

Abstract
The first tissue culture isolates of the unique Anaplasma phagocytophilum strain, Ap-Variant 1, were obtained in the Ixodes scapularis tick-derived cell line ISE6. Two isolates were from goat blood samples: one from a goat infected with I. scapularis ticks from Rhode Island and a second from a goat infected by serial passage of blood from the first infected goat. Eight isolates were made directly from I. scapularis ticks collected from white-tailed deer in Minnesota and represent the first isolations of an Anaplasma species directly from ticks. Each of the 10 isolates had a 16S rRNA gene sequence identical to that previously described for Ap-Variant 1, but differences within the ank gene were found that suggest natural variation. Prevalence of Anaplasma in the Minnesota ticks was 63.9%; 23 of 36 ticks tested by PCR were positive. Six of the tick-derived isolates were obtained from a set of 18 PCR-positive ticks, for a 33.3% isolation success rate. The conservation of host tropism among the Rhode Island and Minnesota isolates of Ap-Variant 1 was examined by use of experimental infections of mice and a goat. A Minnesota tick-derived isolate (MN-61-2) was used to inoculate naïve animals, and this isolate was able to infect a goat but unable to infect each of five mice, confirming that the Minnesota isolates have the same host tropism as Ap-Variant 1 from the northeastern United States. Light and electron microscopy of the Ap-Variant 1 isolate MN-61-2 in ISE6 cells showed cytoplasmic inclusions characteristic of A. phagocytophilum with pleomorphic bacteria in membrane-bound vacuoles and both electron-dense and electron-lucent forms.
Anaplasma phagocytophilum is an obligate intracellular bacterium and the agent of human granulocytic anaplasmosis (HGA). There has been an average of 454 HGA infections per year in the United States between the years 2000 and 2005, with a high of 700 cases reported to the CDC in 2005. The species A. phagocytophilum represents a consolidation of the agents previously referred to as Ehrlichia equi, Ehrlichia phagocytophila, and the agent of human granulocytic ehrlichiosis (7). While all strains of A. phagocytophilum are closely related based on 16S rRNA and groESL sequence data, at least one strain has recently been shown to exhibit unique host tropism (21). This variant strain, designated Ap-Variant 1, has not been associated with human disease and can be distinguished from strains that cause human disease (referred to as Ap-ha) by a 2-bp difference in the 16S rRNA gene (18, 22). The Ap-Variant 1 strain appears to be widespread in nature as the characteristic 16S rRNA gene sequence has previously been PCR amplified from white-tailed deer (WTD) in Maryland, Wisconsin, and Pennsylvania and from Ixodes scapularis ticks in Rhode Island, Connecticut, and Pennsylvania (2, 4, 18, 22). In contrast to Ap-ha strains, for which the white-footed mouse (Peromyscus leucopus) serves as a major reservoir in nature, Ap-Variant 1 has never been detected in this rodent species. In fact, experimental studies have shown that Ap-Variant 1 will not infect the white-footed mouse, inbred laboratory-reared DBA/2 mice, or immunocompromised SCID mice, each of which has been shown to be susceptible to infection by Ap-ha strains (21). While recent studies have shown that WTD are a likely natural reservoir for Ap-Variant 1, to date no studies have shown evidence of Ap-ha in the native deer population (17) even though WTD may be infected experimentally with Ap-ha (33). I. scapularis ticks are the vectors for both AP-Variant 1 and AP-ha (4, 18, 26, 34); however, dually infected ticks are rare in nature, suggesting that ticks may not be able to maintain a simultaneous infection with both agents (18).
While numerous strains of AP-ha have been isolated in human promyelocytic (HL-60) and other cell culture lines, no isolates of AP-Variant 1 exist (9, 11). This lack of a tissue culture isolate has hampered characterization of the Ap-Variant 1 strain. The goal of this study was to obtain an isolate of Ap-Variant 1 in vitro using either the human promyelocytic cell line, HL-60, or the I. scapularis tick-derived ISE6 cell line.
MATERIALS AND METHODS
Tick collection and preparation.
Engorged and partially engorged female I. scapularis ticks were removed from deer harvested by bow hunters on 16 to 25 October 2003 at Camp Ripley, MN (46°5′42′′ to 46°20′6′′N and 94°20′33′′ to 94°30′0′′W). Ticks were surface disinfected within 1 week after collection, and tissues were dissected for organ cultures and DNA extraction. Briefly, ticks were submersed in bleach (1%), followed by soaking in benzalkonium chloride (0.5%) and 70% ethanol, and rinsed in sterile H20(14).
In vitro isolation of anaplasma in tick cell culture.
Organs (gut, salivary glands, and ovaries) were cultured in wells of a 96-well plate containing I. scapularis ISE6 cells (23). One to 2 weeks after initiation, the organ cultures from each tick along with the ISE6 cells were transferred to 24-well plates previously seeded with ISE6 cells. During the third week the tissues from each tick and accompanying ISE6 cells were transferred to 12.5-cm2 culture flasks.
