conclusion was further supported by results of dual-infection/competition experiments that allowed a direct comparison of fitnesses of the encapsulated and unencapsulated strains. In these experiments, HeLa cells were coinfected with equal amounts of B1940 and the B1940 siaD(−C) mutant, and the proportions of the two strains were determined at different time points. After 2, 4, or 6 h, the percentage of unencapsulated meningococci was about 30, 15, and 5%, respectively, confirming the beneficial role of the capsule in the intracellular milieu.
Role of capsule in CAMP resistance.
The intracellular degradation of internalized bacteria may be due to a multitude of mechanisms, including pH and oxidative stress, and the action of lytic enzymes and antimicrobial peptides. In theory, the presence of the capsule may interfere, directly or indirectly, with any of these mechanisms. In particular, there is evidence that the capsular polysaccharide mediates the resistance of K. pneumoniae to CAMPs (3). This finding prompted us to test the protective effect of the meningococcal capsule. Preliminarily, the susceptibility of encapsulated and unencapsulated strains to polymyxin B, a cyclic peptide, was assayed. This substance of microbial origin has been used as a model of CAMP because its permeabilizing action on the outer membrane of gram-negative bacteria is well established (43). In survival experiments, meningococci were exposed for 45 min to different concentrations of polymyxin B; the number of viable bacteria was then determined. Our data confirmed the high levels of intrinsic resistance to polymyxin B previously observed in meningococci (41) and demonstrated that the absence of capsule rendered these bacteria significantly more sensitive to this antimicrobial substance (Fig. 4A).
FIG. 4.

Susceptibilities of encapsulated and unencapsulated meningococcal strains to CAMPs. (A to E) Susceptibilities of B1940 (black columns), the B1940 siaD(−C) mutant (gray columns), and the B1940 cps mutant (white columns) to different concentrations of polymyxin B (A), human defensins (β-defensin 1, β-defensin 2, defensin HNP-1, and defensin HNP-2) (B), porcine protegrin PG-1 (C), human cathelicidin LL-37 (D), and murine cathelicidin-related peptides CRAMP and CRAMP-18 (E). Values are means of three independent experiments made in triplicate with standard errors. Asterisks mark statistically significant differences in survival values between B1940 and the B1940 siaD(−C)/B1940 cps mutant (P value of <0.05).
Sensitivity to human defensins of either epithelial (β-defensin 1 and 2) or macrophagic (defensin HNP-1 and HNP-2) origin was then tested. Defensins are cationic/polar peptides with conserved trisulfide-disulfides linkages and a largely β-sheet structure (12) (see Fig. S2 in the supplemental material). All strains exhibited about 100% survival when exposed to defensin concentrations up to 256 μg ml−1 (data not shown). However, at higher doses (512 μg ml−1) the unencapsulated B1940 siaD(−C) and B1940 cps mutant strains were about 5- to 10-fold more sensitive than the encapsulated strain B1940 (Fig. 4B). The small differences between the B1940 siaD(−C) and B1940 cps mutants were not statistically relevant, suggesting that the absence of capsule and not the absence of LOS outer core was responsible for the increased susceptibility to human defensins.
The scarce sensitivity of meningococci to defensins (MICs > 512 μg ml−1 for all tested strains), consistent with data reported in the literature (27, 33, 41), prompted us to investigate the effects of protegrins and cathelicidins, which are more effective against pathogenic neisseriae than defensins (27, 33, 41).
Considerably smaller than defensins, protegrins are potent CAMPs isolated from porcine leukocytes, which, like tachyplesins (broad-spectrum antibiotic peptides of horseshoe crab hemocytes), contain two intramolecular cysteine disulfide bonds (14) (see Fig. S2 in the supplemental material). The results of our assays confirmed the higher susceptibility of meningococci to protegrin PG-1 than to defensins and confirmed the protective role of the capsule (Fig. 4C). At doses of >8 μg ml−1, the B1940 siaD(−C) and B1940 cps mutants were >10-fold more sensitive than B1940, also suggesting that the integrity of LOS outer core does not affect the sensitivity to protegrins significantly. MICs of PG-1 were 16 μg ml−1 for B1940 and 4 μg ml−1 for both the B1940 siaD(−C) and B1940 cps mutants.
