


Abstract
Mobile genetic elements have significantly contributed to the shaping of mammalian genomes. The RTAnalyzer software tracks sequences of retrotransposed origin by scoring the signature results from an L1-mediated insertion within a genome. More specifically, a sequence of interest is searched for in genomic databases using BLAST. Each hit, along with additional 5′ and 3′ sequences of pre-defined lengths, is saved. RTAnalyzer searches for specific L1 retrotransposition signatures (i.e. target site duplication, endonuclease cleavage site and poly(A)), and then calculates an overall retrotransposition score. This score represents the likelihood of a given sequence originating from a retrotransposition event involving the L1 machinery. RTAnalyzer may be used under GNU public license, and is available at http://www.riboclub.org/rtanalyzer.
INTRODUCTION
Mobile genetic elements have significantly contributed to the shaping of mammalian genomes. In humans, retrotransposons of the long interspersed element (LINE1 or L1) family and their remnants account for ∼17% of the human genome [reviewed in references (1–3)]. L1 contains two ORFs, while the function of the first (ORF1) remains unclear, the second (ORF2) encodes both endonuclease and reverse transcriptase activities (4). These later two proteins have a cis preference for the reverse transcription of the L1 mRNA reverse transcription (4), but are also able to mobilize other RNAs, such as Alu elements or short interspersed nucleotide elements (SINEs), in trans (5,6). L1 are also responsible for the insertion of many processed pseudogenes. The sequences of mature L1 mRNAs devoid of introns and harbouring a polyadenosine tail (poly(A)), can be found throughout the genome and are usually easily recognized as retrotransposed processed pseudogenes (7). Small non-coding RNAs are also a major source of retrotransposed elements. One of the most prolific of these is the Alu element, which is derived from 7SL RNA, with around 1 million copies being present in the human genome (8). A poly(A) tail is normally found at the 3′ end of the Alu insertion, and is generally considered as being important for retrotransposition via the L1 mechanism (6). The L1 endonuclease has a preference for a cleavage site with two pyrimidines followed by four purines. Specifically, it recognizes the TT/AAAA sequence most frequently (cleaving at the position indicated by the slash on the opposite strand) (1). The second DNA strand is typically cleaved 15 nucleotides (nt) away from the first cleavage site, although it may be anywhere from 10 to 25 nt away. The filling of the overhangs produced at the staggered break creates a target site duplication (TSD) flanking the element, another feature of L1 retrotransposition (Figure 1).
Figure 1.

(A) Proposed model for the L1 retrotransposition mechanism. (B) Example of a typical L1 signature.
Currently there are many major sequencing projects of mammalian genomes in progress, and, consequently, many potential new elements retrotransposed by L1 for which to search and analyse. Towards this end the RTAnalyzer software should be an efficient tool, particularly for an indepth analysis of the non-autonomous retrotransposons found in all sequenced mammalian genomes. More specifically it should help us understand the origins and variability of retrotransposition in the context of our evolutionary tree. RTAnalyzer permits anyone to search for a retrotransposition activity of their favourite gene (preferably a small gene of less than 300 bp). The software is designed to search for small non-coding RNAs that possess the signature of a sequence that was inserted in the genome by L1. Depending on the signature, a RetroScore is given to all of the BLAST hits corresponding to the input sequence. Figure 2 shows some screenshots of the software: (A) the user chooses to find retrotransposons; (B) the sequence of interest is submitted; (C) once the search is complete, the user receives a notification by email and he can logon to view his results; (D) the results are sorted in tabular form (an Excel format file can also be downloaded) and (E) details of one hit are viewed. A help link is present on the home page. An example is provided in the supplementary material and in the online help. For the moment, only a few databases are available for searching (human, mouse and rat), but others will be included upon request. As more genomes are sequenced and assembled, more will become available. RTAnalyzer may be used under GNU public license, and is available at http://www.riboclub.org/rtanalyzer. The source code is also available for download at this address.
Figure 2.

Screenshots of RTAnalyzer. (A) The home page showing the links to fetch results, to find retrotransposons and to view help. (B) Fields to input: name of query, e-mail address, genome in which to search for retrotransposed elements and sequence of interest before submitting it. (C) After receiving notification by e-mail, the user inputs his Id and password in order to retrieve his results. (D) View of the table of results (also available for downloading in excel format). (E) View of the details of one hit.
