

Abstract
Immunogenic vaccines against group B Streptococcus (GBS) have been created by coupling the GBS capsular polysaccharides (CPS) to carrier proteins. The GBS beta C protein (BCP) serves as an effective carrier while inducing protective immunity against BCP-expressing strains. BCP also binds human immunoglobulin A (IgA), a characteristic that may be undesirable for use in humans. Here, we examined the immunogenicity and protective efficacy of a recombinant GBS BCP (rBCP), an rBCP modified to eliminate its IgA-binding site (rBCPΔIgA), and their corresponding GBS serotype III CPS conjugates (III-rBCP and III-rBCPΔIgA). Deletion of the IgA-binding site or conjugation to CPS did not alter antigenic BCP epitopes. Recombinant proteins and conjugates elicited specific, high-titered IgG in mice. Antisera to rBCP, rBCPΔIgA, III-rBCP, and III-rBCPΔIgA opsonized GBS strains A909 (Ia/BCP+) and H36B (Ib/BCP+) for killing by HL-60 cells; antiserum to III-rBCP and III-rBCPΔIgA also opsonized strain M781 (III/BCP−). Vaccination of female mice with either rBCP or rBCPΔIgA protected ∼40% of their pups challenged with GBS strain A909. Pups born to III-rBCP- or III-rBCPΔIgA-vaccinated dams survived at rates of 56% and 66%, respectively. Over 90% of pups born to dams that received the type III CPS conjugates survived challenge with GBS strain M781. In summary, rBCP and rBCPΔIgA proteins and the conjugates containing them were immunogenic in mice, inducing both CPS- and protein-specific functional IgG. These results suggest that the rBCPΔIgA could be used as a carrier to augment the immunogenicity of the CPS while expanding coverage to GBS strains bearing BCP.
Over the past two decades, there has been a dramatic reduction in neonatal group B streptococcal disease in the United States. In 1990, group B Streptococcus (GBS) caused an estimated 7,600 cases of serious illness and 310 deaths among infants ≤90 days old; infections among infants aged less than 7 days (i.e., early-onset disease) accounted for approximately 80% of these illnesses (37). During the 1990s, increased use of intrapartum antibiotic prophylaxis (IAP), recommended by the Centers for Disease Control and Prevention and the American College of Obstetricians and Gynecologists, led to an 80% reduction in the incidence rate of early-onset disease (between 1993 and 2005), from 1.7 to 0.3 cases per 1,000 live births (7, 8, 32, 33). Although most early-onset neonatal GBS disease can be prevented through IAP, currently available strategies have had not affected the rate of late-onset (>7 days to 90 days of birth) disease (7). In addition, IAP has had no impact on GBS disease in nonpregnant adults and the elderly, among whom GBS infection rates have increased over the past decade (7). Therefore, the best long-term solution to prevent GBS disease is the development of an effective vaccine (13) that could be administered to all adults, a strategy that would alleviate the limitations of IAP.
The risk of invasive neonatal GBS disease has been correlated with low levels of maternal antibody specific to the GBS capsular polysaccharide (CPS) (3). Investigators have long sought to develop CPS-based vaccines that could stimulate the mother's humoral immune response to bring about the passive transfer of protective immunoglobulin G (IgG) to her offspring in utero. Although human vaccine trials with uncoupled GBS CPS showed low and variable levels of CPS-specific antibodies (5), conjugate vaccines prepared with CPS covalently linked to immunogenic proteins yielded significantly improved CPS-specific antibody responses in phase 1 and 2 human clinical trials (24).
Until recently, low levels of maternal antibody to the CPS were the only documented immunological risk factor for neonatal GBS disease (3). In 2006, Larsson and colleagues reported that low levels of maternal and neonatal antibodies to GBS surface proteins alpha and Rib were associated with invasive neonatal GBS disease caused by Rib-containing strains (16), which strengthens the rationale for inclusion of one or more GBS cell surface proteins in a multivalent vaccine. To date, several GBS proteins that induce protective antibodies in animals have been described, including the alpha and beta C proteins (17, 20), Rib (17, 34), Sip (6), and C5a peptidase (9). The beta C protein (BCP) is a 130-kDa protein found on nearly all strains of GBS serotype Ib, as well as on some isolates of types Ia, II, and V, but almost never on serotype III (19). BCP purified from GBS strain H36B served as an effective carrier for the type Ia, II, and III CPS while simultaneously inducing protective immunity against BCP-containing GBS in mice (21, 22). However, a theoretical drawback of using BCP in a conjugate vaccine is that it binds the Fc portion of human IgA (1, 31) specifically through an MLKKIE-containing motif (12). The ability of the BCP to bind IgA may allow GBS to evade human immune responses that would normally be triggered by the binding of this Ig to its cognate CD89 receptor (30, 35). Although the possible consequences of using an immunogen with the ability to bind human IgA in humans are unclear, it nonetheless would be prudent to avoid this potentially adverse feature.
Here, we describe the synthesis of a recombinant GBS BCP (rBCP) and a rBCP modified to eliminate its IgA-binding site (rBCPΔIgA), expressed separately in Escherichia coli BL21. Conjugate vaccines composed of the type III CPS and either rBCP or rBCPΔIgA were immunogenic in mice, inducing both CPS- and protein-specific IgG, thus demonstrating the ability of the recombinant proteins to act as effective immunogens and carriers for the type III CPS. Moreover, the type III CPS conjugate vaccine prepared with either rBCP or rBCPΔIgA given to female mice provided protection to their newborn pups against lethal challenge with strains of GBS expressing either antigen, demonstrating increased serotype coverage compared to a conjugate prepared with a non-GBS protein carrier.
