


Abstract
Biological robustness, defined as the ability to maintain stable functioning in the face of various perturbations, is an important and fundamental topic in current biology, and has become a focus of numerous studies in recent years. Although structural robustness has been explored in several types of RNA molecules, the origins of robustness are still controversial. Computational analysis results are needed to make up for the lack of evidence of robustness in natural biological systems. The RNA structural robustness evaluator (RSRE) web server presented here provides a freely available online tool to quantitatively evaluate the structural robustness of RNA based on the widely accepted definition of neutrality. Several classical structure comparison methods are employed; five randomization methods are implemented to generate control sequences; sub-optimal predicted structures can be optionally utilized to mitigate the uncertainty of secondary structure prediction. With a user-friendly interface, the web application is easy to use. Intuitive illustrations are provided along with the original computational results to facilitate analysis. The RSRE will be helpful in the wide exploration of RNA structural robustness and will catalyze our understanding of RNA evolution. The RSRE web server is freely available at http://biosrv1.bmi.ac.cn/RSRE/ or http://biotech.bmi.ac.cn/RSRE/.
INTRODUCTION
Biological robustness, a fundamental and ubiquitous phenomenon observed in biological systems, is broadly understood as the ability to maintain stable functioning in the face of various perturbations. Depending on whether the perturbations are inheritable or not, robustness is characterized as genetic (mutational) or environmental robustness (1). Genetic robustness describes insensitivity of a phenotype facing genetic mutations, and the insensitivity to environmental factors is called environmental robustness. Biologists have a long-standing interest in biological robustness, going back to Fisher's work on dominance (2–4) and Waddington's developmental canalization research (5,6). Robustness has become a focus of numerous studies in recent years, and has been found at various levels of biological systems, including gene expression, protein folding, metabolic flux, physiological homeostasis, development and even organism fitness (7). Hiroaki Kitano argued that the requirements for robustness and evolvability are similar, since robustness facilitates evolution and evolution favors robust traits (8). A proper understanding of the origins of robustness in biological systems will catalyze our understanding of evolution (9).
The secondary structure of RNA is a suitable test bed for studying biological robustness. Wagner and Stadler provided evidence that robustness of RNA viruses to mutational changes in secondary structure has evolved (10). Mutational robustness has also been found in viroids (11,12). By examining microRNA genes of serveral species, Borenstein and Ruppin (13) recently showed that the structure of miRNA precursor stem-loops exhibits a significantly high level of genetic robustness, compared with random sequences with similar stem-loop structures as native miRNAs which were generated by inverse folding algorithm, indicating that this excess robustness of miRNA went beyond the intrinsic robustness of the stem-loop hairpin structure. Furthermore, they demonstrated it was not the by-product of a base composition bias. Their findings suggest that the excess robustness of miRNA stem-loops is the result of direct evolutionary pressure toward increased robustness (13).
Although the mechanisms of robustness have been widely explored (13–15), to date, the evolutionary origins of robustness are still controversial, which is partly due to the difficulty in providing evidence for robustness in natural biological systems (16). Addressing this challenge, a convenient computational tool for the structural robustness evaluation is strongly needed.
The RNA structural robustness evaluator (RSRE) presented here is a web tool developed for RNA structural robustness evaluation, both for genetic robustness and environmental robustness. By using classical RNA structural distance measurement methods, the robustness of a given RNA and its control sequences can be evaluated quantitatively based on a generalized definition of neutrality. The RSRE web server will finally give statistical significances of the robustness differences between the given RNA and its control sequences. The RSRE will facilitate wide exploration on the origins of robustness and catalyze our understanding of RNA evolution.