Cathelicidins are amphipathic α-helix peptides with a conserved N-terminal cathelin domain and a variable C-terminal antimicrobial domain. LL-37 (see Fig. S2 in the supplemental material) is the C-terminal part of the human cationic antimicrobial protein (hCAP-18) that is expressed mainly by neutrophils and epithelial cells. Our results confirmed the high sensitivity of meningococci to LL-37 (Fig. 4D). The loss of capsulation resulted in a significant increase (about eightfold at concentrations of >4 μg ml−1) of the bactericidal effect of LL-37. The absence of the LOS outer core further increased (about 10-fold at concentrations of >4 μg ml−1) the susceptibility of unencapsulated strains to cathelicidin. The results of survival experiments were consistent with MICs of LL-37 for B1940 (about 16 μg ml−1), the B1940 siaD(−C) mutant (8 μg ml−1), and the B1940 cps (4 μg ml−1) mutant. Similar results were obtained with CRAMP (Fig. 4E), a murine cathelicidin-related peptide (see Fig. S2 in the supplemental material), and CRAMP-18 (Fig. 4E), a CRAMP-derived peptide exhibiting the same activity as the parental molecule (see Fig. S2 in the supplemental material). MICs of CRAMP were 256, 64, and 32 μg ml−1, respectively, for B1940, the B1940 siaD(−C) mutant, and the B1940 cps mutant. Thus, at variance with defensins and protegrins, sensitivity to cathelicidins was also affected by the presence of an intact LOS.
Transcriptional analysis of capsular genes and those encoding the MtrC-MtrD-MtrE efflux pump during the infectious cycle.
In an attempt to identify meningococcal genes regulated in the intracellular environment, a limited transcriptional analysis was performed by comparing cDNA from intracellular bacteria (after 8 h of infection of HeLa cells) and from bacteria grown for 8 h in culture medium. The strain used for this analysis was B1940. Several DNA sequences that were differently expressed were isolated. One of these sequences, a 1,148-bp Sau3AI fragment, corresponded to lipA, coding for a protein required for the proper translocation and surface expression of the lipidated (α2→8)-linked polysialic acid capsule polymer (42). This gene appeared to be up-regulated in bacteria extracted from the cytoplasm of HeLa cells.
This finding was confirmed by results of real-time RT-PCR experiments that also demonstrated the up-regulation of siaD, coding for a polysialyltransferase involved in the biosynthesis of the (α2→8)-polysialic acid capsule (Fig. 5A). Levels of lipA- and siaD-specific transcripts were normalized to 16S rRNA levels. Normalization with 16S rRNA or rho mRNA, coding for a transcription termination factor subjected to autoregulation in gram-negative bacteria (15, 21), gave very similar results (data not shown). These results demonstrated a three- to fourfold increase in the amounts of siaD- or lipA-specific transcripts in intracellular bacteria compared to control bacteria, indicating that the meningococci up-regulate the genetic loci for capsular polysaccharide biosynthesis, translocation, and surface expression during their permanence in the eukaryotic host cell.
FIG. 5.

Semiquantitative analysis of the siaD, lipA, mtrC, mtrD, and mtrE transcripts in intracellular meningococci. (A) Total RNAs were extracted from meningococci (strain B1940) after 8 h of infection of HeLa cells (intracellular) or control bacteria grown for 8 h in culture medium (DMEM). The amounts of siaD- and lipA-specific transcripts were determined by real-time RT-PCR. The scheme of the genomic region encoding capsular elements is reported above the panel. (B) Total RNAs were extracted as described above (A). The amounts of mtrC-, mtrD-, and mtrE-specific transcripts were determined by real-time RT-PCR. The genetic map of the mtrCDE operon and the regulatory gene mtrR is reported above the panel. In A and B, levels of lipA-, siaD-, mtrC-, mtrD-, and mtrE-specific transcripts were normalized to rho mRNA levels. Transcript levels in control bacteria are arbitrarily assumed to equal 1. Each real-time RT-PCR experiment was repeated five times (with triplicate samples) using distinct cDNA preparations, and means and standard deviations (bars) were determined. Asterisks mark statistically significant differences in transcript levels between intracellular and control bacteria (P value of <0.05).