MATERIALS AND METHODS
Bacterial strains and plasmids.
GBS strains A909 (Ia/BCP+), H36B (Ib/BCP+), and M781 (III/BCP−) were obtained from the Channing Laboratory culture collection. E. coli strains One Shot TOP 10 and BL21(DE3) Star are produced by Invitrogen (Carlsbad, CA). HL-60, a human promyelocytic cell line, was purchased from the American Type Culture Collection (ATCC CCL-240). Plasmids pCR-Blunt (3.5 kb) and pTrcHis A (4.4 kb) are also produced by Invitrogen.
Cloning of GBS beta C protein: rBCP and rBCPΔIgA.
GBS strain H36B was treated with protoplast buffer (20% sucrose, 10 mM MgCl2, 10 mM Tris, 0.05% Triton X-100, and 0.5 U/μl mutanolysin, pH 7.0) at 37°C for 1 h. Supernatant recovered from the protoplast-treated sample was used directly for PCR amplification. The rBCP was constructed by direct PCR with the primer set BamHI-BCP-F/EcoRI-BCP-R (annealing temperature of 60°C), while rBCPΔIgA was constructed by independent PCRs using BamHI-BCP-F/IgA-R (annealing temperature of 50°C) and IgA-F/EcoRI-BCP-R (annealing temperature of 56°C), followed by BamHI-BCP-F/EcoRI-BCP-R amplification (Table 1) to eliminate the MLKKIE site shown to be essential for IgA binding (12). The PCR amplicon was cloned into plasmid pCR-Blunt and transformed into E. coli One Shot TOP 10. To express the rBCP, pCR-Blunt plasmids were purified, digested by BamHI/EcoRI, subcloned into pTrcHis A, and transformed into E. coli BL21(DE3) Star.
TABLE 1.
Primer sequences
| Primer | Sequence |
|---|---|
| BamHI-BCP-F | 5′-GGATCCGACGATAGTGTGAAGACTAC-3′ |
| EcoRI-BCP-R | 5′-GAATTCAAAAGAAAGGACAAAATGCG-3′ |
| IgA-F | 5′-CAAATGAAGATAAAGATTCTGATATTCGTAAACAAGCTCAACAAGC-3′ |
| IgA-R | 5′-GCTTGTTGAGCTTGTTTACGAATATCAGAATCTTTATCTTCATTTG-3′ |
Protein expression and purification.
A 1-ml overnight culture of transformed E. coli BL21(DE3) Star was used to seed 1 liter of Luria-Bertani medium (Difco, Sparks, MD) supplemented with 100 μg/ml ampicillin (Sigma, St. Louis. MO), which was incubated at 37°C with shaking at 200 rpm, and recombinant protein expression was induced at mid-exponential growth phase (optical density at 600 nm [OD600] of ∼0.5 to 0.7) by adding isopropyl-β-d-thiogalactopyranoside to a final concentration of 1 mM. After an additional 3 h of growth, cells were collected by centrifugation, and cell pellets were stored at −20°C. Histidine-tagged rBCPs were liberated by BugBuster Master Mix (Novagen, San Diego, CA) containing Protease Inhibitor Cocktail Set III (Calbiochem, San Diego, CA) and were purified by His-bind Fractogel chromatography (Novagen).
Oxidation of GBS type III CPS.
GBS type III CPS was purified from GBS strain M781 as previously described (36). To perform CPS oxidation, 15.2 mg of CPS was combined with a 2.6 μM concentration of freshly prepared sodium m-periodate (Sigma) in 1 ml of distilled water. The mixture was incubated at room temperature for 2 h in the dark, and excess periodate was consumed by the addition of one drop of ethylene glycol (Sigma). The resulting oxidized CPS was dialyzed against 6 liters of distilled water at 4°C, dried by lyophilization, and stored desiccated at 4°C. The degree of sialic acid oxidation was confirmed with use of a high-performance anion-exchange chromatography system and a pulsed amperometric detector (Dionex, Sunnyvale, CA), as described previously (28).
Conjugation of GBS type III CPS with rBCP.
Five milligrams of oxidized type III CPS was combined with 5 mg of either rBCP or rBCPΔIgA (to yield III-rBCP or III-rBCPΔIgA, respectively) in 1 ml of phosphate-buffered saline (Gibco, Grand Island, NY). Approximately 45 mg of sodium cyanoborohydride (Matreya, Pleasant Gap, PA) was added, and the mixture was incubated at room temperature in the dark for 3 days. The pH of the reaction mixture was monitored and maintained at 9.0 to 9.5 by the addition of 0.1 N NaOH. Conjugation of the oxidized CPS to the protein was confirmed by the formation of a void volume elution volume peak, as determined with use of a Superose 6 PC 3.2/30 gel filtration column (Amersham Biosciences, Piscataway, NJ). The conjugates were separated from the uncoupled components by a HiPrep 16/60 Sephacryl S-300 gel filtration column (Amersham Biosciences) with elution buffer (20 mM sodium phosphate, 0.9% NaCl, and 0.01% thimerosal, pH 7.2). Uncoupled aldehyde groups on the CPS were reduced by the addition of ∼2 mg of sodium borohydride (Sigma) at room temperature for 1 h. The conjugates were dialyzed against 6 liters of distilled water at 4°C, dried by lyophilization, and stored desiccated at 4°C.
Biochemical analysis of III-rBCP and III-rBCPΔIgA conjugates.