EDTA-treated blood samples were collected aseptically from each of two goats at a time when they were PCR positive for the Ap-Variant 1 strain of A. phagocytophilum (17). One goat had been infected by being fed upon by field-collected I. scapularis ticks from Trustom, RI, and the second goat was infected by the transfusion of blood from the first goat. Goat blood (0.2 ml) was placed into a 25-cm2 culture flask that was approximately 75% confluent with ISE6 cells. The culture medium for both tick and goat blood isolates was L15B300 with fetal bovine serum (5%; Harlan, Indianapolis, IN), tryptose phosphate broth (5%; Difco, Detroit, MI), lipoprotein concentrate (0.1%; MP Biomedical, Irvine, CA), 0.25% NaHCO3, and 25 mM HEPES, pH 7.5 (23). Plate cultures were incubated at 34°C in candle jars as described elsewhere (24). Cultures were examined for the presence of anaplasma by use of cytocentrifuge-prepared cells fixed in methanol and stained with either Giemsa or Diff-Quik or by immunofluorescent antibody (IFA) assay. The IFA assay was initially used as a screening tool only on the isolates from the goat bloods. The assay was performed as previously described except that the primary antibody was serum from a horse experimentally infected with A. phagocytophilum (titer, 1/1,024) and the secondary antibody was an optimized dilution (1/100) of fluorescein isothiocyanate-labeled goat anti-horse conjugate specific to the heavy and light chains of immunoglobulin G (Kirkegaard and Perry Laboratories, Gaithersburg, MD) (25). All anaplasma isolates were subsequently maintained by serially transferring infected cell suspensions into fresh ISE6 cell cultures.
Maintenance and animal inoculations of isolate MN-61-2.
Isolate MN-61-2 was obtained from tick gut and ovarian tissues placed into cultured ISE6 cells as described above. Anaplasma-infected cells were first observed in a Giemsa-stained sample taken 33 days postinoculation. This isolate was propagated by transferring cells (100 μl to 250 μl) from infected cultures to fresh noninfected ISE6 cell monolayers containing 5 ml of complete anaplasma medium every 7 to 21 days (23). A culture of this isolate in its 21st passage (319 days postisolation) was inoculated into a goat and five mice by intraperitoneal injection (0.1 ml/mouse; 2.0 ml/goat). Blood samples were collected from the goat and each mouse on days 7, 14, and 21 postinoculation and evaluated for infection by use of a nested PCR assay that amplifies the A. phagocytophilum 16S rRNA gene, as previously described (22).
Transmission electron microscopy.
ISE6 cells infected with isolate MN-61-2 in the 16th passage (258 days postisolation) were gently pipetted off the flask, transferred to a microcentrifuge tube, and centrifuged at low speed (275 × g). The pellet was fixed in modified Ito's Fixative (12) on ice for 1 h and at 4°C for 72 h. Following fixation, the cells were washed three times in cacodylate buffer, postfixed in 1% (wt/vol) osmium tetroxide for 1 h at room temperature, dehydrated in an ascending ethanol series, and embedded in PolyBed 812 (Polysciences, Warrington, PA). Ultrathin sections were collected on carbon-coated collodion copper grids, stained with uranyl acetate and lead citrate, and viewed using a JEOL 1200 EX electron microscope operating at 60 kV.
DNA extraction, PCR primers, and DNA sequencing.
DNA was extracted from internal organs of ticks and from tissue culture-grown isolates using a PureGene DNA Isolation Kit (Gentra Systems, Minneapolis, MN), following the animal tissue protocol as given by the manufacturer. DNA was extracted from goat (200 μl) and mouse (50 μl) bloods using a DNeasy Tissue Kit (QIAGEN, Valencia, CA) and eluted in AE buffer (QIAGEN) in a volume equivalent to the volume of blood used for each extraction. A. phagocytophilum was initially identified in ticks by PCR amplification using the PER1 and PER2 primers as previously described (9). The isolates obtained from tick samples were amplified, sequenced, and identified as Ap-Variant 1 using primers ge3a and ge2 (22). The A. phagocytophilum strains present in the ticks and goat blood and isolated in culture were further characterized based on 16S rRNA gene sequences using a nested PCR assay as previously described (22) or a nested PCR assay that amplifies a 667-bp region of the A. phagocytophilum ank gene. The ank gene assay amplifies the 3′ end of the ankyrin repeat region and the region prior to the 27-, 17-, and 11-amino acid direct repeat regions (20). The primers for the ank gene assay were ANK-F1 (5′-GAAGAAATTACAACTCCTGAAG) and ANK-R1 (5′-CAGCCAGATGCAGTAACGTG) for primary reactions and ANK-F2 (5′-TTGACCGCTGAAGCACTAAC) and ANK-R2 (5′-ACCATTTGCTTCTTGAGGAG) for nested reactions. Thermal cycling conditions were identical to those previously described for the 16S rRNA nested assay (22). To confirm the integrity of the DNA extracted from ticks, a primer set for the rickettsial outer membrane protein A gene (ompA) was used (30). These primers amplify a 533-bp product from a rickettsial endosymbiont associated with I. scapularis ticks (37).