By using the same approach, transcript levels of the operon mtrCDE, coding for an efflux pump involved in CAMP resistance (41), were measured. The results of real-time RT-PCR experiments demonstrated that the mtrC-, mtrD-, or mtrE-specific transcripts were about fourfold more abundant in intracellular bacteria than in control bacteria (Fig. 5B).
We reasoned that the up-regulation of siaD, lipA, and mtrCDE might have been triggered by bacteria sensing intracellular or secreted CAMPs. To verify this hypothesis, we tested the ability of CAMPs to up-regulate these genes. In one experiment, B1940 was grown in GC medium to late logarithmic phase; PG-1 (Fig. 6A) or LL-37 (Fig. 6B) was then added at an MIC of 16 μg ml−1, and cultures were harvested 5 and 15 min later. The results of real-time RT-PCR experiments revealed a moderate increase (ranging between 1.5- and 2-fold) in mtrCDE transcript levels as a function of time, while lipA transcripts remained at a near-constant level. In another experiment, B1940 was grown in the presence of a sublethal concentration (4 or 8 μg ml−1) of PG-1 (Fig. 6C) or LL-37 (Fig. 6D) to the late logarithmic phase. Under these growth conditions, in addition to a moderate increase (up to twofold) in mtrCDE transcript levels, an even more significant increase in the levels of lipA transcripts was observed with PG-1. Such an increase was less marked with LL-37. Altogether, these findings suggest that the up-regulation of the mtrCDE operon in the host cell environment might be directly promoted by the exposure of meningococci to CAMPs. In contrast, the up-regulation of the capsular lipA gene, not detected during the short-term exposure to inhibitory PG-1 levels (Fig. 6A), might be the consequence of a long-term adaptive response to the exposure to sublethal CAMP concentrations.
FIG. 6.

Semiquantitative analysis of the lipA, mtrC, mtrD, and mtrE transcripts in meningococci exposed to PG-1 or LL-37. (A and B) Total RNAs were extracted from meningococci grown to the late logarithmic phase and then exposed to lethal concentrations (16 μg ml−1) of PG-1 (A) or LL-37 (B) for 0, 5, and 15 min. The amounts of specific transcripts were determined by real-time RT-PCR. (C and D) Total RNAs were extracted from meningococci grown to late logarithmic phase in the presence or in the absence of sublethal concentrations (4 or 8 μg ml−1) of PG-1 (C) or LL-37 (D). The amounts of the specific transcripts were determined as described above. In A to D, levels of lipA-, mtrC-, mtrD-, and mtrE-specific transcripts were normalized to rho mRNA levels. Transcript levels in control bacteria are arbitrarily assumed to equal 1. Each real-time RT-PCR experiment was repeated three times (with triplicate samples) using distinct cDNA preparations, and means and standard deviations (bars) were determined. Asterisks mark statistically significant differences in transcript levels with respect to control samples (P value of <0.05).
DISCUSSION
In this study, using in vitro infection assays with both phagocytic and nonphagocytic human cells, we confirm the negative influence of the capsule on meningococcal adhesion and internalization (Fig. 1 and 2) (10, 45). In addition, we provide evidence that the meningococcal capsule confers resistance to degradation in the intracellular environment (Fig. 3). This finding was supported by results of dual infection/competition with encapsulated and unencapsulated strains and by expression data demonstrating that the genetic loci for capsular polysaccharide biosynthesis, translocation, and surface expression are up-regulated by intracellular bacteria (Fig. 5A). These data are consistent with the results of a transcriptome analysis of N. meningitidis that revealed that capsular genes are transcriptionally induced after contact with epithelial (HeLa) cells (5), in contrast to the current concept of the role of the capsule and LOS outer core during the invasion of eukaryotic cells.
One of the mechanisms by which the capsule may protect the meningococci in the intracellular environment is an increased resistance to CAMPs (Fig. 4) that kill gram-negative bacteria by destabilizing the inner and outer membranes (8, 18, 23, 39, 40, 46). Mechanistically, the capsule may protect the bacteria by impairing the interaction of CAMPs with the surface, as shown in K. pneumoniae (3). It should be noted, however, that with respect to K. pneumoniae, N. meningitidis exhibits higher levels of resistance to CAMPs, mostly due to the action of the MtrC-MtrD-MtrE efflux pump (27, 29, 33, 41). In this study, we demonstrated that in addition to the capsular genes, the expression of the mtrCDE operon is up-regulated in the host cell environment (Fig. 5B). The up-regulation of these genetic determinants represents an aspect of the global change in the gene expression pattern following the entry of the bacteria into the hostile environment of the eukaryotic cell. The meningococcal adaptive response might account for the increase in the number of viable intracellular bacteria after the initial decrease during the first 2 to 3 h (Fig. 3).
