


Abstract
DSHIFT is a web server for predicting chemical shifts of DNA sequences in random coil form or double helical B-form. The prediction methods are based on sets of published reference chemical shift values and correction factors which account for shielding or deshielding effects from neighboring nucleotides. Proton, carbon and phosphorus chemical shift predictions are available for random coil DNAs. For double helical B-DNA, only proton chemical shift prediction is available. Results from these predictions will be useful for facilitating NMR resonance assignments and investigating structural features of solution DNA molecules. The URL of this server is: http://www.chem.cuhk.edu.hk/DSHIFT.
INTRODUCTION
Chemical shift contains a wealth of structural information of DNAs. At present, several methods have been established to predict chemical shifts of DNAs in random coil form (1–3) and double helical B-form (4,5). These methods are based on sets of reference chemical shift values and correction factors from experimental measurements, statistical analysis or semi-empirical calculations. Shielding or deshielding contributions from nearest neighbor and/or next-nearest neighbor nucleotides have been included in these prediction methods. To automate these prediction methods, a web server called ‘DSHIFT’ has been established for predicting DNA chemical shifts in this work. This web server is open access to everyone. Through entering a DNA sequence, random coil or double helical B-DNA chemical shifts will be predicted.
DSHIFT results can provide a quick reference guide for resonance assignments based on conventional NOESY and COSY-type experiments, thus facilitating solution structure studies of DNAs. These results can also provide useful information for studying structure–chemical shift relationship, identifying unstructured or right-handed double helical regions, monitoring DNA–drug or DNA–protein binding, and investigating conformational details of special features in DNA structures.
WEB SERVER IMPLEMENTATION AND LAYOUT
DSHIFT is written in HyperText Markup Language (HTML) and Active Server Pages (ASP). Presently, it is implemented on Internet Information Services (IIS) 6.0 using Microsoft Windows Server 2003 operating system. The web server is running on a Dell OptiPlex GX270 Intel Pentium 4 2.4 GHz computer with 512 MB RAM and 80 GB hard drive.
Figure 1 shows the layout of DSHIFT, which is composed of basically three major types of pages, namely, ‘DSHIFT Home’, ‘Sequence Input’ and ‘Prediction Result’. ‘DSHIFT Home’ (Figure 2) is the starting page of DSHIFT which provides access to predict DNA chemical shifts in either random coil form or double helical B-form. ‘Sequence Input’ is the input page of DSHIFT. Sequence content, choices of nucleus and method will be submitted through this page. All input will be validated before submitting to prediction. If the input is valid, sequence information and chemical shift prediction results will appear in ‘Prediction Result’, which is the output page of DSHIFT. Finally, you have the options to either predict another sequence or go back to ‘DSHIFT Home’.
Figure 1.

Layout of DSHIFT.
Figure 2.

DSHIFT Home: starting page of DSHIFT.
SEQUENCE INPUT
Figure 3A shows ‘Sequence Input’ page. You will find the citations of method that you are going to use at the beginning. For random coil chemical shift prediction, you have to (i) enter the sequence and (ii) pick the nucleus that you are interested in. Besides the choice of a particular nucleus, there is also an option ‘Show ALL’ in case you want to predict the chemical shift of all available nuclei. For double helical B-DNA, in addition to the above input items, you have to choose from ‘Altona method’ (5) or ‘Wijmenga method’ (4) as shown in Figure 4A. Due to method limitation, chemical shift predictions of labile protons in double helical B-DNA such as guanine imino (G-NH), thymine imino (T-NH), cytosine bound amino (C-NHB) and free amino (C-NHE) are only available in ‘Altona method’.
Figure 3.

Random coil chemical shift prediction pages: (A) ‘Sequence Input’ (input page) and (B) ‘Prediction Result’ (output page) of DSHIFT.
Figure 4.

