

Abstract
Inner ear dysfunction secondary to chronic otitis media (OM), including high-frequency sensorineural hearing loss or vertigo, is not uncommon. Although chronic middle ear inflammation is believed to cause inner ear dysfunction by entry of OM pathogen components or cytokines from the middle ear into the inner ear, the underlying mechanisms are not well understood. Previously, we demonstrated that the spiral ligament fibrocyte (SLF) cell line up-regulates monocyte chemotactic protein 1 (MCP-1) expression after treatment with nontypeable Haemophilus influenzae (NTHI), one of the most common OM pathogens. We hypothesized that the SLF-derived MCP-1 plays a role in inner ear inflammation secondary to OM that is responsible for hearing loss and dizziness. The purpose of this study was to investigate the signaling pathway involved in NTHI-induced MCP-1 up-regulation in SLFs. Here we show for the first time that NTHI induces MCP-1 up-regulation in the SLFs via Toll-like receptor 2 (TLR2)-dependent activation of NF-κB. TLR2−/−- and MyD88−/−-derived SLFs revealed involvement of TLR2 and MyD88 in NTHI-induced MCP-1 up-regulation. Studies using chemical inhibitors and dominant-negative constructs demonstrated that it is mediated by the IκKβ-dependent IκBα phosphorylation and NTHI-induced NF-κB nuclear translocation. Furthermore, we demonstrated that the binding of NF-κB to the enhancer region of MCP-1 is involved in this up-regulation. In addition, we have identified a potential NF-κB motif that is responsive and specific to certain NTHI molecules or ligands. Further studies are necessary to reveal specific ligands of NTHI that activate host receptors. These results may provide us with new therapeutic strategies for prevention of inner ear dysfunction secondary to chronic middle ear inflammation.
Antibiotics have led to a dramatic decline in the incidence of life-threatening complications of otitis media (OM), such as meningitis or brain abscess (3). However, inner ear dysfunction secondary to chronic OM, including high-frequency sensorineural hearing loss or vertigo, is not uncommon (13, 26, 36, 55, 60). Although chronic middle ear inflammation is believed to cause inner ear dysfunction by entry of OM pathogen components or cytokines from the middle ear into the inner ear, the underlying mechanisms are not well understood (18, 32, 39, 44, 52, 87).
The inner ear is a sensory organ for hearing (cochlea) and equilibrium (vestibule). It consists of a variety of specialized cell types (50, 51), such as sensory hair cells, supporting cells, sulcus cells, and spiral ligament fibrocytes (SLFs), which are the most abundant cell types exposed to the perilymph. The type of inner ear cells that respond to proinflammatory signals entering the inner ear remain unknown. Considering that SLFs are one of the abundant cell types in the cochlea and that they secrete cytokines and chemokines after proinflammatory stimuli (72, 97), we hypothesized that the SLFs are major responders to such signals.
Preliminary studies of human temporal bones with labyrinthitis showed the infiltration of lysozyme-positive round cells with a monomorphic nucleus into the spiral ligament (unpublished data). Also, SLF cell lines (96) showed an induction in monocyte chemotactic protein 1 (MCP-1) expression after treatment with lysate of nontypeable Haemophilus influenzae (NTHI), one of the most common OM pathogens (72). Moreover, it has previously been shown that monocytes can infiltrate cochlea exhibiting chronic middle ear inflammation or acoustic trauma (22, 34, 37). These results led us to focus on MCP-1 as an SLF-derived proinflammatory chemokine attracting effector cells and causing inner ear dysfunction.
MCP-1, also known as the chemokine C-C motif ligand 2, is produced by various cells, including endothelial cells, smooth muscle cells, fibroblasts, and macrophages, in response to cytokines, growth factors, or bacterial components (9, 46, 78). It is encoded by an immediate-early gene (33) and is up-regulated by various stimuli such as bacterial lipopolysaccharide (LPS), interleukin-1 (IL-1), tumor necrosis factor alpha, platelet-derived growth factor, gamma interferon, or oxidized low-density lipoprotein (9, 28, 77). MCP-1 is involved in inflammatory disorders, including rheumatoid arthritis, glomerular disease, pulmonary granulomatous vasculitits, tumor infiltration, psoriasis, and atherosclerosis (14, 16, 20, 45, 54).
NTHI is a small, gram-negative bacterium, existing as a commensal organism in the human nasopharynx (62). Although NTHI rarely causes life-threatening infections, it is nonetheless a clinically important pathogen since it is one of the underlying causes of OM in children and exacerbates chronic obstructive pulmonary disease in adults (21, 73). The organism lacks a polysaccharide capsule, which is used for typing, and it releases a unique endotoxin, lipooligosaccharide, which is structurally different from the LPS of enterobacteria (24). Although NTHI is a gram-negative bacterium, it is believed to express molecules that activate not only Toll-like receptor 4 (TLR4) but also TLR2 (57, 82, 83, 93). The interactions of NTHI antigens with specific host molecules are likely to be involved in the transition of NTHI from a commensal to a pathogenic organism.
TLRs are cell surface receptors that play a role in recognition of pathogen-associated molecular patterns (PAMPs) such as LPS, lipoteichoic acid, and peptidoglycans (2, 6, 65). PAMPs are highly conserved structures present in large groups of microorganisms and are produced only by microbial pathogens, not by their hosts. TLRs are known to transmit signals from the cell surface via activation of NF-κB, mitogen-activated protein kinase p38, and Jun amino-terminal kinase (63). However, it is unknown which signaling pathway is involved in the TLR-dependent MCP-1 up-regulation in cochlear SLFs. It was previously demonstrated that the outer sulcus cells of the cochlea express TLR4, leading to an LPS-induced IL-1β up-regulation in the inner ear (29). However, it is not clear which TLR is involved in recognizing and binding to NTHI molecules and transmitting the signals to the inner ear cells. Therefore, we explored the possibility of TLR2 and/or TLR4 being necessary and required receptors for NTHI-induced MCP-1 up-regulation of SLFs.
To address these questions, we investigated the TLR2- and TLR4-dependent NF-κB signaling pathway. We show that NTHI induces MCP-1 up-regulation in SLFs via TLR2-dependent activation of NF-κB, which in turn is mediated by IκKβ-dependent IκBα phosphorylation. Furthermore, we demonstrate that the binding of NF-κB to the enhancer region of MCP-1 is involved in this up-regulation. In addition, we have also identified a potential NF-κB motif that is responsive and specific to certain NTHI molecules or ligands. These results may provide us with new therapeutic strategies for prevention of inner ear dysfunction secondary to chronic middle ear inflammation.
MATERIALS AND METHODS
Reagents.
Caffeic acid phenetyl ester (CAPE) (an NF-κB inhibitor) and MG132 (a cell-permeative proteasome inhibitor) were purchased from Calbiochem (San Diego, CA). TaqMan primers and probes for rat MCP-1 (Rn00580555_m1), mouse MCP-1 (Mm00441242_m1), rat GAPDH (glyceraldehyde-3-phosphate dehydrogenase) (4352338E), and mouse GAPDH (4352339E) were purchased from Applied Biosystems (Foster City, CA).
Cell culture.
The rat SLF cell line, immortalized with adenovirus type 12-simian virus 40 hybrid virus (96), was maintained in Dulbecco's modified Eagle's medium (DMEM) (Invitrogen, Carlsbad, CA) supplemented with 10% fetal bovine serum, penicillin (100 units/ml), and streptomycin (0.1 mg/ml). Epidermal fibroblasts of Sprague-Dawley rat (CRL-1213) were purchased from the ATCC (Manassas, VA). Primary SLFs were cultured from explants of the cochlear lateral walls of TLR2 and MyD88 gene knockout mice, which were kind gifts from J.-D. Li (University of Rochester, Rochester, NY). C57BL/6, the background strain for both knockout mice, was purchased from Charles River Laboratories, Inc. (Wilmington, MA) and was used as a control. Briefly, 1-day-old mouse pups were euthanatized in a CO2 chamber and then decapitated. All aspects of animal handling were performed according to approved IACUC guidelines. The cochlea was isolated with preservation of its normal structure after dissecting the inner ear from the skull base. After removal of the bony otic capsule, the spiral ligament was dissected away from the surrounding tissue (stria vascularis, organ of Corti, and Reissner's membrane) using fine forceps. Explants of the spiral ligament were plated onto collagen-coated petri dishes in DMEM supplemented with 10% fetal bovine serum. After proliferation of the primary cells, the explant was removed. (See Fig. 3C for dissection micrograph views of these procedures.) Primary cells of passage 5 or less were used in this study. All cells were maintained at 37°C in a humidified atmosphere of 5% CO2 and 95% air.
FIG. 3.

TLR2 and MyD88 are involved in NTHI-induced MCP-1 up-regulation of SLFs. (A) Both conventional RT-PCR and real-time quantitative PCR show that the spiral ligament cell line expresses TLR2 and TLR4. TLR2 is up-regulated only by NTHI treatment in SLFs. Con, control. (B) NTHI-induced MCP-1 up-regulation is inhibited by the dominant-negative TLR2 construct (TLR2_DN) but not by the TLR4 dominant-negative construct (TLR4_DN) (left panel). In contrast, overexpression of wild-type TLR2 (TLR2_WT) enhances NTHI-induced MCP-1 up-regulation. The luciferase assay demonstrates that the TLR2 dominant-negative construct inhibits NTHI-induced activation of the MCP-1 promoter (right panel). MCP1-E vector was cotransfected with the TLR2 dominant-negative construct and pRL-TK vector. pcDNA, mock transfection with a blank vector. (C) Dissection microscopic views demonstrating separation of the cochlear lateral wall from the cochlea of gene-targeted mouse pups. The cochlea is isolated with preservation of its normal structure, such as the round window (arrowhead), after dissecting the inner ear from the skull base (C1). After removal of the bony otic capsule (C2), the cochlear lateral wall is separated (C3) and ready for the explant culture (C4). Apex, cochlear apex; CL, cochlear lateral wall; CoN, cochlear nerve; #, an explant of the cochlear lateral wall. (D) Real-time quantitative PCR shows that targeting TLR2 or MyD88 blocks NTHI-induced MCP-1 up-regulation more than 90% and 100%, respectively. The NTHI lysate was incubated with the primary SLFs derived from wild-type or knockout mice after starvation. Cells were harvested 3 h after treatment, and total RNA was extracted. After synthesizing cDNA, multiplex PCR was performed for real-time quantitation, and the CT values of MCP-1 were normalized to the internal control, GAPDH. Results are expressed as fold induction of mRNA quantity or luciferase activity, taking the value of the nontreated group as 1. The experiments were performed in triplicate and repeated more than twice. Values are given as the means ± standard deviations (n = 3). *, P < 0.05; **, P < 0.01.