The carbohydrate and protein content of conjugates was determined by a modified phenol sulfuric carbohydrate assay (18) and bicinchoninic protein assay (Pierce, Rockford, IL). In the former, 50 μl of sample solution was mixed with 150 μl of concentrated sulfuric acid in a 96-well plate, and the plate was shaken at room temperature for 30 min. After the addition of 30 μl of 5% phenol, the plate was heated at 90°C for 5 min, and the OD490 was measured. The carbohydrate concentration was determined with use of a standard curve constructed with pure GBS type III CPS. The protein concentration was determined with use of a bovine serum albumin (Pierce) standard curve.
Immunoblotting.
Western blot analysis of chromatographically purified rBCP and rBCPΔIgA was performed with mouse anti-Xpress serum (Invitrogen) followed by phosphatase-labeled goat anti-mouse IgG (QED Bioscience, San Diego, CA) or with human myeloma IgA (MP Biomedicals, Solon, OH) followed by phosphatase-labeled goat anti-human IgA (α-chain specific) serum (MP Biomedicals).
ELISA.
Antibodies elicited to the rBCP, the rBCPΔIgA, and the GBS III CPS conjugate were measured by enzyme-linked immunosorbent assay (ELISA). Briefly, microtiter plates (Nalge Nunc, Rochester, NY) were coated with 100 μl of the corresponding primary antigen (0.2 μg/ml rBCP or 1.0 μg/ml III CPS-human serum albumin) in 0.1 M sodium carbonate buffer (pH 9.8) and incubated for 1 h at 37°C. After three rinses with washing buffer (10 mM Tris, 0.05% Brij 35 [Sigma], and 0.85% NaCl, pH 7.4), 150 μl of incubation buffer (10 mM sodium phosphate, 0.05% Brij 35, and 5% newborn calf serum [Gibco]) was added to each well, and plates were incubated for 1 h at 37°C. Plates were then washed and incubated with 100 μl of twofold serially diluted pooled mouse antiserum at 4°C overnight. Phosphatase-labeled goat anti-mouse IgG (diluted 1:3,000) was added after final washes, and the plate was incubated for 1 h at 37°C. ELISA titers were determined as the reciprocal of the highest dilution corresponding to an OD405 of ≥0.2 after 30 min of development with 1 mg/ml phosphatase substrate (Sigma) dissolved in substrate buffer (1 M Tris, 0.3 mM MgCl2, pH 9.8).
Competitive ELISA.
Competitive inhibition of the binding of mouse antiserum (diluted 1:10,000) to rBCP-coated microtiter plates was assessed by using rBCP, rBCPΔIgA, III-rBCP, or III-rBCPΔIgA as inhibitors at concentrations ranging from 1 to 1,000 ng/ml. Percent inhibition was calculated as [(OD405 without inhibitor − OD405 with inhibitor)/OD405 without inhibitor] × 100; the percentage was plotted against the log10 of inhibitor concentration (IC) to generate inhibition curves. The point of 50% inhibition (IC50) was calculated by linear regression analysis.
In vitro opsonophagocytosis assay.
The functional ability of vaccine-induced serum was evaluated with use of an in vitro opsonophagocytosis assay. HL-60 cells (ATCC CCL-240) were grown in RPMI 1640 medium (Gibco) with 20% fetal calf serum (Invitrogen) at 37°C with 5% CO2. Medium was changed every other day until maximum cell density reached 2 × 106 cells/ml. Cells were stimulated with 90 mM N,N-dimethylformamide (Fisher Scientific, Pittsburgh, PA) to cause granulocytic cells to differentiate, a process that was completed after 5 days. A 150-μl volume of saline-washed and differentiated HL-60 cells (∼3.0 × 106 cells) was mixed with 25 μl of phosphate-buffered saline-washed GBS cells (∼1.5 × 106 CFU), 25 μl of rabbit complement (Cedarlane, Burlington, NC), and 50 μl of test antiserum. The titer of pooled mouse antiserum used in the assay was normalized by diluting in minimum essential medium (Eagle; Cambrex, Walkersville, MD), based on the geometric mean titer of type III CPS- or rBCP-specific antibody. Pooled rabbit antiserum raised to GBS type Ia-tetanus toxoid (Ia-TT) (diluted 1:100), Ib-TT (diluted 1:100), or type III CPS-TT (III-TT; diluted 1:1,000) was used as reference serum. Viable GBS cells were enumerated by quantitative plate counts at time zero and after a 60-min incubation at 37°C with mixing. The difference in the number of GBS CFU was calculated and was expressed as the average of two determinations per antiserum.
Neonatal mouse protection.
Eight-week-old female CD-1 outbred mice (Charles River Laboratories, Wilmington, MA) were vaccinated intraperitoneally with either rBCP, rBCPΔIgA, III-rBCP, or III-rBCPΔIgA (10 μg of protein content) mixed with equal amounts of alum (1.3% alhydrogel; Brenntag Biosector, Frederikssund, Denmark) in a total volume of 0.5 ml. Booster doses with alum were administered on days 21 and 42 after the primary dose, and serum was collected before each booster dose. Control dams received III-TT conjugate (∼2 μg of CPS) or saline with alum by the same route and schedule. Mice were bred on day 56. Neonatal mouse pups (<24 h of age) were challenged with GBS at doses determined previously to be lethal for ∼90% of pups of the same age: ∼5 × 104 CFU for strain A909 and ∼8.5 × 103 CFU for strain M781. The challenge was administered intraperitoneally in a total volume of 50 μl of Todd-Hewitt broth (Difco) supplemented with 0.5% yeast extract (Difco). The number of mouse pups that survived GBS infection was assessed 48 h after challenge.
Statistics.