METHODS
Control sequence generation
Random sequences are used to extract statistical significance for properties from biological sequences, providing the ‘back-ground noise’ to differentiate the real biological information (17). However, a simple randomization method of RNA sequence obscures the frequencies of the mononucleotides and dinucleotides, which are biased and crucial for the physical stability of the secondary structure (18–21). It is consequently essential to rule out the bias of base compositions in the robustness analysis. To this end, we can generate additionally four types of random sequences preserving the exact or nearly exact mononucleotide and dinucleotide base compositions as the native sequence, besides the pure random sequences. The five randomization methods used in RSRE are described in detail as follows:
Pure random. This method produces pure random sequences with the same length as the original. The mononucleotide and dinucleotide frequencies are completely distorted using this method.
Shuffling based on zero-Markov model. The mononucleotide frequencies, P(b), for the native biological sequence are calculated and used to generate a random sequence in which bases were simply chosen at random from P(b) until the length of the native sequence is reached.
Mono-shuffling. This type of shuffling is done by permuting the nucleotides of the sequence at random. The dinucleotide frequencies are completely distorted using this method.
Shuffling based on first-Markov model. This method derives as first-order Markov model from the conditional probabilities P(a|b) of nucleotide a given b, which is found from the frequencies of all possible pairs ab in the biological sequences. A random sequence is generated by first choosing a random nucleotide x1, and then, a sequence is generated by choosing each nucleotide xi+1 from the probability P(xi+1|xi). The process will stop when the sequence has exactly the same length as that of the native. This method produces shuffled sequences with dinucleotide frequencies close to the original sequences. Mononucleotide frequencies are not preserved.
Dishuffling. In this method, a sequence is shuffled while keeping the dinucleotide distribution (or frequency) constant. A similar implementation of the Erikson–Altschul algorithm (18,19) was used. The dinucleotide and mononucleotide frequencies are exactly preserved.
Considering that certain secondary structures may be inherently more robust than others, random sequences with both phenotypically similar configurations and similar base compositions as native RNAs are also needed to control the effects of secondary structure in some researches (13). However, it is difficult to provide such control sets by most computational servers, due to the high computational cost (13). With the development of fast RNA inverse folding algorithms, we will find approaches to provide this kind of control sets in the future version of our web server.
Robustness evaluation
Experimental researches have demonstrated that the secondary structure of some RNAs are tolerant to some mutational changes (11–13,22–25). To reflect this flexibility in sequence/structure requirements, at a given threshold Tj, we defined the robustness γj as follows:
| 1 |
where d is the secondary structure distance between the original RNA and its mutant, and Nj(d) is the number of mutants with structure distance lesser than or equal to the threshold Tj. γj is the average of Nj(d) over all 3 × L one-mutant neighbors at the threshold Tj. The maximum value of the secondary structure distances between the random sequences and their mutants was used as a baseline value to evaluate the threshold level of each distance metric (Supplementary Figures S1 and S2). The threshold Tj, j = 0,1,2,…,9 was set to 0, 10, 20,…, 90% of the maximum value of the metric, respectively. At threshold T0, robustness is reduced to the definition of neutrality (13). The larger value of the robustness γj at threshold Tj indicated a relatively higher level of robustness.
A variety of distance measures for secondary structures (26–29) realized by RNAdistance in the Vienna RNA package (version 1.6) (27,30) were used to compare the secondary structures between the wild-type and its mutants, including tree-edit distance, string distance and base-pair distance (27,31,32).
The RNAfold and RNAsubopt (32) in the Vienna RNA package (version 1.6.1) (27,30) were utilized with default parameter values T = 37°C to predict the secondary structures. The former is a variation of the Zuker and Stiegler's (33,34) minimum free energy algorithm, while the latter is for the calculation of all sub-optimal structures within a user-defined energy range above the minimum free energy (MFE). In order to mitigate the uncertainty of the MFE structure, sub-optimal structures of mutants within 1 kcal/mol (the default setting of RNAsubopt) above the MFE are considered. A synthetic estimation method is used to estimate the differences between the structures of the wild-type R and possible structure set of the mutants 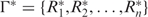 , where
, where 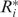 represents the ith predicted structure of the mutant. It is given by summing the contributions of all structures weighted by their Boltzmann probabilities, which is similar to the methods used in other researches (35). In this case, the distance is given by
represents the ith predicted structure of the mutant. It is given by summing the contributions of all structures weighted by their Boltzmann probabilities, which is similar to the methods used in other researches (35). In this case, the distance is given by 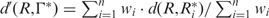 , where
, where 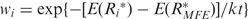 .