MATERIALS AND METHODS
RTAnalyzer searches and scores retrotransposed sequences on whole genome sequences. It was written in Perl version 5.8.8 (9) using freely available CPAN modules (10). The web application runs on a server with apache 2.0.54 (11) and gentoo linux 2.6.17-gentoo-r4 (12). The server communicates with a cluster in order to distribute RTAnalyzer tasks to three different slave nodes.
Search algorithm
In order to locate retrotransposed elements in genomic sequences, RTAnalyzer initially performs a homology search using BLAST (13), generating a first database of potential hits. The user only has to specify the initial sequence to search (ISS), and can set advanced parameters in order to modify the BLAST sensitivity. It is also possible to modify the lengths of the 3′ and 5′ additional sequences to be extracted. After aligning all of the hits into the same polarity, the software performs five steps in order to identify the hits with high retrotransposition probability (Figure 3). Because it is crucial to find the exact positions of 5′ and 3′ extremities of the homologous sequence for the calculation of the RetroScore (see later), the goal of steps 1 and 2 is the accurate determination of these extremities. Although the BLAST analysis will normally determine these extremities properly, sometimes it leaves out up to 10 bp (or even 20) that have a lower percentage identity. In step 1 the program attempts to find the 3' extremity (E3) of the ISS in the retrieved BLAST hit using Matcher [a local alignment program provided by EMBOSS (14)], while in step 2 a similar procedure is applied to the 5′ extremity (E5).
Figure 3.

Schematic representation of the five steps performed by the search algorithm. ‘TSD’ stands for target site duplication in 5′ and 3′, ‘target’ stands for the cleavage recognition site of L1 endonuclease. The ‘E5’, ‘E3’ correspond to the 5′ and 3′ ends of the homologous sequence. REd is the distance between the 5′ TSD and the 5′ extremity.
The next three steps find the signature associated with an L1-based retrotransposition (Figure 3). Step 3 is the search for the TSD. The sequence adjacent to the 5′ extremity of the insertion (5′ TSD mask) is extracted and aligned with the 100 nt (default parameter) following the 3′ end of the insertion. A TSD score is calculated. The operation is then repeated with a different 5′ TSD mask, specifically one shifted by one nucleotide upstream until the maximal distance specified by user is reached. The alignment with the best TSD score (see RetroScore section later) is saved. Step 4 aims to find the poly(A) tail. The sequence between the 3′ end of the insertion and the 5′ end of the putative 3′ TSD sequence is analysed. The poly(A) tail is extended until consecutive non-As are reached. When this point is reached, the upstream sequence is analysed for possible extension of the poly(A) tail (i.e. considering a minimum percentage of additional adenosines). A poly(A) score is then determined. Finally during step 5, the putative endonuclease cleavage site overlapping the 5′ TSD is extracted and compared to a list of pre-defined consensus sequences (15). At each step, only the best signature is saved for analysis in the subsequent steps.
RetroScore algorithm
The equation used to evaluate the RetroScore has been established through extensive testing on a set of 1000 pseudogenes. An arbitrary cutoff of 30 has been chosen in order to prevent RTAnalyzer from validating a signature that could have arisen from random sequence. Even if some TSDs are known to be as short as 2 bp (6), it is not reasonable to consider TSDs that short in a genomic search. This limitation implies that many signatures of real retrotransposed elements will be missed. The cutoff score was thus determined so as to ensure that the probability of the occurrence of positive hits would be very low, except if the sequence analysed was retrotransposed. Conversely, due to the nature of the signature, which is not conserved, but rather mutates during evolution instead, the algorithm and scoring system have to be very flexible.
The RetroScore resulting from these constraints is calculated for each signature according to the following equation (equations for the subscores are based on empirical testing to establish the best compromise between sensitivity and specificity):
in which the TSD and poly(A) are calculated according to:
where AS is the alignment score, ASmax the theoretic maximum alignment score calculated using the EDNAFULL alignment matrix, REd is the distance between 5' TSD and 5’ extremity and PR3d is the distance between the poly(A) tail and the 3' TSD. The poly(A) tail is scored using:
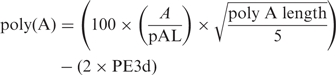 |
where A is the number of As found in the poly(A) tail, pAL the polyA length and PE3d is the distance between 3' end of the insertion and poly(A) start position.