The up-regulation of the mtrCDE operon in the host cell environment might be, at least in part, directly triggered by the exposure of meningococci to intracellular or secreted CAMPs (Fig. 6). The underlying molecular mechanism is currently unknown. It is noteworthy that two structurally unrelated CAMPs, PG-1 and LL-37, elicit the same effects on mtrCDE transcript levels and that these effects could be detected only at CAMP concentrations close to the MIC. These findings make the hypothesis that up-regulation might be promoted by sensing specifically host-produced CAMPs unlikely, suggesting that perturbation of the bacterial inner and/or outer membrane may be the signal that mediates the phenomenon.
Transcription regulation of the mtrCDE operon has been investigated in gonococci and meningococci. In N. gonorrhoeae, the MtrR repressor (whose gene is located upstream of the mtrCDE operon and is transcribed in the opposite direction) negatively regulates the expression of the mtrCDE operon (9, 26). In addition, the inducible mtrCDE-dependent resistance to hydrophobic agents requires the action of the AraC-like protein MtrA (29). In N. meningitidis, regulation of mtrCDE is out of these schemes because of nonsense or missense mutations in mtrR that abrogate MtrR function in all tested strains (30). In addition, the presence of a nemis (Neisseria miniature insertion sequence, or Correia element) in the promoter region of the mtrCDE operon (Fig. 5B) is a typical feature of meningococci. The nemis contains an integration host factor binding site, whose deletion increases mtrCDE transcription (30), and is targeted, at the level of RNA, by RNase III, whose cleavage has been proposed to stabilize the transcript (19, 30). Notably, in E. coli, RNase III fractionates predominantly with the inner membrane (20, 34-36), providing a plausible explanation for the effects of the exposure to inner-membrane-destabilizing agents such as CAMPs on mtrCDE transcript levels. In this context, it would be intriguing to investigate the intracellular location of RNase III under different growth conditions and the role of RNase III, a possible global regulator in Neisseria that is essential for virulence in the infant rat model (19, 37), in mtrCDE up-regulation during the permanence of meningococci in infected cells.
Supplementary Material
Acknowledgments
We are grateful to M. Frosch for the kind gift of B1940, B1940 cps mutant, B1940 siaD(−C) mutant, and B1940 siaD(+C) mutant bacterial strains.
This work was partially supported by grants from Italian MIUR to C.B. and P.A. (60%, COFIN 2004, COFIN 2006). We have no conflicting financial interests.
Editor: J. N. Weiser
Footnotes
Published ahead of print on 30 April 2007.