Double helical B-DNA chemical shift prediction pages: (A) ‘Sequence Input’ (input page) and (B) ‘Prediction Result’ (output page) of DSHIFT.
Sequence input has to be started from the 5′-end. Both upper case and lower case characters of ‘C’, ‘G’, ‘A’ and ‘T’ are accepted. Space is also allowed in sequence input. In case there is a typing mistake in sequence or if nucleus or prediction method (in B-DNA) has not been selected, an error message will appear, detailing the input mistake. Due to serious spectral overlap and broadening of resonance signals, DNA structures containing more than 100 nt are seldom studied by NMR spectroscopy. In DSHIFT, the maximum length of sequence input has been set to 500 nt. An error message will appear if the sequence length is longer than 500 nt.
PREDICTION METHODS
Random coil
In DSHIFT, random coil proton (1), carbon (2) and phosphorus (3) DNA chemical shift predictions are available. For random coil proton prediction, a pentamer model 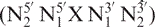 which is based on a set of triplet chemical shift values,
which is based on a set of triplet chemical shift values, 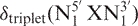 and next nearest neighbor correction factors, has been used (1), i.e.
and next nearest neighbor correction factors, has been used (1), i.e.
| 1 |
where δpred(X) is the predicted chemical shift of X, 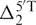 and
and 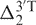 are the 5′- and 3′-next nearest thymine effects on X in the original 17-nt random coil sequences,
are the 5′- and 3′-next nearest thymine effects on X in the original 17-nt random coil sequences, 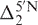 and
and 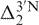 are the real 5′- and 3′-next nearest effects on X in the predicted sequence. Apart from the published data set of H6/H8, H1′, H2′, H2′′ and H3′ which gives a prediction accuracy of 0.02–0.03 p.p.m., adenine H2 (A-H2), cytosine H5 (C-H5) and thymine H7 (T-H7) triplet values from the original 17-nt random coil sequences have also been extracted in this work (Supplementary Data S1). Besides, next nearest neighbor correction factors (Supplementary Data S2) have been derived using the same strategy (1) for these protons and the prediction accuracy has been found to be 0.02 p.p.m.
are the real 5′- and 3′-next nearest effects on X in the predicted sequence. Apart from the published data set of H6/H8, H1′, H2′, H2′′ and H3′ which gives a prediction accuracy of 0.02–0.03 p.p.m., adenine H2 (A-H2), cytosine H5 (C-H5) and thymine H7 (T-H7) triplet values from the original 17-nt random coil sequences have also been extracted in this work (Supplementary Data S1). Besides, next nearest neighbor correction factors (Supplementary Data S2) have been derived using the same strategy (1) for these protons and the prediction accuracy has been found to be 0.02 p.p.m.
Due to the absence of phosphate groups at the 5′- and 3′-termini, random coil proton chemical shifts for nucleotides at penultimate positions are calculated using the following equations:
| 2 |
| 3 |
where 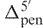 and
and 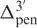 are the 5′- and 3′-penultimate correction factors (Supplementary Data S3), respectively.
are the 5′- and 3′-penultimate correction factors (Supplementary Data S3), respectively.
For random coil carbon chemical shifts, the prediction method is based on a trimer model as only nearest neighbor effect has been found to be significant (2), i.e.
| 4 |
The reported prediction accuracy of this method is 0.09–0.10 p.p.m. Since the absence of phosphate groups at both termini has been found to only affect the predicted C3′ values of the 3′-penultimate nucleotides (2), therefore 3′-penultimate correction of C3′ (Supplementary Data S3) has also been included in this prediction method. In addition to the published values of C6/C8, C1′, C2′ and C3′, triplet values of adenine C2 (A-C2), cytosine C5 (C-C5) and thymine C7 (T-C7) have also been extracted from the original 17-nt random coil sequences (Supplementary Data S1). The prediction accuracy for these carbons has been found to be 0.03–0.04 p.p.m.
For phosphorus, prediction can be made either based on a trimer model (3), i.e.
| 5 |
or a dimer model, i.e.
| 6 |
In both cases, due to the absence of phosphate groups at both 5′- and 3′-termini, end corrections to 5′- and 3′-penultimate nucleotides have been included in these prediction methods. In addition, end correction to 3′-terminal nucleotide is also included in the dimer model. The reported phosphorus chemical shift prediction accuracy is 0.02 and 0.03 p.p.m. for the trimer and dimer models, respectively.
Double helical B-DNA
In DSHIFT, two prediction methods are available for double helical B-DNA. In Altona method, proton chemical shift prediction is based on a trimer model in which an incremental scheme and statistical reference values from experimental results have been used (5). The chemical shift of proton X, δpred(X), in a given central residue X in a triplet N5′XN3′, or at either terminal positions in a doublet E5′XN3′ or N5′XE3′ is predicted as:
| 7 |
| 8 |
| 9 |
where X represents the reference value, N5′ and N3′ represent the incremental shielding or deshielding contributions of the four flanking residues on the 5′- and 3′-side, respectively, E5′ and E3′ represent the effect due to the absence of a phosphate group and attached residue at the 5′- and 3′-termini, respectively. Deshielding corrections have also been made to both 5′- and 3′-penultimate nucleotides and the reported prediction accuracy of this method is 0.01–0.03 p.p.m.