Finally, the target score is equal to 100 if the sequences extracted is found in the target sequence list, or to 0 otherwise.
RESULTS AND DISCUSSION
Validation of results
A comparison of the results obtained with RTAnalyzer and those previously published for hY RNAs pseudogenes gives similar numbers (16). These pseudogenes are derived from the four hY RNAs (non-coding RNAs of approximately 100 nt). This previous study used a very early version of RTAnalyzer; however, in order to ensure that the sequences homologous to hY RNAs were truly retrotransposed, all 1000 of them were inspected. We used this dataset to fine tune the current version of the software. Table 1 shows a comparison of the results from RTAnalyzer with a visual analysis of the Y RNA pseudogenes in the human genome (16). False positives (FP) are pseudogenes identified as having been retrotransposed by RTAnalyzer, but not by visual analysis. The opposite is true for false negatives (FN). The overall number of retrotransposed hits appears to be only slightly underestimated by RTAnalyzer, with a correlation of greater than 75%. There are also a few sequences scored as retrotransposed by RTAnalyzer that did not present a convincing signature, mainly due to the presence of frequent A/T rich sequences both 5′ and 3′ of the sequences that were sometimes mistaken for TSDs. These ‘false positives’ represent less than 10% of the total number of hits. The FP percentages are shown in parentheses, after allowing for the FPs that are in fact good hits, but were classified as FPs in the previous study. Approximately half of the FNs were due to recent Alu insertions between the 3′ TSD and the poly(A) tail from a prior hY retrotransposition event. This means that the TSD was shifted approximately 300 bp away from the extremity of the analysed sequence, thus preventing the software from finding it. On the other hand, this software found a few new signatures that were not correctly evaluated in the previous study. It should be kept in mind that almost all RTAnalyzer parameters can be modified so as to optimize a given retrotransposon search according to the user's needs.
Table 1.
Summary of false positive (FP) and negative (FN) hits based on retrotransposons identified in Perreault et al. (16)
| hY RNA | Manuallya | RTAnalyzera | Correlation | FN | FP |
|---|---|---|---|---|---|
| 1 | 319/369 | 260/352 | 74% | 17% | 9% (7%) |
| 3 | 307/443 | 254/431 | 79% | 10% | 11% (8%) |
| 4 | 109/149 | 84/146 | 80% | 11% | 9% (5%) |
| 5 | 6/9 | 1/9 | 25% | 75% | 0% |
aRetrotransposed hits/total hits.
Even in light of the above discussion, we estimate that an important proportion of the hits with bad scores probably have a retrotransposed origin. The L1 signature degenerates through time and slowly disappears because it has no function and therefore possesses no reason to be conserved. Consequently, it is impossible to accurately detect all retrotransposed sequences, although RTAnalyzer manages to detect the vast majority of them. For example, a sample of MIR elements was analysed and only 1% scored valid for a retrotransposition signature because these elements were retrotransposed over 150 million years ago, in addition of being devoid of a poly(A) tail (17). The sequence of tRNAcys was also searched for with the software, because it is known to produce retropseudogenes lacking a poly(A) tail (18). A first scan with RTAnalyzer using default parameters allowed finding a few tailless pseudogenes. Then, adjusting the parameters (i.e. shorter additional sequences, shorter mask for the repeat and using only the retrotransposed section of the tRNA sequence) in order to fit the specific features of these pseudogenes yielded 9/10 valid scores for the corresponding BLAST hits.
RTAnalyzer was also tested with random sequences using a very high e-value setting in order to obtain large samples of BLAST hits. Overall, less than 1% of the hits had valid retrotransposition signatures, plus, the few valid signatures were only slightly higher than the RetroScore cutoff. In contrast, snoRNA ACA30, which has already been found to be retrotransposed (19), had four copies, three with valid signatures and one was a gene. Alu sequences were also used for testing. Using one Alu query gave ∼55% of valid signatures regardless the family.