Fisher's exact test was used to compare the efficacy of GBS vaccines (Instat, version 3.0a; Graphpad Software, San Diego, CA) at a significance level of 0.05.
RESULTS
Confirmation of rBCP and rBCPΔIgA.
The identities of the His-tagged rBCP and rBCPΔIgA constructs were confirmed by DNA sequencing (data not shown) and Western blot analysis (Fig. 1). The expression of rBCP was examined by immunoblotting with antiserum to pHisTrc inherent Xpress epitope, while the existence of the IgA-binding site (motif MLKKIE) was examined by immunoblotting with human myeloma IgA. Both rBCP and rBCPΔIgA have the Xpress epitope, while rBCPΔIgA no longer bound human myeloma IgA, confirming the elimination of IgA binding in the rBCPΔIgA.
FIG. 1.
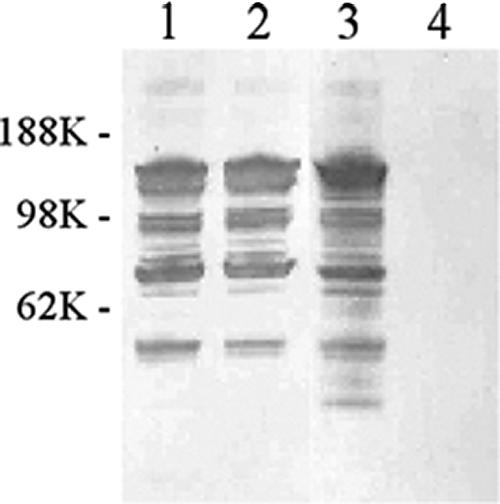
Confirmation of the deletion of the human IgA-binding site of the beta C protein. Western blotting with antiserum against the Xpress epitope (lanes 1 and 2) and human myeloma IgA (lanes 3 and 4). Lanes 1 and 3 each contain 1 μg of rBCP; lanes 2 and 4 each contain 1 μg of rBCPΔIgA.
Characteristics of conjugate vaccines.
Approximately 5% of sialic acid residues on the type III CPS were oxidized by sodium periodate, and the recovery of oxidized type III CPS was 12.4 mg (83%) of the starting material. Oxidized type III CPS was conjugated to either rBCP or rBCPΔIgA. Purified III-rBCP conjugate was composed of 34% (wt/wt) protein and 66% (wt/wt) carbohydrate, while the III-rBCPΔIgA conjugate was composed of 32% (wt/wt) protein and 72% (wt/wt) carbohydrate.
Epitope specificity of uncoupled and coupled rBCP.
An inhibition ELISA with mouse antibody to rBCP was performed to determine whether conjugation affected the antigenicity of the rBCP. Competitive binding curves generated with uncoupled and coupled rBCP were virtually identical (Fig. 2). The IC50 was 53 ng/ml for all inhibitors. The results of ELISA inhibition experiments performed with mouse anti-rBCPΔIgA sera were essentially identical to those obtained with mouse anti-rBCP, with an IC50 of 45 ng/ml for all inhibitors tested (data not shown). Therefore, the deletion of the IgA-binding site or conjugation with type III CPS did not alter the antigenic BCP epitopes.
FIG. 2.

Antigenicity of recombinant proteins and conjugates with the use of an inhibition ELISA with microtiter plates coated with rBCP. Inhibition of mouse antibody to rBCP was achieved with increasing concentrations of rBCP (filled circles), rBCPΔIgA (open circles), III-rBCP (filled triangles), and III-rBCPΔIgA (open triangles). Each point represents the mean of duplicate measurements.
Immunogenicity of rBCP, rBCPΔIgA, III-rBCP, and III-rBCPΔIgA vaccines in mice.
Outbred mice were vaccinated with rBCP, rBCPΔIgA, III-rBCP, III-rBCPΔIgA, III-TT, or saline on days 0, 21, and 42. Mice immunized with either GBS III-rBCP or III-rBCPΔIgA had a final rBCP-specific ELISA geometric mean titer (GMT) of 2,048,000, compared to 512,000 and 1,024,000 for mice immunized with rBCP and rBCPΔIgA, respectively (Table 2). As expected, III-TT and saline did not elicit antibodies to rBCP.
TABLE 2.
Antibody response in mice elicited by immunization with rBCP, rBCPΔIgA, III-rBCP, III-rBCPΔIgA, and III-TT
| Vaccine | n | GMT of rBCP-specific IgG at the indicated time pointa
|
|||
|---|---|---|---|---|---|
| Day 0 | Day 21 | Day 42 | Day 56 | ||
| rBCP | 3 | <100 | 32,000 | 512,000 | 512,000 |
| rBCPΔIgA | 3 | <100 | 32,000 | 512,000 | 1,024,000 |
| III-rBCP | 3 | <100 | 64,000 | 1,024,000 | 2,048,000 |
| III-rBCPΔIgA | 3 | <100 | 128,000 | 1,024,000 | 2,048,000 |
| III-TT | 3 | <100 | <100 | <100 | <100 |
| Saline | 2 | <100 | <100 | <100 | <100 |
Mice were immunized on days 0, 21, and 42. Blood was drawn before the immunization, and ELISA titers were determined by pooled mice sera at each time point. ELISA values are the means of duplicate determinations. n, number of mice.
Antibody specific to coupled GBS type III CPS was also measured (Table 3). The type III CPS-specific GMTs between the two conjugates prepared with rBCP or rBCPΔIgA were similar, with the higher titer elicited by the III-rBCPΔIgA conjugate. As expected, protein alone or saline did not induce type III CPS-specific antibody in mice.
