


Abstract
Knotted proteins are more commonly observed in recent years due to the enormously growing number of structures in the Protein Data Bank (PDB). Studies show that the knot regions contribute to both ligand binding and enzyme activity in proteins such as the chromophore-binding domain of phytochrome, ketol–acid reductoisomerase or SpoU methyltransferase. However, there are still many misidentified knots published in the literature due to the absence of a convenient web tool available to the general biologists. Here, we present the first web server to detect the knots in proteins as well as provide information on knotted proteins in PDB—the protein KNOT (pKNOT) web server. In pKNOT, users can either input PDB ID or upload protein coordinates in the PDB format. The pKNOT web server will detect the knots in the protein using the Taylor's smoothing algorithm. All the detected knots can be visually inspected using a Java-based 3D graphics viewer. We believe that the pKNOT web server will be useful to both biologists in general and structural biologists in particular.
INTRODUCTION
Knotted proteins have become more common in recent years (1–14) due to the enormously growing number of structures deposited in the Protein Data Bank (PDB). The knots in proteins are more than just topological novelties. The knotted regions have been shown to be important in both ligand binding and enzyme activity. For example, the unique knot topology in bacterial phytochrome (6) is common to all red/far-red photochromic phytochrome and is important in stabilizing the chromophore-binding region. The knot regions in TrmD tRNA methyltransferase (MTase) have been shown to be important for S-adenosyl-L-methionine (AdoMet) binding and catalytic activity (3). The deep trefoil knot region in N-acetylornithine transcarbamylase forms part of the active site (10). The figure-eight knot in the mainly α-helical domain of ketol–acid reductoisomerase (KARI) forms most of the keto–acid substrate-binding site (11). In addition, knots in proteins present a challenge in the study of protein folding, for it is hard to image a peptide chain to thread through a hoop to form a knot in a reproducible way (15). Interestingly, a recent study (16) showed that YibK (4), a SpoU MTase containing a deep trefoil knot, is able to fold efficiently and behaves remarkably similar to other proteins.
Though the identification of a general knot is a topologically difficult problem, it is relatively easy to identify knots in proteins. However, there were still many cases of misidentified knots in proteins (17,18) due to the lack of a convenient tool available to general biologists. The causes of the misidentification of knots in proteins may be due to the presence of mobile loops, missing residues or just visual error in tracing out the entangled protein chains. For example, the SET domain was originally identified to have a knot, but later it was pointed out that part of the loop relevant to the formation of the knot is in fact connected through hydrogen bonds (17). As a result, the knot in the SET domain turns out not to be an authentic one. Other examples of misidentified knots are the trefoil knot in clathrin D6 coat protein (19), the left-handed trefoil knot in ubiquitin hydrolase (15) and the figure-eight knot in histone K79 methyltransferase (19). These knots are in fact caused by breaks in the chain and are therefore not authentic knots. A more recent example is the misidentified trefoil knot in the chromophore-binding domain of phytochrome (6), which in fact contains a figure-eight knot.
METHOD AND IMPLEMENTATION
The pKNOT web server detects the knot in a protein by smoothing the protein chain using the Taylor's algorithm (15). The algorithm first fixes both N and C termini in space, then repeatedly smoothes and straightens the protein chain. The chain is reduced in such a way that, with details of the chains eliminated, the knot can be easily detected. If the protein does not contain a knot, the chain will simply shrink into a straight line. The Taylor's algorithm formally goes as follows: Let the protein chain of length N be described by (r1,r2,…,rN), where ri is the coordinate of the i-th Cα atom. A new coordinate 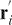 is taken to be
is taken to be 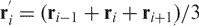 , where 2 ≤ i ≤ N − 1. The termini remain fixed, i.e.