DNA sequencing reactions used fluorescence-labeled dideoxynucleotide technology (Dye Terminator Cycle Sequencing Ready Reaction Kit, version 3.1; Applied Biosystems, Foster City, CA). Sequencing reaction products were separated, and data were collected using an ABI 3100 automated DNA sequencer (Applied Biosystems). The sequence was fully determined for both strands of each DNA template to ensure maximum accuracy of the data. Sequences were edited and assembled using the Staden software programs and aligned using the Pileup program of the Wisconsin Sequence Analysis Package (Genetics Computer Group, Madison, WI) (6). PAUP (version 4.0.0d64) was used for phylogenetic analysis of the optimally aligned protein sequences (32). A heuristic search was done using parsimony as the optimality criterion to create an unrooted tree.
Nucleotide sequence accession numbers.
The partial ank gene sequences for the Ap-Variant 1 strain isolates were deposited in the GenBank database under accession numbers DQ320648 (isolate RI-1), DQ320649 (RI-2), DQ320650 (MN-29-1), DQ320651 (MN-62-6), DQ320652 (MN-61-2), DQ320653 (MN-38-1), and DQ320654 (MN-53-1).
RESULTS
Tick PCR testing and isolation from ticks.
A total of 48 ticks collected from 21 deer were examined for A. phagocytophilum by PCR and/or culture isolation. Thirty-six ticks from 14 deer were tested by PCR using the 16S rRNA gene primers PER1 and PER2, and 23 of them were positive (63.9%) (Fig. 1, top). Nine of the deer (64.3%) had Anaplasma-positive ticks feeding on them. All ticks were positive in control reactions that amplified a rickettsial endosymbiont, demonstrating that there were no inhibitors in the DNA extracts (Fig. 1, bottom).
FIG. 1.

Identification of PCR products of anaplasma (top) and rickettsia (bottom) DNA in ticks removed from hunter-killed deer at Camp Ripley, MN. Primer pair PER1 and PER2 amplifies a 452-bp product from the 16S rRNA gene from anaplasma or ehrlichia DNA. Primer pair Rr190 amplifies a 533-bp product from the rickettsial outer membrane protein A gene from a rickettsial symbiont of I. scapularis. Lane 1 shows the molecular weight standard. Positive controls for the top gel included DNA extracted from Anaplasma marginale (lane A) and Ehrlichia chaffeensis (lane B). Positive control for the rickettsial symbiont was Rickettsia peacockii (lane C).
A total of 44 ticks, from all 21 deer, were used in the cultivation attempt, and isolates were made from 8 of the ticks (18.2%). We noted the presence of Anaplasma in Giemsa-stained preparations 4 to 5 weeks after initiation of cultures (Fig. 2). Six deer (28.6%) had culture-positive ticks feeding on them. The isolates were named MN-29-1, MN-35-1, MN-38-1, MN-53-1, MN-61-2, MN-62-1, MN-62-3, and MN-62-6 (based on deer and tick numbers, respectively).
FIG. 2.
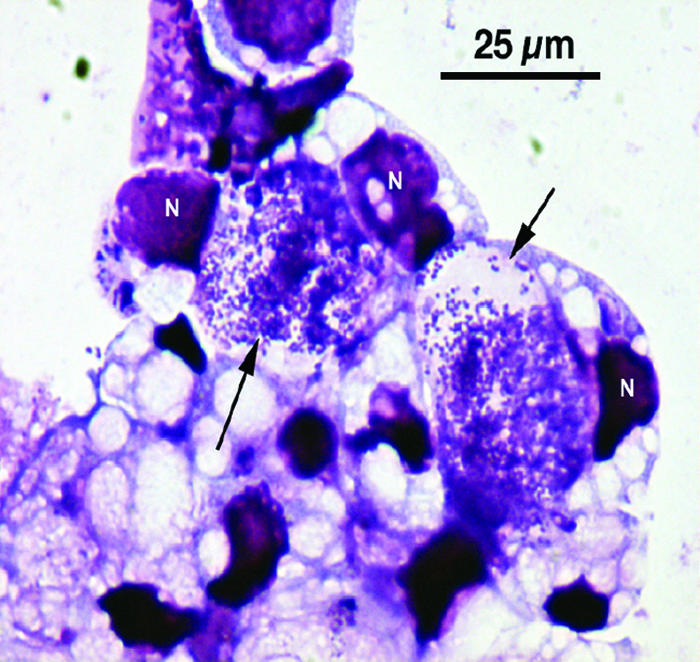
Photomicrograph of Giemsa-stained ISE6 cells infected with A. phagocytophilum strain Ap-Variant 1 isolated from a female I. scapularis tick. Arrows point to large intracellular vacuoles harboring numerous Anaplasma organisms. N, host cell nucleus.