TABLE 3.
Antibody response in mice elicited by immunization with rBCP, rBCPΔIgA, III-rBCP, III-rBCPΔIgA, and III-TT
| Vaccine | n | GMT of type III CPS-specific IgG at the indicated time pointa
|
|||
|---|---|---|---|---|---|
| Day 0 | Day 21 | Day 42 | Day 56 | ||
| rBCP | 3 | <100 | <100 | <100 | <100 |
| rBCPΔIgA | 3 | <100 | <100 | <100 | <100 |
| III-rBCP | 3 | <100 | 2,000 | 8,000 | 64,000 |
| III-rBCPΔIgA | 3 | <100 | 2,000 | 32,000 | 128,000 |
| III-TT | 3 | <100 | 4,000 | 16,000 | 16,000 |
| Saline | 2 | <100 | <100 | <100 | <100 |
Mice were immunized on days 0, 21, and 42. Blood was drawn before the immunization, and ELISA titers were determined by pooled mice sera at each time point. ELISA values are means of duplicate determinations. n, number of mice.
In vitro opsonophagocytosis of GBS strains by serum raised to rBCP, rBCPΔIgA, III-rBCP, and III-rBCPΔIgA vaccines.
The functional activity of mice antisera to rBCP, rBCPΔIgA, III-rBCP, III-rBCPΔIgA, III-TT, and saline was estimated against GBS strains expressing either the BCP or the type III CPS. Live GBS bacteria were incubated with differentiated HL-60 cells in the presence of baby rabbit complement and specific antisera. As shown in Fig. 3, antiserum raised to rBCP, rBCPΔIgA, III-rBCP, and III-rBCPΔIgA induced killing of GBS strains A909 (Ia/BCP+) and H36B (Ib/BCP+); antiserum raised to III-rBCP and III-rBCPΔIgA also induced killing of GBS strain M781 (III/BCP−).
FIG. 3.

In vitro opsonophagocytosis and killing of GBS. The ability of mouse antiserum to opsonize GBS for killing by differentiated HL-60 cells in the presence of complement was determined. Mouse antiserum to rBCP, rBCPΔIgA, III-rBCP, III-rBCPΔIgA, III-TT, or saline was tested. GBS type Ia strain A909, type Ib strain H36B, and type III strain M781 were the bacterial targets. Rabbit reference antiserum specific to each serotype was used as a positive control. Measurements represent the mean of duplicate determinations. ND, not done.
Efficacy of rBCP, rBCPΔIgA, III-rBCP, and III-rBCPΔIgA vaccines in neonatal mice.
Active immunization of mouse dams with rBCP or rBCPΔIgA vaccine resulted in 38% (11 of 29) and 39% (14 of 36) survival, respectively, of neonatal pups challenged with GBS strain A909 (Table 4). Of pups born to III-rBCP- and III-rBCPΔIgA-vaccinated dams, most (56% and 66%, respectively) survived challenge with strain A909, with no significant difference (P = 0.46) in efficacy between these two conjugates. Whether coupled or uncoupled, vaccines prepared with rBCP and rBCPΔIgA were significantly (P < 0.001) more efficacious against strain A909 challenge than III-TT or saline in this model, while III-rBCPΔIgA vaccine was significantly (P = 0.03) more efficacious than unconjugated rBCPΔIgA (Table 4). Most (>90%) neonatal pups born to dams that received the type III CPS conjugate vaccines survived challenge with GBS strain M781 (Table 4). The protective efficiency did not differ between pups born to dams vaccinated with III-rBCP or III-rBCPΔIgA (P = 0.49) or between III-rBCPΔIgA and III-TT (P = 1.0); both vaccines were superior to saline (P < 0.0001).
TABLE 4.
Efficacy of rBCP, rBCPΔIgA, III-rBCP, III-rBCPΔIgA, and III-TT vaccines in neonatal mouse pups born to vaccinated dams
| GBS strain for pup challenge | Dam immunization | No. of dams | No. of surviving pups/ no. of pups challenged (% survival) |
|---|---|---|---|
| A909 (Ia/BCP+) | rBCP | 3 | 11/29 (38)a,b |
| rBCPΔIgA | 3 | 14/36 (39)b,c | |
| III-rBCP | 3 | 18/32 (56)b,d | |
| III-rBCPΔIgA | 3 | 23/35 (66)b | |
| III-TT | 3 | 1/40 (3) | |
| Saline | 2 | 0/23 (0) | |
| M781 (III/BCP−) | III-rBCP | 3 | 28/28 (100)e,f |
| III-rBCPΔIgA | 3 | 30/32 (94)b,g | |
| III-TT | 3 | 27/29 (93) | |
| Saline | 2 | 0/24 (0) |
P = 1.0 compared with rBCPΔIgA.
P ≤ 0.001 compared with saline.
P = 0.03 compared with III-rBCPΔIgA.
P = 0.46 compared with III-rBCPΔIgA.
P = 0.49 compared with III-rBCPΔIgA.
P = 0.49 compared with III-TT.
P = 1.00 compared with III-TT.
DISCUSSION
Vaccines against invasive GBS disease must be safe and sufficiently immunogenic to evoke protective GBS-specific antibodies. Phase 1 and phase 2 human trials have evaluated TT-containing conjugate vaccines with five serotypes of GBS that account for an estimated 98% of GBS-caused invasive disease cases in the United States (14, 26, 27). However, because of the concerns regarding TT overuse (10), proteins including the diphtheria mutant protein cross-reactive material (CRM197) (4), a newly mutated form of diphtheria toxin (25), and the recombinant duck hepatitis B core antigen (25) have been tested as effective conjugate vaccine carriers, with all but CRM197 tested preclinically. Replacing TT with a GBS-protective protein antigen would increase coverage, an obvious goal for a multivalent vaccine.