Bacterial culture and preparation of bacterial lysate.
NTHI strain 12, originally a clinical isolate from the middle ear fluid of a child with acute OM, was used in this study (10). The NTHI lysate was prepared as described previously (48). Briefly, a single colony of NTHI was harvested from a chocolate agar plate, inoculated into 30 ml of brain heart infusion broth supplemented with NAD (3.5 μg/ml), and placed in a shaking incubator overnight. The supernatant was discarded after centrifugation at 10,000 × g for 10 min. The pellet was resuspended in 10 ml of phosphate-buffered saline (PBS) and sonicated to lyse the bacteria. The lysate was then centrifuged at 10,000 × g for 10 min, and the supernatant was collected. The protein concentration in the NTHI lysate was determined using the bicinchoninic acid (BCA) protein assay kit (Pierce Biotechnologies, Rockford, IL) and was in the range of 0.15 mg/ml.
Immunolabeling.
Six 10-week-old male BALB/c mice were used in this experiment. All aspects of animal handling were performed according to approved IACUC guidelines. Ten microliters of live NTHI suspension (108/ml) in saline was transtympanically inoculated in the mouse middle ear after anesthesia with Ketamine (5 mg/100 g). As a control, normal saline was inoculated by the same procedure. Mice were euthanatized by CO2 inhalation and cervical dislocation after the second day postinoculation, and the cochlea was dissected from the skull base. The dissected cochlea was fixed with 4% paraformaldehyde and decalcified with EDTA. After dehydration in a series of graded alcohol and xylene solutions, the cochlea was embedded in paraffin and sectioned through the mid-modiolar plane at a thickness of 10 μm. Endogenous peroxidase activity was quenched with incubation in 0.3% H2O2 for 30 min after tissue sections were deparaffinized and rehydrated through an identical series of graded xylene and alcohol solutions. Nonspecific binding sites were blocked by preincubation with a 1:500 dilution of horse serum for 30 min at room temperature. Polyclonal anti-MCP-1 antibody (1:200; Santa Cruz Biotechnology Inc., Santa Cruz, CA) was incubated with the sample for 1 h at room temperature. The sections were washed with PBS three times and incubated with a 1:500 dilution of biotinylated anti-rabbit immunoglobulin G (IgG) antibody (Vector Laboratories, Burlingame, CA) for 30 min at room temperature. Peroxidase was attached by the avidin-biotin complex method, and signals were detected with diaminobenzidine tetrahydrochloride. For immunofluorescent staining, cells were cultured on four-chamber microscope slides. After overnight starvation, cells were treated with the NTHI lysate for 1 h, fixed with 4% paraformaldehyde, blocked using goat serum, and subsequently incubated in the presence of mouse anti-p65 NF-κB antibody (1:200; Santa Cruz Biotechnology Inc.) for 1 h at room temperature. Primary antibody was detected with rhodamine-conjugated goat anti-mouse IgG (1:500; Santa Cruz Biotechnology Inc.). Samples were then viewed and photographed using a Zeiss Axiophot microscope equipped with an AxioVision image analyzer (Carl Zeiss MicroImaging Inc., Thornwood, NY).
Western blotting and phosphorylation assay.
The cells were grown to 80% confluence in six-well culture plates. After overnight starvation with basal medium, the cells were treated with a 1:20 dilution of the NTHI lysate for 8 h. The cells were lysed with cell lysis buffer (Cell Signaling Technology, Beverly, MA) supplemented with 1 mM phenylmethylsulfonyl fluoride (Calbiochem). The lysates were then centrifuged at 14,000 × g for 15 min, and the supernatants were collected. The protein concentration in the supernatants was measured using the BCA protein assay (Pierce Biotechnologies), and samples were stored at −70°C. An amount equivalent to 20 μg of protein was loaded onto 10% Tricine gels (Invitrogen, Carlsbad, CA) and run using Tris-Tricine-sodium dodecyl sulfate tank buffer (pH 7.4) supplemented with 1 mM sodium orthovanadate (Sigma). After electrophoresis, the proteins were transferred onto polyvinylidene difluoride membranes (0.2 μm; Bio-Rad, Hercules, CA) and washed three times for 5 min each in Tris-buffered saline plus 0.05% Tween 20 (TBST). The membranes were blocked using 5% nonfat dry milk in TBST for 1 h at room temperature and incubated overnight at 4°C in the presence of a 1:1,000 dilution of a polyclonal antibody against MCP-1 (Santa Cruz Biotechnologies Inc.). For phosphorylation assay (70), antibodies against phospho-IκKβ, total IκKβ, phospho-IκBα, or total IκBα (Cell Signaling Technology, Beverly, MA) were used as the primary antibodies. The membranes were washed three times for 5 min each with TBST and incubated for 1 h at room temperature with horseradish peroxidase (HRP)-conjugated secondary antibody in blocking buffer (anti-rabbit IgG; Cell Signaling Technology). The membranes were then washed three times with TBST and incubated in SuperSignal substrate (Pierce Biotechnologies) for 1 min at room temperature. The chemiluminescence signal was detected by exposure to X-ray film and quantitated using Quantity One software (Bio-Rad Laboratories, Hercules, CA).
Protein array.
A solid-phase, sandwich-enzyme-linked immunosorbent assay (ELISA)-format, multiplexed protein assay was performed to simultaneously detect various cytokines released by activated cells (72). Briefly, the culture medium of SLFs was collected 48 h after treatment with either NTHI or PBS. Membranes preblotted with various anticytokine antibodies (RayBio rat cytokine antibody array; RayBiotech Inc., Norcross, GA) were incubated with culture medium for 2 h at room temperature after blocking for nonspecific binding sites as per the manufacturer's recommended instructions. After addition of a biotin-conjugated anticytokine antibody mixture (provided in the kit), the membrane was incubated with HRP-conjugated streptavidin as per the manufacturer's instructions. Signal intensity was quantified with the Quantity One software (Bio-Rad Laboratories). Compared to a vehicle control, blotting with biotin-conjugated IgG produced a positive signal.
Real-time quantitative PCR.
Real-time quantitative PCR was performed as described previously (71). Briefly, 3 h after treatment with NTHI lysate, total RNA was extracted using the RNeasy kit (QIAGEN, Valencia, CA), and cDNA was synthesized using the TaqMan reverse transcription kit (Applied Biosystems, Foster City, CA). Multiplex PCR was performed using the ABI 7500 real-time PCR system (Applied Biosystems) with gene-specific primers, a 6-carboxyfluorescein-conjugated probe for MCP-1, and a VIC-conjugated probe for GAPDH. The cycle threshold (CT) values were determined according to the manufacturer's instructions. The relative quantity of mRNA was also determined using the 2−ΔΔCT method (53). CT values were normalized to the internal control (GAPDH), and the results were expressed as fold induction of mRNA, with the mRNA levels in the nontreated group set as 1.
For reverse transcription-PCR (RT-PCR) analysis, PCR was performed using gene-specific primers after extracting total RNA and synthesizing cDNA. The primers used were as follows; rat TLR2, 5′-TCTCTGTCATGTGATGCTGCTGGT-3′ and 5′-TCCAAGTGTTCAAGACTGCCCAGA-3′ (239 bp); rat TLR4, 5′-AGTGTATCGGTGGTCAGTGTGCTT-3′ and 5′-ATGAAGATGATGCCAGAGCGGCTA-3′ (404 bp); rat 18S rRNA, 5′-GTGGAGCGATTTGTCTGGTT-3′ and 5′-CGCTGAGCCAGTCAGTGTAG-3′ (200 bp); mouse TLR2, 5′-ACGCTGGAGGTGTTGGATGTTAGT-3′ and 5′-AACAAAGTGGTTGTCGCCTGCTTC-3′ (237 bp); mouse MyD88, 5′-TAAGTTGTGTGTGTCCGACCGTGA-3′ and 5′-TGGGAAAGTCCTTCTTCATCGCCT-3′ (230 bp); and mouse vimentin, 5′-ATCATGCGGCTGCGAGAGAAATTG-3′ and 5′-AGCCTCAGAGAGGTCAGCAAACTT-3′ (360 bp). PCR products were analyzed by electrophoresis on 1.2% Tris-acetate-EDTA agarose gels, stained with ethidium bromide, and viewed with UV light.
Plasmids, transfection, and luciferase assay.
The vectors expressing a dominant-negative mutant TLR2 (TLR2_DN), a wild-type TLR2 (TLR2_WT), a dominant-negative mutant TLR4 (TLR4_DN), a dominant-negative mutant IκKβ [IκKβ(K49A)], and a transdominant mutant IκBα [IκBα(S32/36A)] were previously described (38, 83). The luciferase-expressing vectors with 5′ flanking regions of rat MCP-1 were kind gifts from D. L. Eizirik (Brussels University, Brussels, Belgium) (47). The pMCP1-E construct contained NF-κB-binding site 3 (NF-κB3) as well as the NF-κB-binding sites of the enhancer regions (NF-κB1 and NF-κB2). In contrast, the pMCP1 −514 to +53 construct had only NF-κB3. pGL3-B, a blank vector, was purchased from Promega (Madison, WI). Luciferase assay was performed as previously described (70). Cells were seeded into six-well plates at a density of 1.5 × 105 cells/well and transfected at ∼60% confluence. Transfection was performed using the Transit-LT1 transfection reagent (PanVera, Madison, WI) according to the manufacturer's instructions. pRL-TK vector (Promega) was cotransfected to normalize for transfection efficiency. Transfected cells were then starved overnight in serum-free DMEM, followed by exposure to the NTHI lysate for 10 h before harvesting. All transfections were carried out in triplicate. The cells were then washed with PBS, dissolved in 250 μl of cell culture lysis reagent (Promega), and harvested by scraping. Luciferase activity was measured using a luminometer (Pharmingen, La Jolla, CA) after adding the necessary luciferase substrate (Promega). Results were expressed as fold induction of luciferase activity, taking the value of the nontreated group as 1.