Both PCR and culturing were done with 30 ticks from 13 deer. Eighteen (60%) were PCR positive and 6 (20%) were culture positive for Anaplasma species. These results indicate a 33.3% success rate in the culture isolation from PCR-positive ticks using the ISE6 cell line.
Isolation from goat bloods.
Two goats were infected with Ap-Variant 1 as previously described (17). EDTA-treated goat blood samples were inoculated onto I. scapularis-derived (ISE6) and human promyelocytic (HL-60) cultured cells, and the establishment of infection was monitored by light microscopy and by PCR using a nested assay that amplifies the A. phagocytophilum 16S rRNA gene (22). Whereas all attempts to infect HL-60 cells were unsuccessful, the ISE6 cells became infected and were able to maintain the infection for >10 months. An IFA assay was used as a screening tool to identify infected cells within the ISE6 cell monolayer for the isolates from goat bloods (data not shown).
Experimental infections.
The MN-61-2 isolate was used to inoculate a goat and five mice. An EDTA blood sample from each animal was drawn on days 7, 14, and 21, and the extracted DNA was tested by PCR. All mice remained PCR negative throughout the experiment. The goat became PCR positive on day 7, remained positive on day 14, and was negative on day 21, presumably because the infection was cleared. These data confirm that the MN-61-2 isolate exhibits the same host tropism as previously reported for Ap-Variant 1 from the northeastern United States.
Molecular analysis of A. phagocytophilum tissue culture isolates.
DNA sequencing of a PCR fragment amplified from the 16S rRNA gene indicated that each of the tissue culture isolates from both ticks and goat bloods were identical to the 16S rRNA sequence previously described for A. phagocytophilum strain AP-Variant 1 (2, 19). The isolates were further characterized by amplification and sequencing of a 667-bp region of the ank gene that has previously been used to demonstrate variability among North American strains of A. phagocytophilum (20). The amino acid sequence encoded by the portion of the ank gene that was sequenced showed a high degree of conservation, with ≥98% identity among Ap-Variant 1 strains obtained from both the goats and ticks, which represented two diverse geographic locations, Rhode Island and Minnesota, respectively. The sequence encoded by the ank gene for the Ap-Variant 1 strain isolates showed much higher homology to the corresponding sequences from human isolates of Ap-ha strains from the northeastern United States (≥96.5% identity) than to Ap-ha strains from the upper Midwest (88.6 to 89.6% identity). The homology between the Ap-Variant 1 isolates and the northeast Ap-ha isolates is evident in the phylogenetic analysis shown in Fig. 3.
FIG. 3.

Phylogram of the Ap-Variant 1 isolates of A. phagocytophilum based on a 201-amino-acid residue region of the Ank protein. The four identical sequences at the top of the figure are from the two goat blood isolates (RI-1 and RI-2), the sequence obtained directly by PCR from goat blood prior to isolation (Goat blood), and the sequence previously described for Ap-Variant 1 from ticks and WTD (Ap-Variant 1) (19). MN-29-1, MN-62-6, MN-61-2, MN-53-1, and MN-38-1 represent five of the Ap-Variant 1 isolates from Minnesota ticks. NY-Ap-ha and MN-Ap-ha are the Ank protein sequences previously determined for isolates of A. phagocytophilum that cause human infections in the northeastern United States (strain NY8) and the upper Midwest (strain Webster), respectively.
Electron microscopy.
Electron microscopy was used to evaluate the morphology of Ap-Variant 1 in tick cell culture (Fig. 4). Infected ISE6 cells contained cytoplasmic inclusions that varied considerably in both the number of inclusions per cell and the size of individual inclusions. Up to four inclusions could be detected within a single cell (Fig. 4C). While the smaller inclusions primarily contained the reticulate, electron-lucent forms of anaplasma (Fig. 4A), both electron-lucent and electron-dense forms could be found in most of the larger inclusions (Fig. 4C and D). The size of individual bacteria varied, and double-layered membranes could clearly be detected surrounding both electron-lucent and electron-dense forms. The outer membrane of electron-dense forms often appeared rippled and separated from the inner membrane. Infected cells also often contained cytoplasmic inclusions that lacked anaplasma and appeared to be lysosomes (Fig. 4A and B).
FIG. 4.