Performance
The results obtained with RTAnalyzer are comparable to those obtained with a careful manual inspection of the signature. However, what is not comparable is the time to complete this task. The computing time can vary from as little as a few minutes to one-half hour for sequences that are found at hundreds of positions in a genome. Extremely frequent sequences, such as Alu, might require more processing time, but very few characterized examples have that many repeats. Hence, more than 99% of the sequences an user could submit will be analysed sufficiently rapidly so as to provide results by email the same day.
Sequence length
The software was designed to look for rather small retrotransposed pseudogenes, specifically those less than 300 bp. The repertory of such genes is already over a thousand with all tRNA, snRNA, snoRNA, etc., and currently growing very fast with the discovery of many new families (e.g. microRNA). Moreover, a different set of parameters is proposed in the online help and supplementary material in order to optimize the search for longer sequences. For example, tests were performed using ribosomal protein RPL3 (i.e. 1.3 kb) yielding retrieval of valid signatures. This demonstrates that RTAnalyzer can detect retropseudogenes longer than 300 bases (although less efficiently). The site http://www.pseudogene.org (20), or the Hoppsigen database available at http://pbil.univ-lyon1.fr/databases/hoppsigen.html (21), might be more appropriate when searching for large genes. In fact, since the first database is mainly constituted of sequences longer than 100 bp based on sequence homology with known genes that include deleterious mutations, and the second is constituted exclusively of processed pseudogenes for which the original gene possessed introns, RTAnalyzer nicely complements these databases. Indeed, the approach used by our software is very different; it will look for homologous sequences, but will also search for indications that L1 is responsible of the insertion. Moreover, we did not use RTAnalyzer to build whole genome retrogene databases, but rather allow the users to analyse any sequence.
Many small RNAs, like some tRNAs and U snRNAs, have several copies of their genes and pseudogenes in the genome (8). RTAnalyzer permits the recovery of both genes and pseudogenes, and then identifies and annotates the retrotransposition signature of the latter, regardless the number of copies in a genome. Even if small RNAs do not normally possess a poly(A) tail, most corresponding pseudogenes have one (hY RNAs are a good example of this). Moreover, the use of other features of the signature (e.g. TSDs and endonuclease target site) allows the user to recover pseudogenes that could be missing the poly(A) tail. Although this could potentially decrease the number of good scoring hits, it is the result of inevitable compromise between sensitivity and specificity. Our software was also designed to determine the significance of a homologous sequence retrieved with BLAST. A 5′ portion of variable length is often missing from the RNA sequence that was inserted, which can impair the study of pseudogenes. This peculiarity of retrotransposed elements, combined with the accumulated mutations, will often result in its being missed by a common BLAST search. Lowering the BLAST criteria at this level can lead to numerous hits, most of which might represent false positives. In order to overcome these hurdles we developed RTAnalyzer that can be especially efficient in this regard because it takes other characteristics into consideration.
Finally, the software described herein not only permits finding retrotransposons, but also analyzing for the presence of the L1 signature. Systematic examination of various RNAs with RTAnalyzer will help understand the L1 mechanism in non-autonomous retrotransposition and help explain the sharp differences observed between the pseudogene frequencies of different small non-coding RNAs, as well as differences between species. Since L1 is so prominent in mammals, analysis of the numerous mammalian genomes currently being sequenced will benefit from a tool such as RTAnalyzer.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
ACKNOWLEDGEMENTS
We thank Justine Brassard and Jean-Christophe Houde for programming. This work was supported by grants from Genome Québec to J.P.P. and G.B., and by a grant from the Canadian Institute of Health Research (CIHR; EOP-38322) to J.P.P. The RNA group is supported by a grant from the CIHR (PRG-80169) and l’Université de Sherbrooke. J.P. was the recipient of a predoctoral fellowship from FRSQ. J.P.P. holds the Canada Research Chair in Genomics and Catalytic RNA. Funding to pay the Open Access publication charges for this article was provided by CIHR.
Conflict of interest statement. None declared.