REFERENCES
- 1.Billker, O., A. Popp, V. Brinkmann, G. Wenig, J. Schneider, E. Caron, and T. Meyer. 2002. Distinct mechanisms of internalization of Neisseria gonorrhoeae by members of the CEACAM receptor family involving Rac1- and Cdc42-dependent and -independent pathways. EMBO J. 21:560-571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bucci, C., A. Lavitola, P. Salvatore, L. Del Giudice, D. Massardo, C. Bruni, and P. Alifano. 1999. Hypermutation in pathogenic bacteria: frequent phase variation in meningococci is a phenotypic trait of a specialized mutator biotype. Mol. Cell 3:435-445. [DOI] [PubMed] [Google Scholar]
- 3.Campos, M., M. Vargas, V. Regueiro, C. Llompart, S. Alberti, and J. Bengoechea. 2004. Capsule polysaccharide mediates bacterial resistance to antimicrobial peptides. Infect. Immun. 72:7107-7114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Deghmane, A., D. Giorgini, M. Larribe, J. Alonso, and M. Taha. 2002. Down-regulation of pili and capsule of Neisseria meningitidis upon contact with epithelial cells is mediated by CrgA regulatory protein. Mol. Microbiol. 43:1555-1564. [DOI] [PubMed] [Google Scholar]
- 5.Dietrich, G., S. Kurz, C. Hubner, C. Aepinus, S. Theiss, M. Guckenberger, U. Panzner, J. Weber, and M. Frosch. 2003. Transcriptome analysis of Neisseria meningitidis during infection. J. Bacteriol. 185:155-164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Frosch, M., E. Schultz, E. Glenn-Calvo, and T. Meyer. 1990. Generation of capsule-deficient Neisseria meningitidis strains by homologous recombination. Mol. Microbiol. 4:1215-1218. [DOI] [PubMed] [Google Scholar]
- 7.Frosch, M., C. Weisgerber, and T. Meyer. 1989. Molecular characterization and expression in Escherichia coli of the gene complex encoding the polysaccharide capsule of Neisseria meningitidis group B. Proc. Natl. Acad. Sci. USA 86:1669-1673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gidalevitz, D., Y. Ishitsuka, A. Muresan, O. Konovalov, A. Waring, R. Lehrer, and K. Lee. 2003. Interaction of antimicrobial peptide protegrin with biomembranes. Proc. Natl. Acad. Sci. USA 100:6302-6307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hagman, K., and W. Shafer. 1995. Transcriptional control of the mtr efflux system of Neisseria gonorrhoeae. J. Bacteriol. 177:4162-4165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hammerschmidt, S., R. Hilse, J. van Putten, R. Gerardy-Schahn, A. Unkmeir, and M. Frosch. 1996. Modulation of cell surface sialic acid expression in Neisseria meningitidis via a transposable genetic element. EMBO J. 15:192-198. [PMC free article] [PubMed] [Google Scholar]
- 11.Hammerschmidt, S., A. Muller, H. Sillmann, M. Muhlenhoff, R. Borrow, A. Fox, J. van Putten, W. Zollinger, R. Gerardy-Schahn, and M. Frosch. 1996. Capsule phase variation in Neisseria meningitidis serogroup B by slipped-strand mispairing in the polysialyltransferase gene (siaD): correlation with bacterial invasion and the outbreak of meningococcal disease. Mol. Microbiol. 20:1211-1220. [DOI] [PubMed] [Google Scholar]
- 12.Hancock, R. 2001. Cationic peptides: effectors in innate immunity and novel antimicrobials. Lancet Infect. Dis. 1:156-164. [DOI] [PubMed] [Google Scholar]
- 13.Jarvis, G., and N. Vedros. 1987. Sialic acid of group B Neisseria meningitidis regulates alternative complement pathway activation. Infect. Immun. 55:174-180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kokryakov, V., S. Harwig, E. Panyutich, A. Shevchenko, G. Aleshina, O. Shamova, H. Korneva, and R. Lehrer. 1993. Protegrins: leukocyte antimicrobial peptides that combine features of corticostatic defensins and tachyplesins. FEBS Lett. 327:231-236. [DOI] [PubMed] [Google Scholar]
- 15.Kung, H., E. Bekesi, S. Guterman, J. Gray, L. Traub, and D. Calhoun. 1984. Autoregulation of the rho gene of Escherichia coli K-12. Mol. Gen. Genet. 193:210-213. [DOI] [PubMed] [Google Scholar]