Ultrastructure of A. phagocytophilum Ap-Variant 1 strain MN61-2 in tick (ISE6) cell culture. (A and B) Images show reticulated bacteria within membrane-bound endosomes (large arrow) in cells that also contain putative lysosomes (asterisk) with multilammellar bodies. In panel B note the close association of the bacteria with endosomal membrane. N, nucleus; bar, 3 μm. (C) Reticulated, electron-lucent and electron-dense (arrowheads) forms within inclusions that occupy most of the host cell's cytoplasm are visible. Bar, 3 μm. (D) Higher magnification of bacteria within an endosome. Note the reticulated form with infolded membrane (small arrow). Bar, 1.5 μm.
DISCUSSION
I. scapularis ticks are the known vectors of several diseases of humans in North America including Lyme disease, babesiosis, and HGA (formerly human ganulocytic ehrlichiosis). In addition to these known disease-causing agents, I. scapularis ticks are likely to be exposed to many more bacterial, viral, and parasitic agents due to the fact that they will feed on many different animal species including rodents, birds, carnivores, lagomorphs, and reptiles (1, 8). While some of the agents these ticks are exposed to and acquire clearly infect humans, it seems likely that many more of these agents will either not be transmitted or not be infectious to humans. For example, I. scapularis ticks appear to harbor a rickettsial species as an endosymbiont that has not been shown to infect humans. Within the species A. phagocytophilum, several variant strains have been described that have never been associated with human infection, including the Ap-Variant 1 strain (19). While numerous isolates of strains that cause human disease (Ap-ha strains) have been made from infected human blood samples, no isolate of this variant strain has been available. The availability of isolates of variant strains is critical for the further study of these strains, including determination of their pathogenic potential in both humans and animals. The impact of Ap-Variant 1 strains on the epidemiology of human anaplasmosis remains unclear, but the availability of isolates of this strain will permit experimental examination of the hypothesis that Ap-Variant 1 influences the distribution of Ap-ha strains in nature (19). Isolates of Ap-ha strains have been made in several cell lines although most isolates have been obtained in HL-60 cells, a human promyelocytic cell line (9, 11). However, in the current study attempts to infect HL-60 cells with the Ap-Variant 1 strain were unsuccessful. An alternative to the HL-60 line that has been used to obtain isolates of A. phagocytophilum is the ISE6 cell line, which was derived from embryonic I. scapularis ticks (23). In the current study we obtained 10 isolates of the Ap-Variant 1 strain in ISE6 cells (2 isolates from the blood of infected goats and 8 isolates from field-collected ticks feeding on WTD). This represents the first isolates of the Ap-Variant 1 strain and also the first report of direct isolation of Anaplasma sp. from ticks in vitro.
A 2-bp difference in the 16S rRNA gene sequence serves as a marker that allows differentiation of the Ap-Variant 1 and Ap-ha strains (19, 21). While each of the 10 isolates of Ap-Variant 1 contain this 2-bp difference, the question remained as to whether these isolates represent a single strain or multiple strains that happen to share the 2-bp change in their 16S rRNA genes. The ank gene has been used previously to evaluate the relatedness of isolates of A. phagocytophilum from different geographic locations (20, 35). While we cannot answer this question completely without further analysis of the isolates, our present phylogenetic analysis using the ank gene protein coding sequence suggests a close relationship among the isolates, although some variability was noted that may represent natural genetic variation within a population. It was interesting that the Ap-Variant 1 strain isolates grouped phylogenetically with the northeastern Ap-ha strains even though many of the isolates were obtained from ticks collected in the upper Midwest, suggesting that the Ap-Variant 1 is more closely related to northeastern Ap-ha strains than to Ap-ha strains from the upper Midwest.
The prevalence (63.9%) of A. phagocytophilum found in I. scapularis ticks collected from WTD in Camp Ripley, MN, was significantly higher than any previous reports from the upper Midwest, which have ranged from 3.8% to 10.3% positive by PCR (15, 26) although the number of ticks tested in the current study was relatively low. However, a similarly high percentage of positives (49.6%) was found in I. scapularis ticks from southeastern Pennsylvania that were also collected while feeding on WTD (4). Such a high percentage of positives has never been seen when questing flat ticks have been examined. The high rate of Anaplasma-positive ticks feeding on WTD could be attributable to two factors: the acquisition of the AP-Variant 1 strain from infected deer and/or the stimulation of replication of either the AP-Variant 1 or AP-ha agent that was in the tick prior to feeding and had been acquired during a previous feeding stage. The latter has been demonstrated for numerous agents of tick-borne diseases. Tick feeding has been shown to stimulate the expression of endogenous tick genes as well as the rapid replication and gene expression of exogenous parasites such as the Lyme disease agent, Borrelia burgdorferi (5, 29). Babesia and Theileria are found in the salivary gland cells of unfed nymphal ticks as a complex cytoplasmic meshwork until such time as the tick feeds on a vertebrate host, stimulating differentiation and the development of mature infectious sporozoites, which can be found by electron microscopy only after ticks have fed for at least 48 h (3, 13, 28, 31). Reactivation and increase in the virulence of Rickettsia rickettsii upon feeding of the tick vector is well documented (10). A similar mechanism may apply to A. phagocytophilum in that the agent already present in a flat tick but at a level so low that it is undetectable is being amplified during acquisition of the blood meal to a level that can be detected by PCR assays.