In Wijmenga method, proton chemical shift of a specific nucleotide is predicted based on a set of calculated reference shift value (δref) plus the chemical shift effect induced by its own base (δib), its 3′- (δ3′b) and 5′-neighboring bases (δ5′b) (4), i.e.
| 10 |
These calculated values are based on semi-empirical relations derived by Giessner-Prettre and Pullman (6) and the reported proton chemical shift prediction accuracy of this method is 0.17 p.p.m.
PREDICTION RESULT
Figures 3B and 4B show examples of ‘Prediction Result’ pages of random coil and B-DNA, respectively, in which the sequence input, length of sequence and a table summarizing the predicted chemical shifts will be reported. The abbreviation, ‘n.a.’ (means not available), will be given to items that cannot be predicted due to either absence of the nucleus or method limitation. If the ‘Show ALL’ option has been selected instead of a nucleus, prediction results of all available nuclei would be summarized in the resulting table. At the end of this output page, the total number of times that DSHIFT has been successfully used will also be reported.
SCOPE AND LIMITATIONS
DSHIFT serves as a convenient tool for predicting chemical shifts of DNAs in random coil form or double helical B-form. The predicted values are referenced to the most upfield signal of 2,2-dimethyl-2-silapentane-5-sulfonate sodium salt (DSS) and provides a quick reference guide which facilitates the resonance assignment step in solution DNA structure studies. The prediction accuracy of various methods adopted in DSHIFT depends mainly on DNA conformations. Since temperature and solution conditions affect stabilities of DNA structures, it is expected that these factors will also affect the prediction accuracy.
Temperature
For random coil chemical shift prediction, a large deviation between experimental and predicted values will be expected if residual structures are present in DNA sequences. Higher temperatures tend to eliminate residual structures. Since temperature coefficients of random coil chemical shifts have been found to be quite small and they are in the same order of magnitudes as the measurement uncertainties (1–3), the predicted values should agree well with experimental values measured over a wide temperature range.
For double helical B-DNA, higher temperatures lead to denaturation of double helix and thereby a change in DNA conformation. Thus, the prediction accuracy is expected to decrease when the experimental values are obtained at temperatures where the double helix becomes unstable. For prediction of G-NH and T-NH chemical shifts, the predicted values in Altona method have been normalized to values measured at 15°C. For values at other temperatures, corrections have to be made using the temperature coefficients -2.2 × 10−3 and −5.4 × 10−3 p.p.m./°C, respectively (5).
Solution condition
For random coil chemical shift prediction, the values have been tested to represent the fully denatured state (1). Apart from temperature, pH, salt content and whether denaturant is present in solution will also affect the denatured state. If any of these factors favors the presence of residual structures, the predicted values will be expected to deviate greatly from the experimental results.
For double helical B-DNA predication, Altona and co-workers have found that the variations of monovalent salt concentration from 5 to 200 mM or divalent salt concentration from 0 to 20 mM in DNA samples do not lead to significant chemical shift changes (5). Nevertheless, pH and salt content affect the stability of B-form structures. Large deviations between the predicted and experimental values will be expected if pH or salt content does not favor the formation of stable B-DNA structure.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
ACKNOWLEDGEMENTS
I would like to thank the valuable input and suggestions from my students C. K. Kwok, K. F. Lai and L. M. Chi. I would also like to thank K. F. Woo and D. Fenn for setting up the hardware and software for this web server and all people who have tested this server. The work described in this article was partially supported by a grant from the Research Grants Council of the Hong Kong Special Administrative Region (Project No.: 400704) and a direct grant from CUHK Research Committee Funding (Project No.: 2060252). Funding to pay the Open Access publication charge was provided by Research Grants Council of the Hong Kong Special Administrative Region.
Conflict of interest statement. None declared.
REFERENCES
- 1.Lam SL, Ip LN, Cui X, Ho CN. Random coil proton chemical shifts of deoxyribonucleic acids. J. Biomol. NMR. 2002;24:329–337. doi: 10.1023/a:1021671531438. [DOI] [PubMed] [Google Scholar]
- 2.Kwok CW, Ho CN, Chi LM, Lam SL. Random coil carbon chemical shifts of deoxyribonucleic acids. J. Magn. Reson. 2004;166:11–18. doi: 10.1016/j.jmr.2003.10.001. [DOI] [PubMed] [Google Scholar]
- 3.Ho CN, Lam SL. Random coil phosphorus chemical shift of deoxyribonucleic acids. J. Magn. Reson. 2004;171:193–200. doi: 10.1016/j.jmr.2004.08.024. [DOI] [PubMed] [Google Scholar]
- 4.Wijmenga SS, Kruithof M, Hilbers CW. Analysis of 1H chemical shifts in DNA: assessment of the reliability of 1H chemical shift calculations for use in structure refinement. J. Biomol. NMR. 1997;10:337–350. doi: 10.1023/A:1018348123074. [DOI] [PubMed] [Google Scholar]
- 5.Altona C, Faber DH, Westra Hoekzema AJA. Double-helical DNA 1H chemical shifts: an accurate and balanced predictive empirical scheme. Magn. Reson. Chem. 2000;38:95–107. [Google Scholar]
- 6.Giessner-Prettre C, Pullman B. Quantum mechanical calculations of NMR chemical shifts in nucleic acids. Q. Rev. Biophys. 1987;20:113–172. doi: 10.1017/s0033583500004169. [DOI] [PubMed] [Google Scholar]