- 16.Lavitola, A., C. Bucci, P. Salvatore, G. Maresca, C. Bruni, and P. Alifano. 1999. Intracistronic transcription termination in polysialyltransferase gene (siaD) affects phase variation in Neisseria meningitidis. Mol. Microbiol. 33:119-127. [DOI] [PubMed] [Google Scholar]
- 17.Makino, S., J. van Putten, and T. Meyer. 1991. Phase variation of the opacity outer membrane protein controls invasion by Neisseria gonorrhoeae into human epithelial cells. EMBO J. 10:1307-1315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mani, R., S. Cady, M. Tang, A. Waring, R. Lehrer, and M. Hong. 2006. Membrane-dependent oligomeric structure and pore formation of a beta-hairpin antimicrobial peptide in lipid bilayers from solid-state NMR. Proc. Natl. Acad. Sci. USA 103:16242-16247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mazzone, M., E. De Gregorio, A. Lavitola, C. Pagliarulo, P. Alifano, and P. P. Di Nocera. 2001. Whole-genome organization and functional properties of miniature DNA insertion sequences conserved in pathogenic neisseriae. Gene 278:211-222. [DOI] [PubMed] [Google Scholar]
- 20.Miczak, A., R. Srivastava, and D. Apirion. 1991. Location of the RNA-processing enzymes RNase III, RNase E and RNase P in the Escherichia coli cell. Mol. Microbiol. 5:1801-1810. [DOI] [PubMed] [Google Scholar]
- 21.Miloso, M., D. Limauro, P. Alifano, F. Rivellini, A. Lavitola, E. Gulletta, and C. Bruni. 1993. Characterization of the rho genes of Neisseria gonorrhoeae and Salmonella typhimurium. J. Bacteriol. 175:8030-8037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Monaco, C., A. Tala, M. Spinosa, C. Progida, E. De Nitto, A. Gaballo, C. Bruni, C. Bucci, and P. Alifano. 2006. Identification of a meningococcal l-glutamate ABC transporter operon essential for growth in low-sodium environments. Infect. Immun. 74:1725-1740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nagaoka, I., S. Hirota, S. Yomogida, A. Ohwada, and M. Hirata. 2000. Synergistic actions of antibacterial neutrophil defensins and cathelicidins. Inflamm. Res. 49:73-79. [DOI] [PubMed] [Google Scholar]
- 24.Nikulin, J., U. Panzner, M. Frosch, and A. Schubert-Unkmeir. 2006. Intracellular survival and replication of Neisseria meningitidis in human brain microvascular endothelial cells. Int. J. Med. Microbiol. 296:553-558. [DOI] [PubMed] [Google Scholar]
- 25.Pagliarulo, C., P. Salvatore, L. De Vitis, R. Colicchio, C. Monaco, M. Tredici, A. Tala, M. Bardaro, A. Lavitola, C. Bruni, and P. Alifano. 2004. Regulation and differential expression of gdhA encoding NADP-specific glutamate dehydrogenase in Neisseria meningitidis clinical isolates. Mol. Microbiol. 51:1757-1772. [DOI] [PubMed] [Google Scholar]
- 26.Pan, W., and B. Spratt. 1994. Regulation of the permeability of the gonococcal cell envelope by the mtr system. Mol. Microbiol. 11:769-775. [DOI] [PubMed] [Google Scholar]
- 27.Qu, X., S. Harwig, A. Oren, W. Shafer, and R. Lehrer. 1996. Susceptibility of Neisseria gonorrhoeae to protegrins. Infect. Immun. 64:1240-1245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Read, R., S. Zimmerli, C. Broaddus, D. Sanan, D. Stephens, and J. Ernst. 1996. The (α2→8)-linked polysialic acid capsule of group B Neisseria meningitidis modifies multiple steps during interaction with human macrophages. Infect. Immun. 64:3210-3217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rouquette, C., J. Harmon, and W. Shafer. 1999. Induction of the mtrCDE-encoded efflux pump system of Neisseria gonorrhoeae requires MtrA, an AraC-like protein. Mol. Microbiol. 33:651-658. [DOI] [PubMed] [Google Scholar]
- 30.Rouquette-Loughlin, C., J. Balthazar, S. Hill, and W. Shafer. 2004. Modulation of the mtrCDE-encoded efflux pump gene complex of Neisseria meningitidis due to a Correia element insertion sequence. Mol. Microbiol. 54:731-741. [DOI] [PubMed] [Google Scholar]