The host tropism differences previously reported between the Ap-Variant 1 and Ap-ha strains correlate well with the infectivity studies presented in the current study (21). That the MN-61-2 isolate could not infect mice but could infect a goat, as had previously been shown for Ap-Variant 1 from the northeastern United States, suggests that the northeastern and upper Midwest forms of this strain are closely related (18). A. phagocytophilum DNA that has the same 16S rRNA sequence as the Ap-Variant 1 has also been amplified from various animals and Ixodes ricinus ticks in Europe (16, 27, 29, 35, 36); however, the host tropism of these strains has not been examined, and no isolates are available. Therefore, it is impossible to determine the relatedness of the North American Ap-Variant 1 strains to potential Ap-Variant 1 strains from Europe.
While the Ap-ha strain causes human disease and readily infects the human tissue culture cell line HL-60 (9), the Ap-Variant 1 strain could not infect HL-60 cells, and this strain has never been associated with a human infection. However, both strains are transmitted by I. scapularis ticks, and both are able to infect the I. scapularis-derived ISE6 cell line (4, 17, 19, 23, 26). In fact, the morphology of the Ap-Variant 1-infected ISE6 cells appears nearly identical to ISE6 cells infected by Ap-ha, with large cytoplasmic inclusions containing both electron-dense and electron-lucent organisms evident on electron micrographs. This suggests that the strains share the genetic components that allow them to function in the tick environment but differ in the components required for infection and survival in mammalian tissues. Now that isolates of Ap-Variant 1 are available, studies can be designed to address the genetic, proteomic, and biological differences that make these two strains unique and result in the Ap-Variant 1 strain's being noninfectious in murine species and humans (18, 19, 21).
Acknowledgments
We are grateful to Kimetha Slater, Edward Shaw, and Virginia Bain for excellent technical assistance, and Herbert Thompson and Gregory Dasch for review of the manuscript and useful suggestions. We thank the Biotechnology Core Facility of the National Center for Infectious Diseases Scientific Resources Program for the synthesis of reagents used in this study and Thomas Mather (University of Rhode Island) for providing the field-collected ticks from Rhode Island.
The work reported herein was partially supported by a grant to U.G.M. from the National Institutes of Health (grant 5R01-AI042792).
The findings and conclusions in this report are those of the authors and do not necessarily represent the views of the CDC or the Department of Health and Human Services.
Footnotes
Published ahead of print on 2 May 2007.
REFERENCES
- 1.Anderson, J. F. 1988. Mammalian and avian reservoirs for Borrelia burgdorferi. Ann. N. Y. Acad. Sci. 539:180-191. [DOI] [PubMed] [Google Scholar]
- 2.Belongia, E. A., K. D. Reed, P. D. Mitchell, C. P. Kolbert, D. H. Persing, J. S. Gill, and J. J. Kazmierczak. 1997. Prevalence of granulocytic Ehrlichia infection among white-tailed deer in Wisconsin. J. Clin. Microbiol. 35:1465-1468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Binnington, K. C., and D. H. Kemp. 1980. Role of tick salivary glands in feeding and disease transmission. Adv. Parasitol. 18:315-339. [DOI] [PubMed] [Google Scholar]
- 4.Courtney, J. W., R. L. Dryden, J. Montgomery, B. S. Schneider, G. Smith, and R. F. Massung. 2003. Molecular characterization of Anaplasma phagocytophilum and Borrelia burgdorferi in Ixodes scapularis ticks from Pennsylvania. J. Clin. Microbiol. 41:1569-1573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.De Silva, A. M., and E. Fikrig. 1995. Growth and migration of Borrelia burgdorferi in Ixodes ticks during blood feeding. Am. J. Trop. Med. Hyg. 53:397-404. [DOI] [PubMed] [Google Scholar]