, where 2 ≤ i ≤ N − 1. The termini remain fixed, i.e.  and
and  . The iterative procedure will continue to progressively smooth the chain. The main idea is to prevent the chains from passing through each other. This is done by checking that the triangles defined by
. The iterative procedure will continue to progressively smooth the chain. The main idea is to prevent the chains from passing through each other. This is done by checking that the triangles defined by 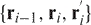 and
and 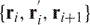 do not intersect any line segments defined by
do not intersect any line segments defined by 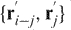 for j < i and
for j < i and 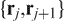 for j > i. In practice, most protein chains reduce to a straight line defined either by two termini or to an obvious knot in less than 50 iterations. However, there are cases that will take 500 or more iterations to converge. Figure 1 shows a typical example of a chain-smoothing procedure from the original structure of the chromophore-binding domain of bacterial phytochrome (1ZTU) to the final smoothed chain that can be easily identified to contain a figure-eight knot.
for j > i. In practice, most protein chains reduce to a straight line defined either by two termini or to an obvious knot in less than 50 iterations. However, there are cases that will take 500 or more iterations to converge. Figure 1 shows a typical example of a chain-smoothing procedure from the original structure of the chromophore-binding domain of bacterial phytochrome (1ZTU) to the final smoothed chain that can be easily identified to contain a figure-eight knot.
Figure 1.

An example of the chain smoothing process. The protein is the chromophore-binding domain of bacterial phytochrome (1ZTU). The X-ray structure of 1ZTU (left) is shown in the cartoon representation and two progressively smoothed chains are shown (center and left). The color is ramped by residues from blue at the N-terminus (labeled by N) to red at the C-terminus (labeled by C). The crossover points are numbered sequentially from the N-terminus. The figure-eight knot is characterized by four crossover points, alternately under and over. The structural pictures are produced using Pymol (Delano Scientific, San Carlos, http://pymol.sourceforge.net/).
Data set and pre-computed knots
To speed up the web server, we pre-computed all proteins in the PDB as of January 12, 2007, which consists of 41 013 proteins comprising 34 971 X-ray structures and 6042 NMR protein structures. The crystal structures of homologous protein chains (even those with identical sequences) as well as the solution structures of the same protein were checked for the presence of knots. The chains with breaks or discontinuities are visually checked for their relevance in knot formation. If the proteins have a missing gap so large that it is improper to simply connect the two ends of the missing fragment to complete the chain, the identified knots will be disregarded. All final smoothed chains that appear to form a knot, i.e. not a simple straight line, were visually examined to decide whether these knots are authentic knots, slipknots or artificial knots caused by large breaks in the chains. The knots in proteins are quite simple in that they can be visually identified, and no sophisticated analysis [such as the Jones polynomials or others (20)] is required. In summary, pKNOT provides information about all knotted proteins, such as their protein classes, their knotted types and the cores and depths of the knotted regions. The core is the smallest region that will remain knotted when the residues are successively deleted from both ends (15), and the depth is the product of the number of residues that must be deleted from both ends in order to free the knot (15).
Users can also upload the protein structure coordinates in the PDB format and the pKNOT server will progressively smooth the chains on the fly and then present the final smoothed chain as well as the original chain in a JAVA-based 3D graphics viewer AstexViewer (21) for users to inspect.
Input format
The web page of the pKNOT web server is shown in Figure 2. The users can either type in the PDB ID or upload a structural file in the PDB format. In the latter case, the default iteration number is set to 500 and the collision threshold, to 0.5 Å. The user can either ignore or preserve the breaks in the chain when smoothing the chain. The former option will close the breaks by using the shortest line segment connecting the breaks, while the latter option preserves the breaks in the chain and smoothes each individual segment, keeping the endpoints of each segment fixed. The default is set to ignore the breaks in the chain. The users can also choose from the pull-down menu the number of iterations to smooth the chain. The collision threshold is the distance threshold to determine whether a line segment will intersect the triangle during the smoothing procedures.
Figure 2.

The web page of the pKNOT web server. The users can either type in the PDB ID or upload a structural file in the PDB format. When submitting the structural file, the user can choose to either ignore or preserve the breaks in the chain. The default iteration number is set to 500 and the collision threshold, to 0.5 Å.
Output format and visualization of chains and knots
Upon query, pKNOT will return a table of the CHAIN, LENGTH, KNOT TYPE and DISPLAY STRUCTURE (Figure 3). When clicking on the column of KNOT TYPE, the server will return a list of all the proteins of the given knot type. pKNOT also provides the molecular viewer AstexViewer so that the users can visualize and manipulate in real time the protein structure and the knot in the protein. Both the original structure and the knot are shown in the same graphics window and the user can toggle on and off one of them for easy inspection.
Figure 3.