BCP is a GBS surface protein found on nearly all strains of GBS serotype Ib, as well as on some strains of types Ia, II, and V (19). Studies of women colonized with BCP-containing GBS strains have shown that vaginal or rectal colonization elicits only low levels of BCP-specific antibodies. For example, in 16 women colonized with BCP-positive GBS, the geometric mean concentration of IgG specific for BCP was 0.76 μg/ml, not significantly different from women colonized with other GBS strains (23). Furthermore, in a separate case-control study, the low levels of BCP-specific IgG associated with colonization in mothers did not correlate with protection from invasive infection in their neonates (15). However, much higher levels of BCP-specific IgG were seen in women who developed GBS bacteremia with BCP-positive strains, demonstrating that the antigen was immunogenic in these women (23).
We have shown that vaccination of female mice with the GBS-purified BCP confers protection to their offspring against lethal challenge with BCP expression strains (20); maternal immunization of mice with GBS type III CPS-BCP conjugate showed that BCP acted as an effective carrier and as a principal immunogen (21). However, a theoretical drawback to the use of BCP as a human vaccine is its ability to bind the Fc portion of human IgA (12). Therefore, we sought to determine whether the E. coli-expressed GBS BCP (rBCP) and its variant rBCPΔIgA would still retain full immunogenicity and protective capacity.
Both rBCP and rBCPΔIgA were expressed as C-terminal degraded proteins in E. coli. Antiserum elicited by either rBCP or rBCPΔIgA showed similar relative binding affinities (as measured by ELISA inhibition) to rBCP, demonstrating that deleting the IgA-binding motif did not alter important antigenic epitopes on the protein. We observed an approximately 40% survival rate against GBS strain A909 (Ia/BCP+) challenge of neonatal mice born to dams that received rBCPs, with no significant difference in efficacy between rBCP and rBCPΔIgA vaccines. The non-IgA-binding BCP variant (rBCPΔIgA) retained full immunogenicity and protective capacity in the mouse model. However, this survival rate was lower than that in the study conducted by Madoff et al. in 1992 (20), in which nearly all neonates in the 10-μg dose group receiving BCP purified directly from GBS survived. It is possible that this difference was due to slight variations in the experimental design of the mouse protection studies or to differences between the cloned and native antigens, for example, in protein processing, folding, or the presence of the His tag.
GBS serotypes Ia, III, and V account for at least 90% of GBS disease in infants and adults in the United States, while most of the severe infections in newborns are caused by GBS serotype III (2, 11, 29). Given the protective effect of the rBCP and rBCPΔIgA against GBS strain A909, an experimental multivalent conjugate vaccine containing GBS CPS III and BCP would theoretically prevent at least one-half of the cases of neonatal and infant GBS infections. Critical to the development of any conjugate vaccine is the maintenance of protective epitopes on both the CPS and the protein components. Mouse antiserum elicited by either coupled or uncoupled rBCP/rBCPΔIgA showed similar relative binding affinities to rBCP, demonstrating that conjugation to the CPS did not alter important antigenic epitopes on the protein.
Antiserum to either III-rBCP or III-rBCPΔIgA conjugate vaccine mediated complement-dependent killing of GBS strain A909 (Ia/BCP+), H36B (Ib/BCP+), and M781 (III/BCP−) by differentiated HL-60 in an opsonophagocytic assay. There is variability among GBS BCP+ strains in the extent of killing induced by the beta-specific antiserum, which may be attributable to factors including capsule size, antigen copy number, or other unmeasured differences between GBS strains (20). The efficacy of the III-rBCP or of the III-rBCPΔIgA vaccines against infection with GBS type III strain M781 did not differ significantly from that obtained with III-TT. This suggests that the rBCP and rBCPΔIgA are strong T-cell-dependent antigens with the capacity to provide help to the coupled CPS moiety.
The protection afforded by III-rBCP or III-rBCPΔIgA vaccines against GBS strain A909 (Ia/BCP+) was superior to either rBCP or rBCPΔIgA vaccines alone and correlated directly to maternal IgG titer and to the functional activity of the maternal serum. CPS-protein conjugate vaccine elicited higher-titered antibodies of greater functional activity than those elicited by the proteins alone, perhaps due to stabilization and/or exposure of immunogenic epitopes on the protein following conjugation, antigen presentation, or a depot effect resulting from the increased mass of the conjugate.
The III-rBCPΔIgA vaccine protected 66% of neonatal mice from GBS strain A909 (Ia/BCP+) and 94% of neonatal mice from GBS strain M781 (III/BCP−), while no significant difference in efficacy was observed between III-rBCP and III-rBCPΔIgA. These findings are consistent with and extend those from prior reports (21), which suggested a 76% protection rate against GBS strain A909 and 93% against GBS strain M781 using GBS-purified BCP in a GBS III-BCP conjugate vaccine.
In summary, rBCPΔIgA is an effective carrier for GBS type III CPS and also improved coverage against infection with GBS strains that express BCP. We conclude that III-rBCPΔIgA may be a useful alternative to III-TT vaccine. It is desirable to develop and to test GBS proteins of known protective efficacy as carrier proteins to cover as many GBS serotypes as possible in one universal vaccine formulation.
Acknowledgments
We thank Francesco Berti (Novartis Vaccines and Diagnostics, Siena, Italy) for measuring the degree of CPS oxidation.