EMSA and transcription factor assay.
Cells were treated with NTHI lysate for 1.5 h after overnight starvation. Nuclear protein was extracted using the NE-PER nuclear extraction reagent (Pierce Biotechnologies), using previously described procedures (86). The protein concentration in extracts was determined using a BCA protein assay kit (Pierce Biotechnologies). The following 5′-biotin-labeled double-stranded oligonucleotide probes were from Integrated DNA Technologies, Inc. (Coralville, IA): rat MCP-1 enhancer NF-κB site 1, 5′-AAGGGTCTGGGAACTTCCAAT-3′; NF-κB site 2, 5′-AGAATGGGAATTTCCACACTCTT-3′. In vitro binding of NF-κB to the MCP-1 enhancer was determined using the LightShift chemiluminescent electrophoretic mobility shift assays (EMSA) kit (Pierce Biotechnologies) according to the manufacturer's instructions. Briefly, nuclear proteins (4 μg) were incubated with a biotin-labeled target DNA probe (20 fmol) in 20 μl of binding buffer containing 1 μg poly(dIdC), 10 mM Tris (pH 7.5), 50 mM KCl, 1 mM dithiothreitol, 10 mM MgCl2, 0.05% NP-40, and 2.5% (vol/vol) glycerol for 20 min at room temperature. Samples were applied to 6% polyacrylamide gels under native conditions in high-ionic-strength buffer, and electrophoresis was performed until the bromophenol blue dye migrated three-fourths down the length of the gel. As positive and negative controls, a biotinylated Epstein-Barr nuclear antigen (EBNA) control DNA (5′-TAGGCATATGCTA-3′) was applied with or without the EBNA extract. Electrophoretic gels were transferred to nylon membranes at 380 mA for 30 min in 0.5× Tris-borate-EDTA buffer cooled to 10°C. Transferred DNA was cross-linked to membranes using a UV lamp for 10 min. To detect biotin-labeled DNA, a 1:300 dilution of streptavidin-HRP conjugate was applied to the membranes for 15 min. After washing, the chemiluminescent substrate was added and the signal was detected with exposure to X-ray films. For transcription factor assay, activated transcription factors were analyzed using the ELISA-based TransFactor kit (Clontech, Mountain View, CA) according to the manufacturer's instructions. In brief, nuclear protein was applied to a 96-well plate coated with oligonucleotides containing the consensus sequence of a different transcription factor, i.e., p65 (5′-GGGGTATTTCC-3′), p50 (5′-GGGGATCCC-3′), c-Rel (5′-GGGGTATTTCC-3′), c-Fos (5′-TGACTCA-3′), CREB-1 (5′-TGACATCA-3′), or ATF2 (5′-TGACATCA-3′). Binding of transcription factors was inhibited with a competitor DNA consisting of the same sequence as in the oligonucleotide-coated wells, in order to demonstrate the binding specificity between DNA and the transcription factor. After the wells were washed, bound transcription factors were labeled with a 1:100 dilution of primary polyclonal antibodies against each transcription factor at room temperature for 1 h. Unbound antibodies were washed out, followed by incubation with a 1:1,000 dilution of anti-rabbit IgG conjugated with HRP at room temperature for 30 min. One hundred microliters of tetramethylbenzidine substrate was added to each well and incubated at room temperature for 10 min. The absorbance was measured at 655 nm with a microtiter plate reader.
Statistics.
All experiments were carried out in triplicate. Results are expressed as means ± standard deviations. Statistical analysis was performed using Student's t test, with significance considered to be a P value of <0.01 or <0.05.
RESULTS
NTHI highly induces MCP-1 in SLFs.
We hypothesized that SLFs recognize inflammatory signals and express chemokines and cytokines. Up-regulation of inner-ear-derived proinflammatory chemokines and cytokines probably leads to attraction of effector cells, resulting in inner ear dysfunction. Our preliminary data showed that mononuclear cells infiltrated in the spiral ligament of the human cochlea with labyrinthitis, combined with OM, which is consistent with our hypothesis (data not shown). We also previously reported that the SLF cell line up-regulates various chemokines and cytokines upon exposure to lysate of NTHI, a common OM pathogen (72). Most significantly, a predominant induction of MCP-1 after treatment with the NTHI lysate was shown. To determine whether NTHI up-regulates MCP-1 in SLFs of animal models, we transtympanically inoculated live NTHI in the mouse middle ear and performed immunolabeling of the cochlear lateral wall. The results showed up-regulation of MCP-1 expression in the NTHI-treated group (Fig. 1D) compared to a control (Fig. 1C). Western blotting demonstrated that the SLF cell line induces MCP-1 upon exposure to NTHI lysate (Fig. 1E). The MCP-1 band (16.3 kDa), which is hardly seen in the control, suggests that SLFs do not express measurable levels of MCP-1 in the unexposed state.
FIG. 1.

SLFs up-regulate MCP-1 upon exposure to NTHI. (A and B) Illustrations of the cochlear three-dimensional structure (A) and the cochlear lateral wall (B). Mo, modiolus; SV, scala vestibuli; ST, scala tympani; *, scala media; CoN, cochlear nerve; CL, cochlear lateral wall. (C and D) Immunolabeling of the cochlear lateral wall shows up-regulation of MCP-1 expression in the NTHI-treated group (D) compared to a control (C). Live NTHI was inoculated in the mouse middle ear, and the cochlea was dissected after 2 days. As a control, normal saline was inoculated with a same procedure. After decalcification and sectioning, immunolabeling was performed using a polyclonal anti-MCP-1 antibody. (E) Western blotting and its densitinogram show that SLFs up-regulate MCP-1 upon exposure to the NTHI lysate, compared to the PBS-treated control (Con). The spiral ligament cell line was treated with two different culture batches (1 and 2) of NTHI lysate for 8 h, and the cells were harvested. Soluble protein was extracted by gently lysing the cells in the presence of protease inhibitors, followed by centrifugation. The soluble protein fraction was separated using 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis and subsequently transferred onto polyvinylidene difluoride membranes and labeled with a polyclonal anti-MCP-1 antibody. Signal was detected by exposure to X-ray film, and the relative density of MCP-1 was measured with normalization to β-tubulin levels.
To evaluate if SLFs release MCP-1, we performed a solid-phase multiplexed protein assay in a sandwich ELISA format in culture medium that was collected 48 h after exposure to the NTHI lysate. The results indicate that MCP-1 is released from SLFs after NTHI exposure, compared to a PBS-treated control (Fig. 2A). Interestingly, the release of IL-1β after NTHI exposure is not obvious. Real-time quantitative PCR was performed to examine the mRNA level of MCP-1. MCP-1 mRNA was highly up-regulated with exposure to NTHI lysate in a time-dependent manner, reaching saturation at between 4 h and 8 h postexposure (Fig. 2B). To rule out that this up-regulation is not a cell-specific phenomenon, primary fibroblasts of the epidermis were compared in parallel. The fold induction of MCP-1 mRNA by exposure to the NTHI lysate was much higher than that of IL-1β in both SLF cell lines and primary epidermal fibroblasts (Fig. 2C).
FIG. 2.

SLFs release MCP-1, but not Il-1β, upon exposure to NTHI lysate. (A) The protein array and its densitinogram show that the spiral ligament cells release MCP-1 after treatment with a lysate of NTHI, compared with the PBS-treated control. Cell culture medium was collected 48 h after treatment, and a solid-phase multiplexed protein assay in a sandwich ELISA format was performed. The chemiluminescence signal was detected by exposure to X-ray film and quantitated using Quantity One software. MCP-1 is released from SLFs after treatment with a lysate of NTHI, while release of IL-1β is not obvious. Con, PBS-treated control; Pos, positive signal produced by blotting of biotin-conjugated IgG; Neg, negative signal produced by blotting of vehicle. (B) Real-time quantitative PCR demonstrates a time-dependent up-regulation in the levels of MCP-1 mRNA after treatment with NTHI lysate, reaching saturation at between 4 h and 8 h. (C) MCP-1 expression levels are much higher than IL-1β levels in both the spiral ligament cell line and primary epidermal fibroblasts (CRL). For real-time quantitative PCR, multiplex PCR was performed and the CT values of MCP-1 were normalized to the internal control, GAPDH. Results are expressed relative to the fold induction of mRNA levels, taking the value of the nontreated group as 1. The experiments were performed in triplicate and repeated more than twice. Values are given as means ± standard deviations (n = 3). *, P < 0.05; **, P < 0.01.
TLR-2 and MyD88 are involved in NTHI-induced MCP-1 up-regulation.