- 6.Devereux, J., P. Haeberli, and O. Smithies. 1984. A comprehensive set of sequence analysis programs for the VAX. Nucleic Acids Res. 12:387-395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dumler, J. S., A. F. Barbet, C. P. Bekker, G. A. Dasch, G. H. Palmer, S. C. Ray, Y. Rikihisa, and F. R. Rurangirwa. 2001. Reorganization of genera in the families Rickettsiaceae and Anaplasmataceae in the order Rickettsiales: unification of some species of Ehrlichia with Anaplasma, Cowdria with Ehrlichia and Ehrlichia with Neorickettsia, descriptions of six new species combinations and designation of Ehrlichia equi and “HGE agent” as subjective synonyms of Ehrlichia phagocytophila. Int. J. Syst. Evol. Microbiol. 51:2145-2165. [DOI] [PubMed] [Google Scholar]
- 8.Durden, L. A., J. H. Oliver, Jr., C. W. Banks, and G. N. Vogel. 2002. Parasitism of lizards by immature stages of the blacklegged tick, Ixodes scapularis (Acari, Ixodidae). Exp. Appl. Acarol. 26:257-266. [DOI] [PubMed] [Google Scholar]
- 9.Goodman, J. L., C. Nelson, B. Vitale, J. E. Madigan, J. S. Dumler, T. J. Kurtti, and U. G. Munderloh. 1996. Direct cultivation of the causative agent of human granulocytic ehrlichiosis. N. Engl. J. Med. 334:209-215. [DOI] [PubMed] [Google Scholar]
- 10.Hayes, S. F., and W. Burgdorfer. 1982. Reactivation of Rickettsia rickettsii in Dermacentor andersoni ticks: an ultrastructural analysis. Infect. Immun. 37:779-785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Horowitz, H. W., M. E. Aguero-Rosenfeld, D. F. McKenna, D. Holmgren, T. C. Hsieh, S. A. Varde, S. J. Dumler, J. M. Wu, I. Schwartz, Y. Rikihisa, and G. P. Wormser. 1998. Clinical and laboratory spectrum of culture-proven human granulocytic ehrlichiosis: comparison with culture-negative cases. Clin. Infect. Dis. 27:1314-1317. [DOI] [PubMed] [Google Scholar]
- 12.Ito, S., J. W. Vinson, and T. J. McGuire, Jr. 1975. Murine typhus rickettsiae in the Oriental rat flea. Ann. N. Y. Acad. Sci. 266:35-60. [DOI] [PubMed] [Google Scholar]
- 13.Karakashian, S. J., M. A. Rudzinska, A. Spielman, S. Lewengrub, J. Piesman, and N. Shoukrey. 1983. Ultrastructural studies on sporogony of Babesia microti in salivary gland cells of the tick Ixodes dammini. Cell Tissue Res. 231:275-287. [DOI] [PubMed] [Google Scholar]
- 14.Kurtti, T. J., U. G. Munderloh, C. A. Hughes, S. M. Engstrom, and R. C. Johnson. 1996. Resistance to tick-borne spirochete challenge induced by Borrelia burgdorferi strains that differ in expression of outer surface proteins. Infect. Immun. 64:4148-4153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Layfield, D., and P. Guilfoile. 2002. The prevalence of Borrelia burgdorferi (Spirochaetales:Spirochaetaceae) and the agent of human granulocytic ehrlichiosis (Rickettsiaceae:Ehrlichiae) in Ixodes scapularis (Acari:Ixodidae) collected during 1998 and 1999 from Minnesota. J. Med. Entomol. 39:218-220. [DOI] [PubMed] [Google Scholar]
- 16.Liz, J. S., J. W. Sumner, K. Pfister, and M. Brossard. 2002. PCR detection and serological evidence of granulocytic ehrlichial infection in roe deer (Capreolus capreolus) and chamois (Rupicapra rupicapra). J. Clin. Microbiol. 40:892-897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Massung, R. F., J. W. Courtney, S. L. Hiratzka, V. E. Pitzer, G. Smith, and R. L. Dryden. 2005. Anaplasma phagocytophilum in white-tailed deer. Emerg. Infect. Dis. 11:1604-1606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Massung, R. F., T. N. Mather, and M. L. Levin. 2006. Reservoir competency of goats for the Ap-Variant 1 strain of Anaplasma phagocytophilum. Infect. Immun. 74:1373-1375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Massung, R. F., M. J. Mauel, J. H. Owens, N. Allan, J. W. Courtney, K. C. Stafford, III, and T. N. Mather. 2002. Genetic variants of Ehrlichia phagocytophila, Rhode Island and Connecticut. Emerg. Infect. Dis. 8:467-472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Massung, R. F., J. H. Owens, D. Ross, K. D. Reed, M. Petrovec, A. Bjoersdorff, R. T. Coughlin, G. A. Beltz, and C. I. Murphy. 2000. Sequence analysis of the ank gene of granulocytic ehrlichiae. J. Clin. Microbiol. 38:2917-2922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Massung, R. F., R. A. Priestley, N. J. Miller, T. N. Mather, and M. L. Levin. 2003. Inability of a variant strain of Anaplasma phagocytophilum to infect mice. J. Infect. Dis. 188:1757-1763. [DOI] [PubMed] [Google Scholar]