REFERENCES
- 1.Ostertag EM, Kazazian HH., Jr Biology of mammalian L1 retrotransposons. Annu. Rev. Genet. 2001;35:501–538. doi: 10.1146/annurev.genet.35.102401.091032. [DOI] [PubMed] [Google Scholar]
- 2.Kazazian HH., Jr Mobile elements: drivers of genome evolution. Science. 2004;303:1626–1632. doi: 10.1126/science.1089670. [DOI] [PubMed] [Google Scholar]
- 3.Feng Q, Moran JV, Kazazian HH, Jr, Boeke JD. Human L1 retrotransposon encodes a conserved endonuclease required for retrotransposition. Cell. 1996;87:905–916. doi: 10.1016/s0092-8674(00)81997-2. [DOI] [PubMed] [Google Scholar]
- 4.Wei W, Gilbert N, Ooi SL, Lawler JF, Ostertag EM, Kazazian HH, Boeke JD, Moran JV. Human L1 retrotransposition in the human population. Mol. Cell. Biol. 2001;21:1429–1439. doi: 10.1128/MCB.21.4.1429-1439.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Weiner AM. Do all SINEs lead to LINEs? Nat. Genet. 2000;24:332–333. doi: 10.1038/74135. [DOI] [PubMed] [Google Scholar]
- 6.Dewannieux M, Esnault C, Heidmann T. LINE-mediated retrotransposition of marked Alu sequences. Nat. Genet. 2003;35:41–48. doi: 10.1038/ng1223. [DOI] [PubMed] [Google Scholar]
- 7.Esnault C, Maestre J, Heidmann T. Human LINE retrotransposons generate processed pseudogenes. Nat. Genet. 2000;24:363–367. doi: 10.1038/74184. [DOI] [PubMed] [Google Scholar]
- 8.International Human Genome Sequencing Consortium. Initial sequencing and analysis of the human genome. Nature. 2001;409:860–921. doi: 10.1038/35057062. [DOI] [PubMed] [Google Scholar]
- 9. The Perl directory. Available at http://www.perl.org/.
- 10.CPAN: Comprehensive Perl archive network. Available at http://cpan.org/.
- 11.The Apache Software Foundation. Available at http://www.apache.org/.
- 12.Gentoo linux. Available at http://www.gentoo.org/.
- 13.Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rice P, Longden I, Bleasby A. EMBOSS: the European Molecular Biology Open Software Suite. Trends Genet. 2000;16:276–277. doi: 10.1016/s0168-9525(00)02024-2. [DOI] [PubMed] [Google Scholar]
- 15.Jurka J. Sequence patterns indicate an enzymatic involvement in integration of mammalian retroposons. Proc. Natl Acad. Sci. U.S.A. 1997;95:2083–2088. doi: 10.1073/pnas.94.5.1872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Perreault J, Noël JF, Brière F, Cousineau B, Lucier JF, Perreault JP, Boire G. Retropseudogenes derived from the human Ro/SS-A autoantigen-associated hY RNAs. Nucleic Acids Res. 2005;33:2032–2041. doi: 10.1093/nar/gki504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Smit AF, Riggs AD. MIRs are classic, tRNA-derived SINEs that amplified before the mammalian radiation. Nucleic Acids Res. 1995;23:98–102. doi: 10.1093/nar/23.1.98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schmitz J, Churakov G, Zischler H, Brosius J. A novel class of mammalian-specific tailless retropseudogenes. Genome Res. 2004;14:1911–1915. doi: 10.1101/gr.2720104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Weber JM. Mammalian small Nucleolar RNAs are mobile genetic elements. PLoS Genet. 2006;2:1984. doi: 10.1371/journal.pgen.0020205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Karro JE, Yan Y, Zheng D, Zhang Z, Carriero N, Cayting P, Harrison P, Gerstein M. Pseudogene.org: a comprehensive database and comparison platform for pseudogene annotation. Nucleic Acids Res. 2007;35(database issue):D55–D60. doi: 10.1093/nar/gkl851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Khelifi A, Duret L, Mouchiroud D. HOPPSIGEN: a database of human and mouse processed pseudogenes. Nucleic Acids Res. 2005;33(database issue):D59–D66. doi: 10.1093/nar/gki084. [DOI] [PMC free article] [PubMed] [Google Scholar]