It is unknown which cell surface receptors of SLFs are involved in recognizing specific molecules of NTHI lysates and transmitting signals to the cytoplasm. TLRs have been implicated in playing a role in the recognition of PAMPs such as LPS, peptidoglycans, and lipoproteins (2, 64). Based on the evidence that TLR2 plays a critical role in the cellular response to bacterial products (83, 95), we first investigated the expression of TLR2 in the rat SLF cell line. Conventional RT-PCR showed that TLR2 and TLR4 are expressed in SLFs (Fig. 3A). Interestingly, real-time quantitative PCR demonstrated that TLR2, but not TLR4, is up-regulated when treated with NTHI, indicating the possible involvement of TLR2 in the NTHI-induced signaling pathway. To evaluate if TLR2 is required for NTHI-induced MCP-1 up-regulation, dominant-negative mutant constructs of TLR2 and TLR4 were transfected in SLFs. The results show that NTHI-induced MCP-1 up-regulation was inhibited by transfection of SLFs with a dominant-negative mutant construct of TLR2 by 80 to 90%, but not by TLR4 (Fig. 3B, left panel). Moreover, overexpression of wild-type TLR2 enhanced NTHI-induced MCP-1 up-regulation by two- to threefold. The luciferase assay showed that NTHI-induced activation of the MCP-1 promoter decreased by dominant-negative inhibition of TLR2 (Fig. 3B, right panel).
We next examined the involvement of TLR2 and MyD88 by using gene knockout mice (1, 88). MyD88 is a general adaptor/regulator molecule for the Toll/IL-1 receptor family of receptors such as TLR2 and TLR4 (67). The spiral ligaments was dissected from the inner ears of the gene knockout mice, and primary SLFs were cultured from explanted pieces of the spiral ligament (Fig. 3C). RT-PCR identified gene expression profiles of primary SLFs derived from gene knockout mice (data not shown). Neither TLR2 nor MyD88 mRNA was expressed in TLR2 or MyD88 gene knockout mice, respectively. In contrast, vimentin mRNA was expressed in all of the wild-type and knockout mice, regardless of NTHI treatment. As shown in Fig. 3D, knocking out TLR2 blocks NTHI-induced MCP-1 up-regulation by more than 90%. Interestingly, in the case of MyD88-targeted SLFs, MCP-1 remained unchanged when exposed to the NTHI lysate, indicating that MyD88 may be involved in TLR2-independent signaling pathways. Taken together, the results suggest that the TLR2-MyD88 signaling pathway is involved in the NTHI-induced up-regulation of MCP-1 expression.
Activation of NF-κB via IκKβ-dependent IκBα phosphorylation is required for NTHI-induced MCP-1 up-regulation.
The transcription factor NF-κB plays a critical role in inflammatory responses (8, 49). Since the original classification of NF-κB as a nuclear factor that bound to the κB enhancer motif of the Ig κ chain gene (81, 85), five other NF-κB subunits (p65, c-Rel, RelB, p50, and p52) have been identified, which function as homo- and heterodimers (30). In addition, since NF-κB activation is required for up-regulation of MCP-1 in other systems, such as kidney (79), blood vessel (61), and pancreas (47), we sought to explore the involvement of NF-κB in NTHI-induced MCP-1 up-regulation of SLFs. Nuclear translocation is a key step for activation of NF-κB and is controlled mainly by IκBα, an inhibitory subunit. Once IκBα is phosphorylated and degraded by proteasomes, NF-κB is released from the NF-κB-IκBα complex and is free to translocate to the nucleus (75). Immunolabeling of SLFs showed that NF-κB translocates to the nucleus upon exposure to NTHI lysate (Fig. 4A). Pretreatment with CAPE or MG-132 (a cell-permeative proteasome inhibitor) resulted in partial inhibition of NF-κB translocation by NTHI treatment. CAPE and MG-132 are chemical inhibitors which block NF-κB translocation without affecting IκBα degradation (74) and prevent degradation of IκBα (75), respectively.
FIG. 4.

Activation of the IκKβ-IκBα-NF-κB signaling pathways is required for NTHI-induced MCP-1 up-regulation. (A) NF-κB is translocated from the cytoplasm to the nucleus after NTHI treatment. However, CAPE and MG132 (a cell-permeative proteasome inhibitor) block the NTHI-induced NF-κB translocation. Cells were exposed to the NTHI lysate with or without pretreatment with CAPE or MG132. Cells were immunolabeled using a polyclonal anti-p65 antibody to localize cytoplasmic or nuclear NF-κB. Con, PBS-treated control. (B) Real-time quantitative PCR shows that CAPE and MG132 inhibit NTHI-induced MCP-1 up-regulation. NTHI lysate was added to the spiral ligament cell line with or without pretreatment with CAPE or MG132. Cells were harvested after 3 h, and total RNA was extracted. Multiplex PCR was performed using specific primers and probes to MCP-1 and GAPDH for real-time quantitative PCR. (C) Upstream components of the NF-κB pathway are involved in NTHI-induced MCP-1 up-regulation. Phosphorylation assay demonstrates that IκBα and IκKβ are phosphorylated by treatment with the NTHI lysate. Real-time quantitative PCR shows that transfecting a dominant-negative construct of IκBα and IκKβ inhibits NTHI-induced MCP-1 up-regulation in a dose-dependent manner. (D) The luciferase assay demonstrates that a dominant-negative construct of IκBα and IκKβ inhibits NTHI-induced activation of the MCP-1 promoter. MCP1-E vector was cotransfected with the dominant-negative constructs and pRL-TK vector. pcDNA, mock transfection with a blank vector. Results are expressed as fold induction of mRNA levels or luciferase activity, taking the value of the nontreated group as 1. The experiments were performed in triplicate and repeated more than twice. Values are given as the means ± standard deviations (n = 3). *, P < 0.05; **, P < 0.01.
To determine if activation of NF-κB is involved in NTHI-induced MCP-1 up-regulation, SLFs were pretreated with CAPE or MG-132 before NTHI treatment. Real-time quantitative PCR demonstrated that NTHI-induced MCP-1 up-regulation was inhibited, in a dose-dependent manner, by 30% to 40% in cells pretreated with CAPE and by 80% to 90% in cells pretreated with MG-132 (Fig. 4B). IκB is phosphorylated by its upstream kinase, IκK, which is itself activated by phosphorylation via another upstream kinase (30, 40). Phosphorylation assay demonstrated that both IκBα and IκK are phosphorylated upon exposure to the NTHI lysate but not in the control (Fig. 4C). Moreover, NTHI-induced MCP-1 up-regulation was inhibited by transfection of SLFs with a dominant-negative mutant construct of IκKβ [IκKβ(K49A)] or a transdominant mutant form of IκBα [IκBα(S32/36A)], by 85 to 95% or 70 to 90%, respectively. IκBα(S32/36A) was mutated in two critical serine residues which are required for IκK-mediated phosphorylation. In addition, the luciferase assay demonstrated that inhibition of IκKβ and IκBα decreased NTHI-induced activation of the MCP-1 promoter (Fig. 4D). These results suggest that this up-regulation requires the activation of NF-κB, mediated by the IκKβ-dependent IκBα signaling pathway in the SLFs.
Binding of NF-κB to the enhancer region of MCP-1 is involved in NTHI-induced MCP-1 up-regulation.
To determine the 5′ flanking promoter region of the MCP-1 gene, which is responsive to the NTHI lysate, promoter activity was measured and compared among the promoter fragments from nucleotide −514 to +53 relative to the transcription start site, with or without the enhancer region between −2,180 and −2,478. The promoter constructs were transfected in SLFs, and the luciferase activity was measured after treatment with the NTHI lysate. The promoter construct with the enhancer region induced luciferase activity more than fivefold with NTHI treatment, whereas the promoter construct without the enhancer region showed no significant response to NTHI treatment (Fig. 5A). This result indicates a potentially important role for NF-κB binding sites within the enhancer region (NF-κB sites 1 and 2), but not in the promoter region (NF-κB site 3).
FIG. 5.

Identification of NF-κB subunits and their associated binding sites involved in NTHI-induced MCP-1 up-regulation. (A) 5′ flanking regions of rat MCP-1 were subcloned into a luciferase-expressing vector. The pMCP1-E construct contained the NF-κB-binding site 3 (NF-κB3) as well as the NF-κB-binding sites of the enhancer region (NF-κB1 and NF-κB2). In contrast, the pMCP1 construct had only NF-κB3. Cells were first transfected with pGL3-B (a blank vector without a promoter region), pMCP1, or pMCP1-E and subsequently treated with NTHI lysate, and the effects measured using a luciferase assay. The assay shows that the enhancer region containing NF-κB1 and NF-κB2 is of importance for NTHI-induced MCP-1 up-regulation. Results are expressed as fold-induction of luciferase activity, taking the value of the nontreated group as 1. (B) NF-κB-binding sites of the enhancer region are conserved in mammals, i.e., mouse, rat, and human. EMSA was performed with oligonucleotides of the rat MCP-1 enhancer region, spanning the region from −2,272 to −2,297 for NF-κB site 1 and from −2,242 to −2,266 for NF-κB site 2. (C) Nuclear extract of NTHI-treated cells binds to NF-κB site 1 but not to NF-κB site 2. Two complexes (arrows) are induced in NTHI-treated cells. Lanes 1 and 4, NTHI-treated cells; lanes 2 and 5, non-NTHI-treated cells; lanes 3 and 6, oligonucleotide only; PC, positive control (biotinylated EBNA control DNA plus EBNA extract); NC, negative control (biotinylated EBNA control DNA only). (D) ELISA-based transcription factor assay shows that the p65 subunit is activated and translocated to the nucleus by treatment with the NTHI lysate. Nuclear protein was applied to a 96-well plate coated with oligonucleotides containing the consensus sequence of a different transcription factor (p65, p50, c-Rel, c-Fos, CREB-1 or ATF2). Binding of transcription factors was inhibited with a competitor DNA sequence similar to that in the oligonucleotide-coated wells, demonstrating the binding specificity between DNA and the transcription factor. Bound transcription factors were labeled first with primary polyclonal antibodies specific for each transcription factor and then with a secondary antibody, an anti-rabbit IgG antibody conjugated with HRP. After incubation with the tetramethylbenzidine substrate, the absorbance was measured at 655 nm with a microtiter plate reader. OD, optical density. The experiments were performed in triplicate and repeated more than twice. Values are given as the means ± standard deviations (n = 3). **, P < 0.01.