.
To explore the evolutionary origins of genetic robustness, we also examined the thermodynamic stability of RNAs in an analogous manner to the method used in previous researches (18,19,36), due to the possible correlation between the thermodynamic stability (environmental robustness) of the minimum free energy structure of a given sequence and its genetic robustness (32).
Statistical significance analysis of robustness
At each threshold Tj, we evaluated the robustness γi of the inputting sequence and  of the corresponding control sequence set X (N is the number of sequences in the control set X), and then compared γi with ϒj. The Z-score and P-value were then computed to determine whether the secondary structure of the inputting RNA molecular showed significantly more robustness than the control sequences. The Z-score is defined as:
of the corresponding control sequence set X (N is the number of sequences in the control set X), and then compared γi with ϒj. The Z-score and P-value were then computed to determine whether the secondary structure of the inputting RNA molecular showed significantly more robustness than the control sequences. The Z-score is defined as:
| 2 |
where 〈 · 〉 and σ(·)denote the mean and the standard deviation of ϒj, respectively. The P-value of γj is the fraction of sequences in X having robustness greater than the inputting RNA molecular, defined as:
| 3 |
where M is the number of sequences with more robustness than the inputting RNA molecular in X.
The statistical significance analysis of environmental robustness was similar to that done for genetic robustness, in which the robustness γj at threshold Tj was replaced by free energy of the sequences.
IMPLEMENTATION
The core module of RSRE is written in C++ and the web interface is implemented in PHP and JavaScript. RSRE runs on two work stations with dual AMD X64 CPUs, 4G memory and Linux operating system.
Input and options
With a step-by-step style input interface (Figure 1), the RSRE web server is easy to use. A valid email address is required for each job. The sequence of an RNA molecule can be inputted either by pasting raw sequence or by uploading sequence file in FASTA format. The sequence should be a string of unmodified RNA/DNA bases (A, U/T, G and C), any other character in the sequence will be edited out. Multi-FASTA (MFA) format sequence file is also supported to facilitate users. The inputting limit is set to 10 sequences for a job and 200 bases for each sequence. The analysis scheme is designed to be custom-built for users. The methods for using the sub-optimal structures can be selected by users. Users can also choose any one of the randomization methods described above and the number of control sequences according to their analysis requirements. Evaluation of either type of robustness (environmental robustness and genetic robustness) or both of them can be selected by the user. In the case of genetic robustness, users can select the algorithms for computing structure distance.
Figure 1.

Web interface of RSRE.
Output
To illustrate how our web applications can be helpful to the evaluation of the RNA structural robustness, the Caenorhabditis elegans let-7 microRNA precursor, one of the founding members of the microRNA family (37,38), was submitted to RSRE. A notification email containing a URL linked to the output page (Figure 2A) was sent to the user when the job was completed. This URL remains valid for 48 h. To make the analysis results intuitive, the statistical distributions of free energy and robustness value γj at threshold Tj, j = 0,1,2,…,9 are calculated and illustrated as histograms. By selecting the content item and clicking the ‘view’ button on the output page, the details of the results can be viewed as graphic representations. Figure 2B is the distribution histogram of free energy of cel-let-7 with its corresponding control sequences preserving the dinucleotide frequencies. Figure 2C is the distribution histograms of the robustness values at different threshold levels. With a hyperlink located at the bottom of the output page (Figure 2A), the output page offers download of the results as a single packed file in ‘.gz’ format for off-line analysis. In addition to the robustness distribution histograms (in ‘PNG’image format), the corresponding P-value and Z-score of let-7 at different thresholds (in ‘TXT’text format), the corresponding control sequences (in MFA format) and the robustness values at all the 10 threshold levels of let-7 and its corresponding 1000 control sequences (in ‘TXT’ text format) are also included in the result file (Figure 2D). The result file name is in the form ‘yymmddhhmmss.no’, where ‘yy’ is year, ‘mm’ is month, ‘dd’ is day, ‘hh’ is hour, ‘mm’ is minute, ‘ss’ is second and ‘no’ is serial number.