This work was supported by PHS grants AI-060603 and AI-38424 from the National Institute of Allergy and Infectious Diseases.
Editor: A. Camilli
Footnotes
Published ahead of print on 30 April 2007.
REFERENCES
- 1.Areschoug, T., M. Stalhammar-Carlemalm, I. Karlsson, and G. Lindahl. 2002. Streptococcal β protein has separate binding sites for human factor H and IgA-Fc. J. Biol. Chem. 277:12642-12648. [DOI] [PubMed] [Google Scholar]
- 2.Baker, C. J., and M. S. Edwards. 2003. Group B streptococcal conjugate vaccines. Arch. Dis. Child. 88:375-378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Baker, C. J., and D. L. Kasper. 1976. Correlation of maternal antibody deficiency with susceptibility to neonatal group B streptococcal infection. N. Engl. J. Med. 294:753-756. [DOI] [PubMed] [Google Scholar]
- 4.Baker, C. J., L. C. Paoletti, M. A. Rench, H.-K. Guttormsen, M. S. Edwards, and D. L. Kasper. 2004. Immune response of healthy women to 2 different group B streptococcal type V capsular polysaccharide-protein conjugate vaccines. J. Infect. Dis. 189:1103-1112. [DOI] [PubMed] [Google Scholar]
- 5.Baker, C. J., M. A. Rench, M. S. Edwards, R. J. Carpenter, B. M. Hays, and D. L. Kasper. 1988. Immunization of pregnant women with a polysaccharide vaccine of group B Streptococcus. N. Engl. J. Med. 319:1180-1185. [DOI] [PubMed] [Google Scholar]
- 6.Brodeur, B. R., M. Boyer, I. Charlebois, J. Hamel, F. Couture, C. R. Rioux, and D. Martin. 2000. Identification of group B streptococcal Sip protein, which elicits cross-protective immunity. Infect. Immun. 68:5610-5618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Centers for Disease Control and Prevention. 2006. Active Bacterial Core Surveillance report. Group B Streptococcus 2005. Division of Bacterial and Mycotic Diseases, Centers for Disease Control and Prevention, Atlanta, GA. http://www.cdc.gov/ncidod/dbmd/abcs/survreports.htm.
- 8.Centers for Disease Control and Prevention. 2004. Diminishing racial disparities in early-onset neonatal group B streptococcal disease—United States, 2000-2003. Morb. Mortal. Wkly. Rep. 53:502-505. [PubMed] [Google Scholar]
- 9.Cheng, Q., B. Carlson, S. Pillai, R. Eby, L. Edwards, S. B. Olmsted, and P. Cleary. 2001. Antibody against surface-bound C5a peptidase is opsonic and initiates macrophage killing of group B streptococci. Infect. Immun. 69:2302-2308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dagan, R., J. Eskola, C. Leclerc, and O. Leroy. 1998. Reduced response to multiple vaccines sharing common protein epitopes that are administered simultaneously to infants. Infect. Immun. 66:2093-2098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Harrison, L. H., J. A. Elliott, and D. M. Dwyer. 1998. Serotype distribution of invasive group B streptococcal isolates in Maryland: implications for vaccine formulation. J. Infect. Dis. 177:998-1002. [DOI] [PubMed] [Google Scholar]
- 12.Jerlstrom, P., S. Talay, P. Valentin-Weigand, K. Timmis, and G. Chhatwal. 1996. Identification of an immunoglobulin A binding motif located in the beta-antigen of the c protein complex of group B streptococci. Infect. Immun. 64:2787-2793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Johri, A. K., L. C. Paoletti, P. Glaser, M. Dua, P. K. Sharma, G. Grandi, and R. Rappuoli. 2006. Group B Streptococcus: global incidence and vaccine development. Nat. Rev. Microbiol. 4:932-942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kasper, D. L., L. C. Paoletti, M. R. Wessels, H. K. Guttormsen, V. J. Carey, H. J. Jennings, and C. J. Baker. 1996. Immune response to type III group B streptococcal polysaccharide-tetanus toxoid conjugate vaccine. J. Clin. Investig. 98:2308-2314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lachenauer, C. S., C. J. Baker, M. J. Baron, D. L. Kasper, C. Gravekamp, and L. C. Madoff. 2002. Quantitative determination of immunoglobulin G specific for group B streptococcal β C protein in human maternal serum. J. Infect. Dis. 185:368-374. [DOI] [PubMed] [Google Scholar]
- 16.Larsson, C., M. Lindroth, P. Nordin, M. Stalhammar-Carlemalm, G. Lindahl, and I. Krantz. 2006. Association between low concentrations of antibodies to protein alpha and Rib and invasive neonatal group B streptococcal infection. Arch. Dis. Child. Fetal Neonatal Ed. 91:F403-F408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Larsson, C., M. Stalhammar-Carlemalm, and G. Lindahl. 1996. Experimental vaccination against group B streptococcus, an encapsulated bacterium, with highly purified preparations of cell surface proteins Rib and alpha. Infect. Immun. 64:3518-3523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lee, C. H., and C. E. Frasch. 2001. Quantification of bacterial polysaccharides by the purpald assay: measurement of periodate-generated formaldehyde from glycol in the repeating unit. Anal. Biochem. 296:73-82. [DOI] [PubMed] [Google Scholar]