In order to identify the potential NF-κB-binding site(s) in vitro, EMSAs were performed using complementary single-stranded DNA probes spanning two of the NF-κB motifs, NF-κB site 1 (−2,272 to −2,297) and NF-κB site 2 (−2,242 to −2,266). Two complexes were apparent upon exposure to the NTHI lysate in the presence of the NF-κB site 1 probe, but none were formed with the NF-κB site 2 probe (Fig. 5B). This was unexpected since based on a previous report (47), which used IL-1β as a stimulant, NF-κB site 2 is a potential NF-κB-binding motif that is responsive to NTHI treatment. This result suggests that the activation of the NF-κB-binding motif is stimulant specific. We next sought to identify the subunits of NF-κB/Rel dimers involved, using an ELISA-based transcription factor assay. Nuclear protein was extracted after NTHI treatment and applied to a 96-well plate coated with oligonucleotides containing either the consensus sequence of NF-κB subunits (p65, p50, and c-Rel) or control transcription factors (c-Fos, CREB-1, and ATF2). NTHI treatment activated the p65 subunit but not the other NF-κB subunits (p50 or c-Rel) or control transcription factors (Fig. 5C). Binding of the p65 subunit was inhibited with a competitor DNA sequence, demonstrating the specificity of binding. This result suggests that the NF-κB activated by NTHI exposure is a p65 homodimer or a p65 heterodimer with another subunit other than p50 and c-Rel (89).
DISCUSSION
In this study, we demonstrated that SLFs up-regulate MCP-1 upon exposure to NTHI lysate (Fig. 6). We have shown that this up-regulation requires the TLR2-dependent activation of NF-κB, mediated by IκKβ-dependent IκBα phosphorylation. We also showed the presence of a potential NF-κB promoter motif that is sensitive to NTHI-specific molecules/ligands. Additionally, NF-κB binding to the enhancer region of MCP-1 is required in NTHI-induced MCP-1 up-regulation.
FIG. 6.
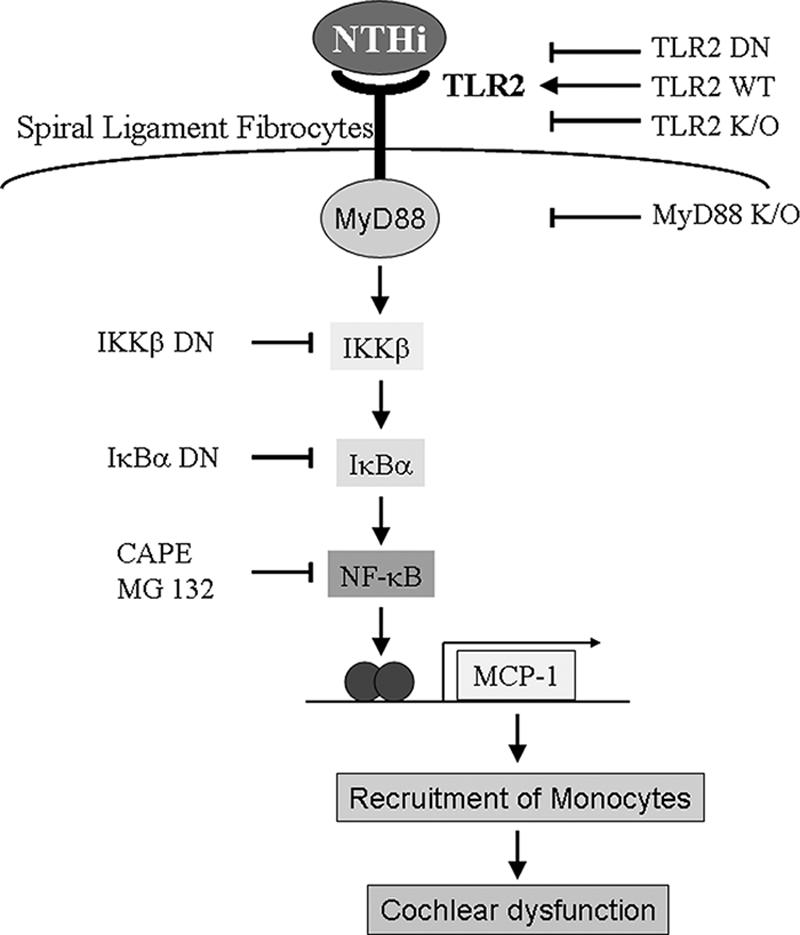
Schematic representation of the signaling pathways involved in NTHI-induced MCP-1 up-regulation. As indicated, NTHI up-regulates MCP-1 expression via TLR2-dependent NF-κB activation. It is hypothesized that the up-regulation of MCP-1 results in recruitment and attraction of effector cells, leading to cochlear dysfunction and subsequent sensorineural hearing loss.
NTHI, a gram-negative bacterium is a normal pharyngeal flora as well as an important human respiratory pathogen, which selectively adheres to mucus and mucosal epithelium in the Eustachian tube and the middle ear (69) and induces various proinflammatory cytokines (68). Nasopharyngeal colonization of NTHI may persist for prolonged periods of time, since the bacteria undergo antigenic variation by way of point mutation, gene amplification, phase variation, or horizontal gene transfer and homologous recombination (25). The organism releases its outer membrane fragments during growth (27) or after treatment with antibiotics (17). NTHI is nontypeable since it lacks a polysaccharide capsule. The polysaccharide capsule of H. influenzae type b contains porin, a ligand for TLR2 (23), whereas TLR4 mediates innate immune responses to H. influenzae type b (90). Although NTHI activates both TLR2 and TLR4 (57, 82, 83, 93), our results demonstrated that TLR2, and not TLR4, is required for NTHI-induced MCP-1 up-regulation. Partially investigated molecules of NTHI, such as the lipoprotein P6, a specific ligand to TLR2, activate a specific receptor upon interaction with their hosts (15). Although TLR4 has been shown to be involved in clearing NTHI from the mouse lung (93), the decrease in lipooligosaccharide acylation in NTHI mutants has made this clearance more TLR-2 dependent (57). Further studies are needed to better understand the interaction between NTHI molecules and specific host receptors.
The Toll family of receptors is defined by homology to the Drosophila Toll receptor protein, which plays important roles in providing antifungal defenses as well as controlling the dorso-ventral polarity in developing embryos (11, 31, 66). Targeting of TLRs makes the host insensitive to bacterial components and delays bacterial clearance (58, 93). TLRs can be grouped into subfamilies according to the types of ligands they recognize (76). In addition to recognizing PAMPs, it is also known that TLRs are activated by endogenous ligands such as heat shock proteins and necrotic cells (7, 42). TLRs enable the host to distinguish between the different types of infections in order to induce the appropriate effector responses. TLR2 and TLR4 have been shown to recognize lipid-based molecules. TLR4 recognizes LPS of gram-negative bacteria, while TLR2 is important for recognition of molecules of gram-positive bacteria such as lipoproteins and lipoteichoic acid (4). In addition, TLR2 recognizes a broader range of microbial molecules than other TLRs. It acts as a receptor for the peptidoglycans of gram-positive bacteria (80), bacterial lipoproteins (12), and even atypical LPS of Leptospira and Porphyromonas (35, 92). TLR2 functions as a heterodimer with either TLR1 or TLR6, which may explain the broader range of ligand specificity for TLR2.
In our study, NTHI-induced MCP-1 up-regulation was blocked completely by targeting of the MyD88 gene but incompletely by targeting of TLR2. Although our results demonstrated that TLR2 is a major receptor involved in NTHI-induced MCP-1 up-regulation, involvement of alternate TLR2-independent, MyD88-dependent signaling pathways cannot be ruled out. In addition, since MyD88 is a ubiquitous adaptor protein for membrane receptors such as IL-1 receptor, TLR2, and TLR4 (5, 56), it is expected that targeting the MyD88 gene will result in more efficient inhibition of signaling pathways generated from NTHI molecules than blocking of a single receptor, such as TLR2. It has been demonstrated that both MyD88-dependent and -independent pathways mediate signaling in response to LPS (41, 43). Compared to the MyD88-dependent signaling cascade, the activation of NF-κB via a MyD88-independent signaling is delayed. Our results did not show MyD88-independent pathways with delayed kinetics, since we evaluated only early signaling in response to NTHI. However, the temporal activation of interferon-related signaling via the Toll/IL-1 receptor domain-containing adaptor inducing beta interferon (TRIF) can be fast (94), suggesting that TRIF and interferon-related factors may not be involved in MCP-1 regulation. Moreover, the NTHI lysate, which contains a mixture of bacterial products, may activate more than one receptor simultaneously or sequentially. Considering that epithelial cell-derived IL-1 synergistically enhances NTHI-induced DEFB4 (previously human β-defensin 2) up-regulation (71), it is also possible that NTHI-induced secondary molecules of the host (i.e., cytokines) may be involved in the activation of TLR2-independent signaling pathways. These results point to the complexity of the signaling pathways involved in controlling the expression of MCP-1.
The induction of MCP-1 in other cell types requires activation of multiple signaling molecules, including protein kinase C, protein tyrosine kinase, mitogen-activated protein kinase, and calmodulin (19, 84, 98). Moreover, activation of NF-κB is important in the transcriptional regulation of MCP-1, where the activator protein (AP-1) and sequence-specific transcription factor (Sp-1) are essential for basal expression (47, 89, 91). Our results suggest that NTHI-induced MCP-1 up-regulation requires activation of NF-κB, mediated by IκKβ-dependent IκBα phosphorylation in the SLFs of the cochlea. Both p65 and p50 of the NF-κB subunits are known to be involved in IL-1β-induced MCP-1 up-regulation (47), but in this study, we showed that NTHI activates only the p65 subunit of NF-κB. This may explain why NTHI-activated NF-κB binds to a different sequence motif on the promoter region than does IL-1β-activated NF-κB. p65 may form homodimers in complex with IκB, potentially changing the binding specificity (59) and resulting in an increased promiscuity compared to heterodimers due to conformational and structural shifts that occur in one of the dimer partners (15). However, further studies are necessary to elucidate the other subunit of NF-κB involved in forming a dimer.