Upon query, the pKNOT server will return a table of the CHAIN, LENGTH, KNOT TYPE, CORE, DEPTH and DISPLAY STRUCTURE (upper center). When clicking on the column of KNOT TYPE, the server will return a list of all the proteins of the given knot type in the database of the pKNOT server (lower left). PKNOT also provides a JAVA-based 3D molecular viewer AstexViewer and the users can visualize and manipulate in real time the protein structure and the knot in the protein (lower right).
RESULTS
The knotted proteins come from the following protein classes: (1) methyltransferase, (2) transcarbamylase, (3) carbonic anhydrase, (3) ketol–acid reductosiomerase, (4) ubiquitin hydrolase, (5) methionine adenosyl transferase, (6) the chromophore-binding domain of bacterial phytochrome and (7) the inner core shell component protein of bluetongue virus. In addition, we also identified two knotted NMR structures: 1POQ and 1J2O. However, it is not clear whether these knots are authentic or due to incorrect structural refinement, since only one knotted model is identified among all NMR models for each protein (model 7 in 1POQ and model 14 in1J2O).
The knot types in proteins
There are three types of knot (up to the mirror image) identified in the PDB: the trefoil knot, the figure-eight knot and the knot with five crossings(15,19).
The trefoil knot
The trefoil knot (also called the threefoil or overhand knot) is the simplest knot of all, which is characterized by three crossings. It is mathematically denoted as a 31 knot. The proteins with a trefoil knot are (1) methyltransferase, (2) transcarbamylase, (3) methionine adenosyltransferase, (4) carbonic anhydrase and (5) YMPa superantigen (NMR).
The figure-eight knot
The figure-eight knot is characterized by four crossover points, alternately under and over. There is only one prime knot with four crossings and is denoted as the 41 knot. The proteins with a 41 knot are (1) the chromophore-binding domain of bacterial phytochrome, (2) the core protein of bluetongue virus, (3) ketol–acid reductoisomerase and (4) a LIM-ldbl-LID chimeric protein (NMR).
The 52 knots
There are two types of knot with five crossings: the 51 and 52 knots. Only the 52 knot has been identified in the protein structure and, as of writing, no proteins with six or more crossings have been identified in the PDB. The only protein family with a 52 knot is ubiquitin c-terminal hydrolase (1).
Comparison with other work
It will be interesting to compare our results with those of the recent work by Lua and Grosberg (19). For example, they identified 19 knot proteins using the RANDOM method from the PDB-REPRDB data set (22) comprising 4716 representative protein. However, 5 of the identified 19 knotted proteins (1T0H:B, 1GKU:B, 1U2Z:C, 1M72:B and 1XI4:C) are questionable, since all of them have very large gaps in their structures due to missing residues. These knots arise either from the artificial virtual bonds that are used to connect the gaps or from the nonstandard PDB format. For example, 1T0H:B(23) has missing residues 414–424. A knot will form only if a virtual bond of length 32 Å connects the structural gap; 1U2Z:C has missing residues 570–573 and 575. The total distance of the structural gaps is around 52 Å. If these chain breaks were connected by virtual bonds, there will be a 41 knot. However, we notice that there is a chain in the complex (i.e. 1U2Z:A), which has identical sequence with 1U2Z:C and does not have a knot even if the structural gaps are connected by virtual bonds.
CONCLUSION
Here we have presented the first web server to detect knots in proteins. With an increasing number of proteins with knots deposited in PDB, we believe that the pKNOT web server will be useful to both biologists in general and structural biologists in particular.
ACKNOWLEDGEMENTS
This work is supported in part by the National Science Council, the National Research Program of Genomic Medicine, and the MoE ATU plan. We are grateful to both the hardware and software supports of the Structural Bioinformatics Core Facility at National Chiao Tung University. We are also grateful to Dr. W. R. Taylor for providing the knot-detecting codes. Funding to pay the Open Access publication charge was provided by the MoE ATU plan.
Conflict of interest statement. None declared.