Figure 2.

Robustness analysis results of Caenorhabditis elegans let-7 microRNA precursor. Both the environmental robustness and genetic robustness with base-pair distance metric were evaluated. The number of control sequences that preserved the dinucleotide frequency with let-7 is 1000. (A) Output page of RSRE. (B) Free energy distribution histogram. (C) Robustness distribution histograms at different threshold levels. (D) In addition to the histogram figures, the Z-score and P-value of let-7 at different threshold levels (in ‘TXT’ text format), the corresponding 1000 control sequences (in ‘MFA’ format), and the robustness values at all 10 levels of let-7 and its corresponding 1000 control sequences (in ‘TXT’ text format) can be downloaded through a hyperlink located at the bottom of the output page.
Performance of the web server
To test the computational efficiency of RSRE, 10 groups of random sequences with 8 different lengths (from 25 to 200 with step 25) were submitted. All types of structure distance measurement are used in these tests. The CPU time of the 10 groups’ tests is illustrated in Supplementary Figure S3. Since June 2006, the two sites have been active for several months and served over 1000 submissions.
CONCLUSION
The RSRE web server we presented here provides a freely available online tool for RNA structural robustness evaluation. The sufficient control data and the widely accepted definition of neutrality give high reliability to the estimation results. The sub-optimal predicted RNA structures can also be optionally involved to mitigate the uncertainty of secondary structure prediction. Intuitive illustrations are provided along with the original computational results in the output page of RSRE to facilitate analysis. RSRE will facilitate a wide range of studies on RNA structural robustness, and therefore, will be helpful in RNA evolution exploration, artificial RNA design and other related research.
FUTURE PLANS
To provide a wide basis for RNA robustness exploration, our future works will focus on increasing the computational ability of the web server. By using a supercomputing blade system, the limit of inputting sequence length will be eased to meet the need of ncRNA robustness analysis in more cases. Also, in the future, we will provide more randomization methods, including the method-generating random sequences with both phenotypically similar configurations and similar base compositions as native RNAs.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
ACKNOWLEDGEMENTS
The authors would like to thank the Super Biomed Computation Center at the Beijing Institute of Health Administration and Medicine Information for providing computing resources. We thank Rujia Liu (Tsinghua University) for help in the programming of the web interface. This work was supported by a grant from the National Nature Science Foundation of China (No. 30600120) and a grant from the National High Technology Research and Development Program (863 program) of China (No. 2006AA02Z304). Funding to pay the Open Access publication charges for this article was provided by 863 program.
Conflict of Interest. None declared.