- 19.Lindahl, G., M. Stalhammar-Carlemalm, and T. Areschoug. 2005. Surface proteins of Streptococcus agalactiae and related proteins in other bacterial pathogens. Clin. Microbiol. Rev. 18:102-127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Madoff, L. C., J. L. Michel, E. W. Gong, A. K. Rodewald, and D. L. Kasper. 1992. Protection of neonatal mice from group B streptococcal infection by maternal immunization with beta C protein. Infect. Immun. 60:4989-4994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Madoff, L. C., L. C. Paoletti, J. Y. Tai, and D. L. Kasper. 1994. Maternal immunization of mice with group B streptococcal type III polysaccharide-beta C protein conjugate elicits protective antibody to multiple serotypes. J. Clin. Investig. 94:286-292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Michon, F., P. C. Fusco, A. J. D'Ambra, M. Laude-Sharp, K. Long-Rowe, M. S. Blake, and J. Y. Tai. 1997. Combination conjugate vaccines against multiple serotypes of group B streptococci, p. 847-850. In T. Horaud, A. Bouvet, R. Leclercq, H. de Montclos, and M. Sicard (ed.), Streptococci and the host. Plenum Press, New York, NY. [DOI] [PubMed]
- 23.Pannaraj, P. S., J. K. Kelly, L. C. Madoff, M. A. Rench, C. S. Lachenauer, M. S. Edwards, and C. J. Baker. 2007. Group B Streptococcus bacteremia elicits beta C protein-specific IgM and IgG in humans. J. Infect. Dis. 195:353-356. [DOI] [PubMed] [Google Scholar]
- 24.Paoletti, L. C., and D. L. Kasper. 2003. Glycoconjugate vaccines to prevent group B streptococcal infections. Expert Opin. Biol. Ther. 3:975-984. [DOI] [PubMed] [Google Scholar]
- 25.Paoletti, L. C., D. L. Peterson, R. Legmann, and R. J. Collier. 2001. Preclinical evaluation of group B streptococcal polysaccharide conjugate vaccines prepared with a modified diphtheria toxin and a recombinant duck hepatitis B core antigen. Vaccine 20:370-376. [DOI] [PubMed] [Google Scholar]
- 26.Paoletti, L. C., J. Pinel, K. D. Johnson, B. Reinap, R. A. Ross, and D. L. Kasper. 1999. Synthesis and preclinical evaluation of glycoconjugate vaccines against group B Streptococcus types VI and VIII. J. Infect. Dis. 180:892-895. [DOI] [PubMed] [Google Scholar]
- 27.Paoletti, L. C., M. A. Rench, and D. L. Kasper. 2001. Effects of alum adjuvant or a booster dose on immunogenicity during clinical trials of group B streptococcal type III conjugate vaccines. Infect. Immun. 69:6696-6701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Paoletti, L. C., R. A. Ross, and K. D. Johnson. 1996. Cell growth rate regulates expression of group B Streptococcus type III capsular polysaccharide. Infect. Immun. 64:1220-1226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Persson, E., S. Berg, B. Trollfors, P. Larsson, E. Ek, E. Backhaus, B. E. B. Claesson, L. Jonsson, G. Radberg, T. Ripa, and S. Johansson. 2004. Serotypes and clinical manifestations of invasive group B streptococcal infections in western Sweden, 1998-2001. Clin. Microbiol. Infect. 10:791-796. [DOI] [PubMed] [Google Scholar]
- 30.Pleass, R. J., T. Areschoug, G. Lindahl, and J. M. Woof. 2001. Streptococcal IgA-binding proteins bind in the Ca2-Ca3 interdomain region and inhibit binding of IgA to human CD89. J. Biol. Chem. 276:8197-8204. [DOI] [PubMed] [Google Scholar]
- 31.Russell-Jones, G., E. Gotschlich, and M. Blake. 1984. A surface receptor specific for human IgA on group B streptococci possessing the Ibc protein antigen. J. Exp. Med. 160:1467-1475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Schrag, S. J., S. Zywicki, M. M. Farley, A. L. Reingold, L. H. Harrison, L. B. Lefkowitz, J. L. Hadler, R. Danila, Cieslak, P. R., and A. Schuchat. 2000. Group B streptococcal disease in the era of intrapartum antibiotic prophylaxis. N. Engl. J. Med. 342:15-20. [DOI] [PubMed] [Google Scholar]
- 33.Schrag, S., R. Gorwitz, K. Fultz-Butts, and A. Schuchat. 2002. Prevention of perinatal group B streptococcal disease. Revised guidelines from CDC. Morb. Mortal. Wkly. Recomm. Rep. 51(RR-11):1-22. [PubMed] [Google Scholar]
- 34.Stalhammar-Carlemalm, M., L. Stenberg, and G. Lindahl. 1993. Protein Rib: a novel group B streptococcal cell surface protein that confers protective immunity and is expressed by most strains causing invasive infections. J. Exp. Med. 177:1593-1603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.van Egmond, M., E. van Garderen, A. B. van Spriel, C. A. Damen, E. S. van Amersfoort, G. van Zandbergen, J. van Hattum, J. Kuiper, and J. G. van de Winkel. 2000. FcalphaRI-positive liver Kupffer cells: reappraisal of the function of immunoglobulin A in immunity. Nat. Med. 6:680-685. [DOI] [PubMed] [Google Scholar]
- 36.Wessels, M., L. Paoletti, D. Kasper, J. DiFabio, F. Michon, K. Holme, and H. Jennings. 1990. Immunogenicity in animals of a polysaccharide-protein conjugate vaccine against type III group B Streptococcus. J. Clin. Investig. 86:1428-1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zanqwill, K. M., A. Schuchat, and J. D. Wenger. 1992. Group B streptococcal disease in the United States, 1990: report from a multistate active surveillance system. Morb. Mortal. Wkly. Rep. Surveill. Summ. 41:25-32. [PubMed] [Google Scholar]