- 22.Massung, R. F., K. Slater, J. H. Owens, W. L. Nicholson, T. N. Mather, V. B. Solberg, and J. G. Olson. 1998. Nested PCR assay for detection of granulocytic ehrlichiae. J. Clin. Microbiol. 36:1090-1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Munderloh, U. G., S. D. Jauron, V. Fingerle, L. Leitritz, S. F. Hayes, J. M. Hautman, C. M. Nelson, B. W. Huberty, T. J. Kurtti, G. G. Ahlstrand, B. Greig, M. A. Mellencamp, and J. L. Goodman. 1999. Invasion and intracellular development of the human granulocytic ehrlichiosis agent in tick cell culture. J. Clin. Microbiol. 37:2518-2524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Munderloh, U. G., J. E. Madigan, J. S. Dumler, J. L. Goodman, S. F. Hayes, J. E. Barlough, C. M. Nelson, and T. J. Kurtti. 1996. Isolation of the equine granulocytic ehrlichiosis agent, Ehrlichia equi, in tick cell culture. J. Clin. Microbiol. 34:664-670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nicholson, W. L., J. A. Comer, J. W. Sumner, C. Gingrich-Baker, R. T. Coughlin, L. A. Magnarelli, J. G. Olson, and J. E. Childs. 1997. An indirect immunofluorescence assay using a cell culture-derived antigen for detection of antibodies to the agent of human granulocytic ehrlichiosis. J. Clin. Microbiol. 35:1510-1516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pancholi, P., C. P. Kolbert, P. D. Mitchell, K. D. Reed, Jr., J. S. Dumler, J. S. Bakken, S. R. Telford III, and D. H. Persing. 1995. Ixodes dammini as a potential vector of human granulocytic ehrlichiosis. J. Infect. Dis. 172:1007-1012. [DOI] [PubMed] [Google Scholar]
- 27.Petrovec, M., A. Bidovec, J. W. Sumner, W. L. Nicholson, J. E. Childs, and T. Avsic Zupanc. 2002. Infection with Anaplasma phagocytophila in cervids from Slovenia: evidence of two genotypic lineages. Wien. Klin. Wochenschr. 114:641-647. [PubMed] [Google Scholar]
- 28.Piesman, J., S. J. Karakashian, S. Lewengrub, M. A. Rudzinska, and A. Spielman. 1986. Development of Babesia microti sporozoites in adult Ixodes dammini. Int. J. Parasitol. 16:381-385. [DOI] [PubMed] [Google Scholar]
- 29.Piesman, J., B. S. Schneider, and N. S. Zeidner. 2001. Use of quantitative PCR to measure density of Borrelia burgdorferi in the midgut and salivary glands of feeding tick vectors. J. Clin. Microbiol. 39:4145-4148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Regnery, R. L., C. L. Spruill, and B. D. Plikaytis. 1991. Genotypic identification of rickettsiae and estimation of intraspecies sequence divergence for portions of two rickettsial genes. J. Bacteriol. 173:1576-1589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Reid, G. D., and L. J. Bell. 1984. The development of Theileria annulata in the salivary glands of the vector tick Hyalomma anatolicum anatolicum. Ann. Trop. Med. Parasitol. 78:409-421. [DOI] [PubMed] [Google Scholar]
- 32.Swofford, D. L. 2002. PAUP*. Phylogenetic analysis using parsimony (* and other methods), version 4. Sinauer Associates, Sunderland, MA.
- 33.Tate, C. M., D. G. Mead, M. P. Luttrell, E. W. Howerth, V. G. Dugan, U. G. Munderloh, and W. R. Davidson. 2005. Experimental infection of white-tailed deer with Anaplasma phagocytophilum, etiologic agent of human granulocytic anaplasmosis. J. Clin. Microbiol. 43:3595-3601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Telford, S. R., III, J. E. Dawson, P. Katavolos, C. K. Warner, C. P. Kolbert, and D. H. Persing. 1996. Perpetuation of the agent of human granulocytic ehrlichiosis in a deer tick-rodent cycle. Proc. Natl. Acad. Sci. USA 93:6209-6214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.von Loewenich, F. D., B. U. Baumgarten, K. Schröppel, W. Geiβdörfer, M. Röllinghoff, and C. Bogdan. 2003. High diversity of ankA sequences of Anaplasma phagocytophilum among Ixodes ricinus ticks in Germany. J. Clin. Microbiol. 41:5033-5040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.von Stedingk, L. V., M. Gürtelschmid, H. S. Hanson, R. Gustafson, L. Dotevall, E. Olsson, and M. Granström. 1997. The human granulocytic ehrlichiosis (HGE) agent in Swedish ticks. Clin. Microbiol. Infect. 3:573-574. [DOI] [PubMed] [Google Scholar]
- 37.Weller, S. J., G. D. Baldridge, U. G. Munderloh, H. Noda, J. Simser, and T. J. Kurtti. 1998. Phylogenetic placement of rickettsiae from the ticks Amblyomma americanum and Ixodes scapularis. J. Clin. Microbiol. 36:1305-1317. [DOI] [PMC free article] [PubMed] [Google Scholar]