In conclusion, our studies demonstrate that NTHI, a common human pathogen of OM and obstructive pulmonary disease, induces up-regulation of MCP-1 via a TLR2-dependent NF-κB activation pathway in the SLFs of the cochlea. As far as we know, this is the first report elucidating the involvement of TLR2 in MCP-1 up-regulation by bacterial molecules in the SLFs of the cochlea. Our findings may lead to the development of a new model for molecular inner ear defense mechanism and inner ear dysfunction. TLR activation by the specific ligands entering inner ear may result in up-regulation of SLF-derived proinflammatory chemokines, leading to attraction of effector cells and causing inner ear dysfunction. Further studies are needed to better understand the molecular pathogenesis of inner ear dysfunction secondary to OM, such as sensorineural hearing loss and dizziness.
Acknowledgments
We acknowledge Jian-Dong Li for providing TLR2 and MyD88 gene knockout mice and dominant-negative constructs and Décio L. Eizirik for the luciferase-expressing vectors with 5′ flanking regions of rat MCP-1. We also thank Ali Andalibi for critical review and Arsen Akopyan for technical assistance.
This work was supported in part by grants 5R01 DC005025-05 and P30 DC006276-04 from the NIH, NIDCD.
Editor: J. N. Weiser
Footnotes
Published ahead of print on 23 April 2007.
REFERENCES
- 1.Adachi, O., T. Kawai, K. Takeda, M. Matsumoto, H. Tsutsui, M. Sakagami, K. Nakanishi, and S. Akira. 1998. Targeted disruption of the MyD88 gene results in loss of IL-1- and IL-18-mediated function. Immunity 9:143-150. [DOI] [PubMed] [Google Scholar]
- 2.Aderem, A., and R. J. Ulevitch. 2000. Toll-like receptors in the induction of the innate immune response. Nature 406:782-787. [DOI] [PubMed] [Google Scholar]
- 3.Agrawal, S., M. Husein, and D. MacRae. 2005. Complications of otitis media: an evolving state. J. Otolaryngol 34(Suppl. 1):S33-S39. [PubMed] [Google Scholar]
- 4.Akira, S., and K. Takeda. 2004. Toll-like receptor signalling. Nat. Rev. Immunol. 4:499-511. [DOI] [PubMed] [Google Scholar]
- 5.Akira, S., M. Yamamoto, and K. Takeda. 2003. Role of adapters in Toll-like receptor signalling. Biochem. Soc. Trans. 31:637-642. [DOI] [PubMed] [Google Scholar]
- 6.Anderson, K. V. 2000. Toll signaling pathways in the innate immune response. Curr. Opin. Immunol. 12:13-19. [DOI] [PubMed] [Google Scholar]
- 7.Andrade, C. F., T. K. Waddell, S. Keshavjee, and M. Liu. 2005. Innate immunity and organ transplantation: the potential role of toll-like receptors. Am. J. Transplant. 5:969-975. [DOI] [PubMed] [Google Scholar]
- 8.Baeuerle, P. A. 1998. IkappaB-NF-kappaB structures: at the interface of inflammation control. Cell 95:729-731. [DOI] [PubMed] [Google Scholar]
- 9.Baggiolini, M., and C. A. Dahinden. 1994. CC chemokines in allergic inflammation. Immunol. Today 15:127-133. [DOI] [PubMed] [Google Scholar]
- 10.Barenkamp, S. J., and E. Leininger. 1992. Cloning, expression, and DNA sequence analysis of genes encoding nontypeable Haemophilus influenzae high-molecular-weight surface-exposed proteins related to filamentous hemagglutinin of Bordetella pertussis. Infect. Immun. 60:1302-1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Belvin, M. P., and K. V. Anderson. 1996. A conserved signaling pathway: the Drosophila toll-dorsal pathway. Annu. Rev. Cell Dev. Biol. 12:393-416. [DOI] [PubMed] [Google Scholar]
- 12.Brightbill, H. D., D. H. Libraty, S. R. Krutzik, R. B. Yang, J. T. Belisle, J. R. Bleharski, M. Maitland, M. V. Norgard, S. E. Plevy, S. T. Smale, P. J. Brennan, B. R. Bloom, P. J. Godowski, and R. L. Modlin. 1999. Host defense mechanisms triggered by microbial lipoproteins through toll-like receptors. Science 285:732-736. [DOI] [PubMed] [Google Scholar]
- 13.Casselbrant, M. L., J. M. Furman, E. Rubenstein, and E. M. Mandel. 1995. Effect of otitis media on the vestibular system in children. Ann. Otol. Rhinol. Laryngol. 104:620-624. [DOI] [PubMed] [Google Scholar]
- 14.Charo, I. F., and W. Peters. 2003. Chemokine receptor 2 (CCR2) in atherosclerosis, infectious diseases, and regulation of T-cell polarization. Microcirculation 10:259-264. [DOI] [PubMed] [Google Scholar]
- 15.Chen, R., J. H. Lim, H. Jono, X. X. Gu, Y. S. Kim, C. B. Basbaum, T. F. Murphy, and J. D. Li. 2004. Nontypeable Haemophilus influenzae lipoprotein P6 induces MUC5AC mucin transcription via TLR2-TAK1-dependent p38 MAPK-AP1 and IKKbeta-IkappaBalpha-NF-kappaB signaling pathways. Biochem. Biophys. Res. Commun. 324:1087-1094. [DOI] [PubMed] [Google Scholar]
- 16.Deleuran, M., L. Buhl, T. Ellingsen, A. Harada, C. G. Larsen, K. Matsushima, and B. Deleuran. 1996. Localization of monocyte chemotactic and activating factor (MCAF/MCP-1) in psoriasis. J. Dermatol. Sci. 13:228-236. [DOI] [PubMed] [Google Scholar]
- 17.DeMaria, T. F., R. B. Prior, B. R. Briggs, D. J. Lim, and H. G. Birck. 1984. Endotoxin in middle-ear effusions from patients with chronic otitis media with effusion. J. Clin. Microbiol. 20:15-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Engel, F., R. Blatz, J. Kellner, M. Palmer, U. Weller, and S. Bhadki. 1995. Breakdown of the round window membrane permeability barrier evoked by streptolysin O: possible etiologic role in development of sensorineural hearing loss in acute otitis media. Infect. Immun. 63:1305-1310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fietta, A. M., M. Morosini, F. Meloni, A. M. Bianco, and E. Pozzi. 2002. Pharmacological analysis of signal transduction pathways required for Mycobacterium tuberculosis-induced IL-8 and MCP-1 production in human peripheral monocytes. Cytokine 19:242-249. [PubMed] [Google Scholar]
- 20.Flory, C. M., M. L. Jones, and J. S. Warren. 1993. Pulmonary granuloma formation in the rat is partially dependent on monocyte chemoattractant protein 1. Lab. Investig. 69:396-404. [PubMed] [Google Scholar]
- 21.Foxwell, A. R., J. M. Kyd, and A. W. Cripps. 1998. Nontypeable Haemophilus influenzae: pathogenesis and prevention. Microbiol. Mol. Biol. Rev. 62:294-308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fredelius, L., and H. Rask-Andersen. 1990. The role of macrophages in the disposal of degeneration products within the organ of corti after acoustic overstimulation. Acta Otolaryngol. 109:76-82. [DOI] [PubMed] [Google Scholar]
- 23.Galdiero, M., M. Galdiero, E. Finamore, F. Rossano, M. Gambuzza, M. R. Catania, G. Teti, A. Midiri, and G. Mancuso. 2004. Haemophilus influenzae porin induces Toll-like receptor 2-mediated cytokine production in human monocytes and mouse macrophages. Infect. Immun. 72:1204-1209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gibson, B. W., W. Melaugh, N. J. Phillips, M. A. Apicella, A. A. Campagnari, and J. M. Griffiss. 1993. Investigation of the structural heterogeneity of lipooligosaccharides from pathogenic Haemophilus and Neisseria species and of R-type lipopolysaccharides from Salmonella typhimurium by electrospray mass spectrometry. J. Bacteriol. 175:2702-2712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gilsdorf, J. R. 1998. Antigenic diversity and gene polymorphisms in Haemophilus influenzae. Infect. Immun. 66:5053-5059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Golz, A., A. Netzer, B. Angel-Yeger, S. T. Westerman, L. M. Gilbert, and H. Z. Joachims. 1998. Effects of middle ear effusion on the vestibular system in children. Otolaryngol. Head Neck Surg. 119:695-699. [DOI] [PubMed] [Google Scholar]
- 27.Gu, X. X., C. M. Tsai, M. A. Apicella, and D. J. Lim. 1995. Quantitation and biological properties of released and cell-bound lipooligosaccharides from nontypeable Haemophilus influenzae. Infect. Immun. 63:4115-4120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Han, K. H., and O. Quehenberger. 2000. Ligands for peroxisome proliferator-activated receptor inhibit monocyte CCR2 expression stimulated by plasma lipoproteins. Trends Cardiovasc Med. 10:209-216. [DOI] [PubMed] [Google Scholar]
- 29.Hashimoto, S., P. Billings, J. P. Harris, G. S. Firestein, and E. M. Keithley. 2005. Innate immunity contributes to cochlear adaptive immune responses. Audiol. Neurootol. 10:35-43. [DOI] [PubMed] [Google Scholar]