REFERENCES
- 1.Wagner GP, Booth G, Bagheri-Chaichian H. A population genetic theory of canalization. Evolution. 1997;v51:329–347. doi: 10.1111/j.1558-5646.1997.tb02420.x. [DOI] [PubMed] [Google Scholar]
- 2.Fisher RA. The possible modifications of the response of the wild type to recurrent mutations. Am. Nat. 1928;62:115–116. [Google Scholar]
- 3.Fisher RA. Two further notes on the origin of dominance. Am. Nat. 1928;62:571–574. [Google Scholar]
- 4.Fisher RA. The evolution of dominance. Biological reviews. 1931;6:345–368. [Google Scholar]
- 5.Waddington CH. The genetic assimilation of an acquired character. Evolution. 1953;7:118–126. [Google Scholar]
- 6.Waddington CH. The strategy of the genes. New York: MacMillan; 1957. [Google Scholar]
- 7.de Visser JA, Hermisson J, Wagner GP, Ancel ML, Bagheri-Chaichian H, Blanchard JL, Chao L, Cheverud JM, Elena SF, et al. Perspective: evolution and detection of genetic robustness. Evolution Int. J. Org. Evolution. 2003;57:1959–1972. doi: 10.1111/j.0014-3820.2003.tb00377.x. [DOI] [PubMed] [Google Scholar]
- 8.Kitano H. Biological robustness. Nat. Rev. Genet. 2004;5:826–837. doi: 10.1038/nrg1471. [DOI] [PubMed] [Google Scholar]
- 9.Wagner A. Robustness and Evolvability in Living Systems (Princeton Studies in Complexity) Princeton University Press, Princeton; 2005. [Google Scholar]
- 10.Wagner A, Stadler PF. Viral RNA and evolved mutational robustness. J. Exp. Zool. 1999;285:119–127. [PubMed] [Google Scholar]
- 11.Sanjuan R, Forment J, Elena SF. In silico predicted robustness of viroids RNA secondary structures. I. The effect of single mutations. Mol. Biol. Evol. 2006;23:1427–1436. doi: 10.1093/molbev/msl005. [DOI] [PubMed] [Google Scholar]
- 12.Sanjuan R, Forment J, Elena SF. In silico predicted robustness of viroids RNA secondary structures. II. Interaction between mutation pairs. Mol. Biol. Evol. 2006;23:2123–2130. doi: 10.1093/molbev/msl083. [DOI] [PubMed] [Google Scholar]
- 13.Borenstein E, Ruppin E. Direct evolution of genetic robustness in microRNA. Proc. Natl Acad. Sci. USA. 2006;103:6593–6598. doi: 10.1073/pnas.0510600103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Elena SF, Carrasco P, Daros JA, Sanjuan R. Mechanisms of genetic robustness in RNA viruses. EMBO Rep. 2006;7:168–173. doi: 10.1038/sj.embor.7400636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Montville R, Froissart R, Remold SK, Tenaillon O, Turner PE. Evolution of mutational robustness in an RNA virus. PLoS Biol. 2005;3:e381. doi: 10.1371/journal.pbio.0030381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gibson G, Wagner G. Canalization in evolutionary genetics: a stabilizing theory? Bioessays. 2000;22:372–380. doi: 10.1002/(SICI)1521-1878(200004)22:4<372::AID-BIES7>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- 17.Ponty Y, Termier M, Denise A. GenRGenS: software for generating random genomic sequences and structures. Bioinformatics. 2006;22:1534–1535. doi: 10.1093/bioinformatics/btl113. [DOI] [PubMed] [Google Scholar]
- 18.Bonnet E, Wuyts J, Rouze P, Van de PY. Evidence that microRNA precursors, unlike other non-coding RNAs, have lower folding free energies than random sequences. Bioinformatics. 2004;20:2911–2917. doi: 10.1093/bioinformatics/bth374. [DOI] [PubMed] [Google Scholar]
- 19.Clote P, Ferre F, Kranakis E, Krizanc D. Structural RNA has lower folding energy than random RNA of the same dinucleotide frequency. RNA. 2005;11:578–591. doi: 10.1261/rna.7220505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Katz L, Burge CB. Widespread selection for local RNA secondary structure in coding regions of bacterial genes. Genome Res. 2003;13:2042–2051. doi: 10.1101/gr.1257503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Workman C, Krogh A. No evidence that mRNAs have lower folding free energies than random sequences with the same dinucleotide distribution. Nucleic Acids Res. 1999;27:4816–4822. doi: 10.1093/nar/27.24.4816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lee Y, Ahn C, Han J, Choi H, Kim J, Yim J, Lee J, Provost P, Radmark O, et al. The nuclear RNase III Drosha initiates microRNA processing. Nature. 2003;425:415–419. doi: 10.1038/nature01957. [DOI] [PubMed] [Google Scholar]