- 31.Salvatore, P., C. Bucci, C. Pagliarulo, M. Tredici, R. Colicchio, G. Cantalupo, M. Bardaro, L. Del Giudice, D. Massardo, A. Lavitola, C. Bruni, and P. Alifano. 2002. Phenotypes of a naturally defective recB allele in Neisseria meningitidis clinical isolates. Infect. Immun. 70:4185-4195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Salvatore, P., C. Pagliarulo, R. Colicchio, P. Zecca, G. Cantalupo, M. Tredici, A. Lavitola, C. Bucci, C. Bruni, and P. Alifano. 2001. Identification, characterization, and variable expression of a naturally occurring inhibitor protein of IS1106 transposase in clinical isolates of Neisseria meningitidis. Infect. Immun. 69:7425-7436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shafer, W., X. Qu, A. Waring, and R. Lehrer. 1998. Modulation of Neisseria gonorrhoeae susceptibility to vertebrate antibacterial peptides due to a member of the resistance/nodulation/division efflux pump family. Proc. Natl. Acad. Sci. USA 95:1829-1833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Srivastava, N., and R. Srivastava. 1996. Expression, purification and properties of recombinant E. coli ribonuclease III. Biochem. Mol. Biol. Int. 39:171-180. [DOI] [PubMed] [Google Scholar]
- 35.Srivastava, R., A. Miczak, and D. Apirion. 1990. Maturation of precursor 10Sa RNA in Escherichia coli is a two-step process: the first reaction is catalyzed by RNase III in presence of Mn2+. Biochimie 72:791-802. [DOI] [PubMed] [Google Scholar]
- 36.Srivastava, R., N. Srivastava, and D. Apirion. 1991. RNA processing enzymes RNase III, E and P in Escherichia coli are not ribosomal enzymes. Biochem. Int. 25:57-65. [PubMed] [Google Scholar]
- 37.Sun, Y., S. Bakshi, R. Chalmers, and C. Tang. 2000. Functional genomics of Neisseria meningitidis pathogenesis. Nat. Med. 6:1269-1273. [DOI] [PubMed] [Google Scholar]
- 38.Tettelin, H., N. Saunders, J. Heidelberg, A. Jeffries, K. Nelson, J. Eisen, K. Ketchum, D. Hood, J. Peden, R. Dodson, W. Nelson, M. Gwinn, R. DeBoy, J. Peterson, E. Hickey, D. Haft, S. Salzberg, O. White, R. Fleischmann, B. Dougherty, T. Mason, A. Ciecko, D. Parksey, E. Blair, H. Cittone, E. Clark, M. Cotton, T. Utterback, H. Khouri, H. Qin, J. Vamathevan, J. Gill, V. Scarlato, V. Masignani, M. Pizza, G. Grandi, L. Sun, H. Smith, C. Fraser, E. Moxon, R. Rappuoli, and J. Venter. 2000. Complete genome sequence of Neisseria meningitidis serogroup B strain MC58. Science 287:1809-1815. [DOI] [PubMed] [Google Scholar]
- 39.Tossi, A., M. Scocchi, B. Skerlavaj, and R. Gennaro. 1994. Identification and characterization of a primary antibacterial domain in CAP18, a lipopolysaccharide binding protein from rabbit leukocytes. FEBS Lett. 339:108-112. [DOI] [PubMed] [Google Scholar]
- 40.Turner, J., Y. Cho, N. Dinh, A. Waring, and R. Lehrer. 1998. Activities of LL-37, a cathelin-associated antimicrobial peptide of human neutrophils. Antimicrob. Agents Chemother. 42:2206-2214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tzeng, Y., K. Ambrose, S. Zughaier, X. Zhou, Y. Miller, W. Shafer, and D. Stephens. 2005. Cationic antimicrobial peptide resistance in Neisseria meningitidis. J. Bacteriol. 187:5387-5396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tzeng, Y., A. Datta, C. Strole, M. Lobritz, R. Carlson, and D. Stephens. 2005. Translocation and surface expression of lipidated serogroup B capsular polysaccharide in Neisseria meningitidis. Infect. Immun. 73:1491-1505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Vaara, M. 1992. Agents that increase the permeability of the outer membrane. Microbiol. Rev. 56:395-411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.van Putten, J., and S. Paul. 1995. Binding of syndecan-like cell surface protoglycan receptors is required for Neisseria gonorrhoeae entry into human mucosal cells. EMBO J. 14:2144-2154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Virji, M., K. Makepeace, D. Ferguson, M. Achtman, and E. Moxon. 1993. Meningococcal Opa and Opc proteins: their role in colonization and invasion of human epithelial and endothelial cells. Mol. Microbiol. 10:499-510. [DOI] [PubMed] [Google Scholar]
- 46.Yu, K., K. Park, S. Kang, S. Shin, K. Hahm, and Y. Kim. 2002. Solution structure of a cathelicidin-derived antimicrobial peptide, CRAMP as determined by NMR spectroscopy. J. Pept. Res. 60:1-9. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.