- 30.Hatada, E. N., D. Krappmann, and C. Scheidereit. 2000. NF-kappaB and the innate immune response. Curr. Opin. Immunol. 12:52-58. [DOI] [PubMed] [Google Scholar]
- 31.Heguy, A., C. T. Baldari, G. Macchia, J. L. Telford, and M. Melli. 1992. Amino acids conserved in interleukin-1 receptors (IL-1Rs) and the Drosophila toll protein are essential for IL-1R signal transduction. J. Biol. Chem. 267:2605-2609. [PubMed] [Google Scholar]
- 32.Hellstrom, S., P. O. Eriksson, Y. J. Yoon, and U. Johansson. 1997. Interactions between the middle ear and the inner ear: bacterial products. Ann. N. Y. Acad. Sci. 830:110-119. [DOI] [PubMed] [Google Scholar]
- 33.Herschman, H. R. 1991. Primary response genes induced by growth factors and tumor promoters. Annu. Rev. Biochem. 60:281-319. [DOI] [PubMed] [Google Scholar]
- 34.Hirose, K., C. M. Discolo, J. R. Keasler, and R. Ransohoff. 2005. Mononuclear phagocytes migrate into the murine cochlea after acoustic trauma. J. Comp. Neurol. 489:180-194. [DOI] [PubMed] [Google Scholar]
- 35.Hirschfeld, M., J. J. Weis, V. Toshchakov, C. A. Salkowski, M. J. Cody, D. C. Ward, N. Qureshi, S. M. Michalek, and S. N. Vogel. 2001. Signaling by Toll-like receptor 2 and 4 agonists results in differential gene expression in murine macrophages. Infect. Immun. 69:1477-1482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hunter, L. L., R. H. Margolis, J. R. Rykken, C. T. Le, K. A. Daly, and G. S. Giebink. 1996. High frequency hearing loss associated with otitis media. Ear Hear. 17:1-11. [DOI] [PubMed] [Google Scholar]
- 37.Jokay, I., Z. Papp, G. Soos, I. Sziklai, and B. Dezso. 2001. The effect of chronic otitis media on the immunoreactivity of human inner ear. Eur. Arch. Otorhinolaryngol. 258:529-532. [DOI] [PubMed] [Google Scholar]
- 38.Jono, H., T. Shuto, H. Xu, H. Kai, D. J. Lim, J. R. Gum, Jr., Y. S. Kim, S. Yamaoka, X. H. Feng, and J. D. Li. 2002. Transforming growth factor-beta-Smad signaling pathway cooperates with NF-kappa B to mediate nontypeable Haemophilus influenzae-induced MUC2 mucin transcription. J. Biol. Chem. 277:45547-45557. [DOI] [PubMed] [Google Scholar]
- 39.Juhn, S. K., T. T. Jung, J. Lin, and C. K. Rhee. 1997. Effects of inflammatory mediators on middle ear pathology and on inner ear function. Ann. N. Y. Acad. Sci. 830:130-142. [DOI] [PubMed] [Google Scholar]
- 40.Karin, M. 1999. How NF-kappaB is activated: the role of the IkappaB kinase (IKK) complex. Oncogene 18:6867-6874. [DOI] [PubMed] [Google Scholar]
- 41.Kawai, T., O. Adachi, T. Ogawa, K. Takeda, and S. Akira. 1999. Unresponsiveness of MyD88-deficient mice to endotoxin. Immunity 11:115-122. [DOI] [PubMed] [Google Scholar]
- 42.Kawai, T., and S. Akira. 2005. Toll-like receptor downstream signaling. Arthritis Res. Ther. 7:12-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kawai, T., O. Takeuchi, T. Fujita, J. Inoue, P. F. Muhlradt, S. Sato, K. Hoshino, and S. Akira. 2001. Lipopolysaccharide stimulates the MyD88-independent pathway and results in activation of IFN-regulatory factor 3 and the expression of a subset of lipopolysaccharide-inducible genes. J. Immunol. 167:5887-5894. [DOI] [PubMed] [Google Scholar]
- 44.Kawauchi, H., T. F. DeMaria, and D. J. Lim. 1989. Endotoxin permeability through the round window. Acta Otolaryngol. Suppl. 457:100-115. [DOI] [PubMed] [Google Scholar]
- 45.Koch, A. E., S. L. Kunkel, L. A. Harlow, B. Johnson, H. L. Evanoff, G. K. Haines, M. D. Burdick, R. M. Pope, and R. M. Strieter. 1992. Enhanced production of monocyte chemoattractant protein-1 in rheumatoid arthritis. J. Clin. Investig. 90:772-779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kunkel, S. L., T. Standiford, K. Kasahara, and R. M. Strieter. 1991. Stimulus specific induction of monocyte chemotactic protein-1 (MCP-1) gene expression. Adv. Exp. Med. Biol. 305:65-71. [DOI] [PubMed] [Google Scholar]
- 47.Kutlu, B., M. I. Darville, A. K. Cardozo, and D. L. Eizirik. 2003. Molecular regulation of monocyte chemoattractant protein-1 expression in pancreatic beta-cells. Diabetes 52:348-355. [DOI] [PubMed] [Google Scholar]
- 48.Lee, H. Y., A. Andalibi, P. Webster, S. K. Moon, K. Teufert, S. H. Kang, J. D. Li, M. Nagura, T. Ganz, and D. J. Lim. 2004. Antimicrobial activity of innate immune molecules against Streptococcus pneumoniae, Moraxella catarrhalis and nontypeable Haemophilus influenzae. BMC Infect. Dis. 4:12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Li, Q., and I. M. Verma. 2002. NF-kappaB regulation in the immune system. Nat. Rev. Immunol. 2:725-734. [DOI] [PubMed] [Google Scholar]
- 50.Lim, D. J. 1986. Functional structure of the organ of Corti: a review. Hear. Res. 22:117-146. [DOI] [PubMed] [Google Scholar]
- 51.Lim, D. J. 1971. Vestibular sensory organs. A scanning electron microscopic investigation. Arch. Otolaryngol. 94:69-76. [DOI] [PubMed] [Google Scholar]
- 52.Lim, D. J., H. Kawauchi, and T. F. DeMaria. 1990. Role of middle ear endotoxin in inner ear inflammatory response and hydrops: long-term study. Ann. Otol. Rhinol. Laryngol. Suppl. 148:33-34. [DOI] [PubMed] [Google Scholar]
- 53.Livak, K. J., and T. D. Schmittgen. 2001. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-delta delta C(T)) method. Methods 25:402-408. [DOI] [PubMed] [Google Scholar]
- 54.Lloyd, C. M., M. E. Dorf, A. Proudfoot, D. J. Salant, and J. C. Gutierrez-Ramos. 1997. Role of MCP-1 and RANTES in inflammation and progression to fibrosis during murine crescentic nephritis. J. Leukoc. Biol. 62:676-680. [DOI] [PubMed] [Google Scholar]
- 55.Lopponen, H., M. Sorri, R. Pekkala, and J. Penna. 1992. Secretory otitis media and high-frequency hearing loss. Acta Otolaryngol. Suppl. 493:99-107. [PubMed] [Google Scholar]
- 56.Lord, K. A., B. Hoffman-Liebermann, and D. A. Liebermann. 1990. Nucleotide sequence and expression of a cDNA encoding MyD88, a novel myeloid differentiation primary response gene induced by IL6. Oncogene 5:1095-1097. [PubMed] [Google Scholar]
- 57.Lorenz, E., D. C. Chemotti, A. L. Jiang, and L. D. McDougal. 2005. Differential involvement of Toll-like receptors 2 and 4 in the host response to acute respiratory infections with wild-type and mutant Haemophilus influenzae strains. Infect. Immun. 73:2075-2082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.MacArthur, C. J., S. H. Hefeneider, J. B. Kempton, and D. R. Trune. 2006. C3H/HeJ mouse model for spontaneous chronic otitis media. Laryngoscope 116:1071-1079. [DOI] [PubMed] [Google Scholar]
- 59.Malek, S., D. B. Huang, T. Huxford, S. Ghosh, and G. Ghosh. 2003. X-ray crystal structure of an IkappaBbeta × NF-kappaB p65 homodimer complex. J. Biol. Chem. 278:23094-23100. [DOI] [PubMed] [Google Scholar]
- 60.Margolis, R. H., G. L. Saly, and L. L. Hunter. 2000. High-frequency hearing loss and wideband middle ear impedance in children with otitis media histories. Ear Hear. 21:206-211. [DOI] [PubMed] [Google Scholar]
- 61.Martin, T., P. M. Cardarelli, G. C. Parry, K. A. Felts, and R. R. Cobb. 1997. Cytokine induction of monocyte chemoattractant protein-1 gene expression in human endothelial cells depends on the cooperative action of NF-kappa B and AP-1. Eur. J. Immunol. 27:1091-1097. [DOI] [PubMed] [Google Scholar]
- 62.Maxson, S., and T. Yamauchi. 1996. Acute otitis media. Pediatr. Rev. 17:191-196. [DOI] [PubMed] [Google Scholar]
- 63.McGettrick, A. F. and L. A. O'Neill. 2004. The expanding family of MyD88-like adaptors in Toll-like receptor signal transduction. Mol. Immunol. 41:577-582. [DOI] [PubMed] [Google Scholar]
- 64.Medzhitov, R., and C. Janeway, Jr. 2000. Innate immunity. N. Engl. J. Med. 343:338-344. [DOI] [PubMed] [Google Scholar]
- 65.Medzhitov, R., and C. Janeway, Jr. 2000. The Toll receptor family and microbial recognition. Trends Microbiol. 8:452-456. [DOI] [PubMed] [Google Scholar]
- 66.Medzhitov, R., P. Preston-Hurlburt, and C. A. Janeway, Jr. 1997. A human homologue of the Drosophila Toll protein signals activation of adaptive immunity. Nature 388:394-397. [DOI] [PubMed] [Google Scholar]
- 67.Medzhitov, R., P. Preston-Hurlburt, E. Kopp, A. Stadlen, C. Chen, S. Ghosh, and C. A. Janeway, Jr. 1998. MyD88 is an adaptor protein in the hToll/IL-1 receptor family signaling pathways. Mol. Cell 2:253-258. [DOI] [PubMed] [Google Scholar]