- 23.Zeng Y, Wagner EJ, Cullen BR. Both natural and designed micro RNAs can inhibit the expression of cognate mRNAs when expressed in human cells. Mol. Cell. 2002;9:1327–1333. doi: 10.1016/s1097-2765(02)00541-5. [DOI] [PubMed] [Google Scholar]
- 24.Zeng Y, Cullen BR. Structural requirements for pre-microRNA binding and nuclear export by Exportin 5. Nucleic Acids Res. 2004;32:4776–4785. doi: 10.1093/nar/gkh824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zeng Y, Cullen BR. Sequence requirements for micro RNA processing and function in human cells. RNA. 2003;9:112–123. doi: 10.1261/rna.2780503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fontana W, Konings DA, Stadler PF, Schuster P. Statistics of RNA secondary structures. Biopolymers. 1993;33:1389–1404. doi: 10.1002/bip.360330909. [DOI] [PubMed] [Google Scholar]
- 27.Hofacker IL, Fontana W, Stadler PF, Bonhoeffer LS, Tacker M, Schuster P. Fast folding and comparison of RNA secondary structures. Monatshefte fur Chemie/Chemical Monthly. 1994;125:167–188. [Google Scholar]
- 28.Shapiro BA. An algorithm for comparing multiple RNA secondary structures. Comput. Appl. Biosci. 1988;4:387–393. doi: 10.1093/bioinformatics/4.3.387. [DOI] [PubMed] [Google Scholar]
- 29.Shapiro BA, Zhang KZ. Comparing multiple RNA secondary structures using tree comparisons. Comput. Appl. Biosci. 1990;6:309–318. doi: 10.1093/bioinformatics/6.4.309. [DOI] [PubMed] [Google Scholar]
- 30.Hofacker IL. Vienna RNA secondary structure server. Nucleic Acids Res. 2003;31:3429–3431. doi: 10.1093/nar/gkg599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hogeweg P, Hesper B. Energy directed folding of RNA sequences. Nucl. Acids Res. 1984;12:67–74. doi: 10.1093/nar/12.1part1.67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wuchty S, Fontana W, Hofacker IL, Schuster P. Complete suboptimal folding of RNA and the stability of secondary structures. Biopolymers. 1999;49:145–165. doi: 10.1002/(SICI)1097-0282(199902)49:2<145::AID-BIP4>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- 33.Zuker M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 2003;31:3406–3415. doi: 10.1093/nar/gkg595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zuker M, Stiegler P. Optimal computer folding of large RNA sequences using thermodynamics and auxiliary information. Nucleic Acids Res. 1981;9:133–148. doi: 10.1093/nar/9.1.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shu W, Bo X, Liu R, Zhao D, Zheng Z, Wang S. RDMAS: a web server for RNA deleterious mutation analysis. BMC Bioinformatics. 2006;7:404. doi: 10.1186/1471-2105-7-404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Seffens W, Digby D. mRNAs have greater negative folding free energies than shuffled or codon choice randomized sequences. Nucleic Acids Res. 1999;27:1578–1584. doi: 10.1093/nar/27.7.1578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Reinhart BJ, Slack FJ, Basson M, Pasquinelli AE, Bettinger JC, Rougvie AE, Horvitz HR, Ruvkun G. The 21-nucleotide let-7 RNA regulates developmental timing in Caenorhabditis elegans. Nature. 2000;403:901–906. doi: 10.1038/35002607. [DOI] [PubMed] [Google Scholar]
- 38.Slack FJ, Basson M, Liu Z, Ambros V, Horvitz HR, Ruvkun G. The lin-41 RBCC gene acts in the C. elegans heterochronic pathway between the let-7 regulatory RNA and the LIN-29 transcription factor. Mol. Cell. 2000;5:659–669. doi: 10.1016/s1097-2765(00)80245-2. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.