- 68.Melhus, A., and A. F. Ryan. 2000. Expression of cytokine genes during pneumococcal and nontypeable Haemophilus influenzae acute otitis media in the rat. Infect. Immun. 68:4024-4031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Miyamoto, N., and L. O. Bakaletz. 1996. Selective adherence of non-typeable Haemophilus influenzae (NTHi) to mucus or epithelial cells in the chinchilla eustachian tube and middle ear. Microb. Pathog. 21:343-356. [DOI] [PubMed] [Google Scholar]
- 70.Moon, S. K., H. Y. Lee, J. D. Li, M. Nagura, S. H. Kang, Y. M. Chun, F. H. Linthicum, T. Ganz, A. Andalibi, and D. J. Lim. 2002. Activation of a Src-dependent Raf-MEK1/2-ERK signaling pathway is required for IL-1alpha-induced upregulation of beta-defensin 2 in human middle ear epithelial cells. Biochim. Biophys. Acta 1590:41-51. [DOI] [PubMed] [Google Scholar]
- 71.Moon, S. K., H. Y. Lee, H. Pan, T. Takeshita, R. Park, K. Cha, A. Andalibi, and D. J. Lim. 2006. Synergistic effect of interleukin 1 alpha on nontypeable Haemophilus influenzae-induced up-regulation of human beta-defensin 2 in middle ear epithelial cells. BMC Infect. Dis. 6:12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Moon, S. K., R. Park, H. Y. Lee, G. J. Nam, K. Cha, A. Andalibi, and D. J. Lim. 2006. Spiral ligament fibrocytes release chemokines in response to otitis media pathogens. Acta Otolaryngol. 126:564-569. [DOI] [PubMed] [Google Scholar]
- 73.Murphy, T. F. 2000. Haemophilus influenzae in chronic bronchitis. Semin. Respir. Infect. 15:41-51. [DOI] [PubMed] [Google Scholar]
- 74.Natarajan, K., S. Singh, T. R. Burke, Jr., D. Grunberger, and B. B. Aggarwal. 1996. Caffeic acid phenethyl ester is a potent and specific inhibitor of activation of nuclear transcription factor NF-kappa B. Proc. Natl. Acad. Sci. USA 93:9090-9095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Neish, A. S., A. T. Gewirtz, H. Zeng, A. N. Young, M. E. Hobert, V. Karmali, A. S. Rao, and J. L. Madara. 2000. Prokaryotic regulation of epithelial responses by inhibition of IkappaB-alpha ubiquitination. Science 289:1560-1563. [DOI] [PubMed] [Google Scholar]
- 76.O'Neill, L. A. 2006. How Toll-like receptors signal: what we know and what we don't know. Curr. Opin. Immunol. 18:3-9. [DOI] [PubMed] [Google Scholar]
- 77.Oppenheim, J. J., W. J. Murphy, O. Chertox, V. Schirrmacher, and J. M. Wang. 1997. Prospects for cytokine and chemokine biotherapy. Clin. Cancer Res. 3:2682-2686. [PubMed] [Google Scholar]
- 78.Rollins, B. J. 1991. JE/MCP-1: an early-response gene encodes a monocyte-specific cytokine. Cancer Cells 3:517-524. [PubMed] [Google Scholar]
- 79.Rovin, B. H., J. A. Dickerson, L. C. Tan, and C. A. Hebert. 1995. Activation of nuclear factor-kappa B correlates with MCP-1 expression by human mesangial cells. Kidney Int. 48:1263-1271. [DOI] [PubMed] [Google Scholar]
- 80.Schwandner, R., R. Dziarski, H. Wesche, M. Rothe, and C. J. Kirschning. 1999. Peptidoglycan- and lipoteichoic acid-induced cell activation is mediated by toll-like receptor 2. J. Biol. Chem. 274:17406-17409. [DOI] [PubMed] [Google Scholar]
- 81.Sen, R., and D. Baltimore. 1986. Multiple nuclear factors interact with the immunoglobulin enhancer sequences. Cell 46:705-716. [DOI] [PubMed] [Google Scholar]
- 82.Shuto, T., A. Imasato, H. Jono, A. Sakai, H. Xu, T. Watanabe, D. D. Rixter, H. Kai, A. Andalibi, F. Linthicum, Y. L. Guan, J. Han, A. C. Cato, D. J. Lim, S. Akira, and J. D. Li. 2002. Glucocorticoids synergistically enhance nontypeable Haemophilus influenzae-induced Toll-like receptor 2 expression via a negative cross-talk with p38 MAP kinase. J. Biol. Chem. 277:17263-17270. [DOI] [PubMed] [Google Scholar]
- 83.Shuto, T., H. Xu, B. Wang, J. Han, H. Kai, X. X. Gu, T. F. Murphy, D. J. Lim, and J. D. Li. 2001. Activation of NF-kappa B by nontypeable Hemophilus influenzae is mediated by toll-like receptor 2-TAK1-dependent NIK-IKK alpha /beta-I kappa B alpha and MKK3/6-p38 MAP kinase signaling pathways in epithelial cells. Proc. Natl. Acad. Sci. USA 98:8774-8779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Shyy, Y. J., L. L. Wickham, J. P. Hagan, H. J. Hsieh, Y. L. Hu, S. H. Telian, A. J. Valente, K. L. Sung, and S. Chien. 1993. Human monocyte colony-stimulating factor stimulates the gene expression of monocyte chemotactic protein-1 and increases the adhesion of monocytes to endothelial monolayers. J. Clin. Investig. 92:1745-1751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Singh, H., R. Sen, D. Baltimore, and P. A. Sharp. 1986. A nuclear factor that binds to a conserved sequence motif in transcriptional control elements of immunoglobulin genes. Nature 319:154-158. [DOI] [PubMed] [Google Scholar]
- 86.Smirnova, I. V., D. C. Bittel, R. Ravindra, H. Jiang, and G. K. Andrews. 2000. Zinc and cadmium can promote rapid nuclear translocation of metal response element-binding transcription factor-1. J. Biol. Chem. 275:9377-9384. [DOI] [PubMed] [Google Scholar]
- 87.Spandow, O., M. Anniko, and S. Hellstrom. 1989. Inner ear disturbances following inoculation of endotoxin into the middle ear. Acta Otolaryngol. 107:90-96. [DOI] [PubMed] [Google Scholar]
- 88.Takeuchi, O., K. Hoshino, T. Kawai, H. Sanjo, H. Takada, T. Ogawa, K. Takeda, and S. Akira. 1999. Differential roles of TLR2 and TLR4 in recognition of gram-negative and gram-positive bacterial cell wall components. Immunity 11:443-451. [DOI] [PubMed] [Google Scholar]
- 89.Ueda, A., Y. Ishigatsubo, T. Okubo, and T. Yoshimura. 1997. Transcriptional regulation of the human monocyte chemoattractant protein-1 gene. Cooperation of two NF-kappaB sites and NF-kappaB/Rel subunit specificity. J. Biol. Chem. 272:31092-31099. [DOI] [PubMed] [Google Scholar]
- 90.Wang, X., C. Moser, J. P. Louboutin, E. S. Lysenko, D. J. Weiner, J. N. Weiser, and J. M. Wilson. 2002. Toll-like receptor 4 mediates innate immune responses to Haemophilus influenzae infection in mouse lung. J. Immunol. 168:810-815. [DOI] [PubMed] [Google Scholar]
- 91.Wang, Y., G. K. Rangan, B. Goodwin, Y. C. Tay, and D. C. Harris. 2000. Lipopolysaccharide-induced MCP-1 gene expression in rat tubular epithelial cells is nuclear factor-kappaB dependent. Kidney Int. 57:2011-2022. [DOI] [PubMed] [Google Scholar]
- 92.Werts, C., R. I. Tapping, J. C. Mathison, T. H. Chuang, V. Kravchenko, I. Saint Girons, D. A. Haake, P. J. Godowski, F. Hayashi, A. Ozinsky, D. M. Underhill, C. J. Kirschning, H. Wagner, A. Aderem, P. S. Tobias, and R. J. Ulevitch. 2001. Leptospiral lipopolysaccharide activates cells through a TLR2-dependent mechanism. Nat. Immunol. 2:346-352. [DOI] [PubMed] [Google Scholar]
- 93.Wieland, C. W., S. Florquin, N. A. Maris, K. Hoebe, B. Beutler, K. Takeda, S. Akira, and T. van der Poll. 2005. The MyD88-dependent, but not the MyD88-independent, pathway of TLR4 signaling is important in clearing nontypeable Haemophilus influenzae from the mouse lung. J. Immunol. 175:6042-6049. [DOI] [PubMed] [Google Scholar]
- 94.Yamamoto, M., S. Sato, H. Hemmi, K. Hoshino, T. Kaisho, H. Sanjo, O. Takeuchi, M. Sugiyama, M. Okabe, K. Takeda, and S. Akira. 2003. Role of adaptor TRIF in the MyD88-independent toll-like receptor signaling pathway. Science 301:640-643. [DOI] [PubMed] [Google Scholar]
- 95.Yang, R. B., M. R. Mark, A. Gray, A. Huang, M. H. Xie, M. Zhang, A. Goddard, W. I. Wood, A. L. Gurney, and P. J. Godowski. 1998. Toll-like receptor-2 mediates lipopolysaccharide-induced cellular signalling. Nature 395:284-288. [DOI] [PubMed] [Google Scholar]
- 96.Yian, C., S. K. Moon, S. Jin, P. Webster, J. S. Rhim, A. Andalibi, and D. J. Lim. 2006. Characterization of rat spiral ligament cell line immortalized by Adeno12-SV40 hybrid virus. Ann. Otol. Rhinol. Laryngol. 115:930-938. [DOI] [PubMed] [Google Scholar]
- 97.Yoshida, K., I. Ichimiya, M. Suzuki, and G. Mogi. 1999. Effect of proinflammatory cytokines on cultured spiral ligament fibrocytes. Hear. Res. 137:155-159. [DOI] [PubMed] [Google Scholar]
- 98.Zeng, X. K., Y. F. Guan, D. G. Remick, and X. Wang. 2005. Signal pathways underlying homocysteine-induced production of MCP-1 and IL-8 in cultured human whole blood. Acta Pharmacol. Sin. 26:85-91. [DOI] [PubMed] [Google Scholar]