REFERENCES
- 1.Ahn HJ, Eom SJ, Yoon HJ, Lee BI, Cho H, Suh SW. Crystal structure of class I acetohydroxy acid isomeroreductase from Pseudomonas aeruginosa. J. Mol. Biol. 2003;328:505–515. doi: 10.1016/s0022-2836(03)00264-x. [DOI] [PubMed] [Google Scholar]
- 2.Ahn HJ, Kim HW, Yoon HJ, Lee BI, Suh SW, Yang JK. Crystal structure of tRNA(m1G37)methyltransferase: insights into tRNA recognition. EMBO J. 2003;22:2593–2603. doi: 10.1093/emboj/cdg269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Elkins PA, Watts JM, Zalacain M, van Thiel A, Vitazka PR, Redlak M, Andraos-Selim C, Rastinejad F, Holmes WM. Insights into catalysis by a knotted TrmD tRNA methyltransferase. J. Mol. Biol. 2003;333:931–949. doi: 10.1016/j.jmb.2003.09.011. [DOI] [PubMed] [Google Scholar]
- 4.Lim K, Zhang H, Tempczyk A, Krajewski W, Bonander N, Toedt J, Howard A, Eisenstein E, Herzberg O. Structure of the YibK methyltransferase from Haemophilus influenzae (HI0766): a cofactor bound at a site formed by a knot. Proteins. 2003;51:56–67. doi: 10.1002/prot.10323. [DOI] [PubMed] [Google Scholar]
- 5.Mosbacher TG, Bechthold A, Schulz GE. Structure and function of the antibiotic resistance-mediating methyltransferase AviRb from Streptomyces viridochromogenes. J. Mol. Biol. 2005;345:535–545. doi: 10.1016/j.jmb.2004.10.051. [DOI] [PubMed] [Google Scholar]
- 6.Wagner JR, Brunzelle JS, Forest KT, Vierstra RD. A light-sensing knot revealed by the structure of the chromophore-binding domain of phytochrome. Nature. 2005;438:325–331. doi: 10.1038/nature04118. [DOI] [PubMed] [Google Scholar]
- 7.Liu J, Wang W, Shin DH, Yokota H, Kim R, Kim SH. Crystal structure of tRNA (m1G37) methyltransferase from Aquifex aeolicus at 2.6 A resolution: a novel methyltransferase fold. Proteins. 2003;53:326–328. doi: 10.1002/prot.10479. [DOI] [PubMed] [Google Scholar]
- 8.Forouhar F, Shen J, Xiao R, Acton TB, Montelione GT, Tong L. Functional assignment based on structural analysis: crystal structure of the yggJ protein (HI0303) of Haemophilus influenzae reveals an RNA methyltransferase with a deep trefoil knot. Proteins. 2003;53:329–332. doi: 10.1002/prot.10510. [DOI] [PubMed] [Google Scholar]
- 9.Badger J, Sauder JM, Adams JM, Antonysamy S, Bain K, Bergseid MG, Buchanan SG, Buchanan MD, Batiyenko Y, et al. Structural analysis of a set of proteins resulting from a bacterial genomics project. Proteins. 2005;60:787–796. doi: 10.1002/prot.20541. [DOI] [PubMed] [Google Scholar]
- 10.Shi D, Morizono H, Yu X, Roth L, Caldovic L, Allewell NM, Malamy MH, Tuchman M. Crystal structure of N-acetylornithine transcarbamylase from Xanthomonas campestris: a novel enzyme in a new arginine biosynthetic pathway found in several eubacteria. J. Biol. Chem. 2005;280:14366–14369. doi: 10.1074/jbc.C500005200. [DOI] [PubMed] [Google Scholar]
- 11.Tyagi R, Duquerroy S, Navaza J, Guddat LW, Duggleby RG. The crystal structure of a bacterial class II ketol-acid reductoisomerase: domain conservation and evolution. Protein Sci. 2005;14:3089–3100. doi: 10.1110/ps.051791305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Misaghi S, Galardy PJ, Meester WJ, Ovaa H, Ploegh HL, Gaudet R. Structure of the ubiquitin hydrolase UCH-L3 complexed with a suicide substrate. J. Biol. Chem. 2005;280:1512–1520. doi: 10.1074/jbc.M410770200. [DOI] [PubMed] [Google Scholar]
- 13.Das C, Hoang QQ, Kreinbring CA, Luchansky SJ, Meray RK, Ray SS, Lansbury PT, Ringe D, Petsko GA. Structural basis for conformational plasticity of the Parkinson's disease-associated ubiquitin hydrolase UCH-L1. Proc. Natl Acad. Sci. USA. 2006;103:4675–4680. doi: 10.1073/pnas.0510403103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Komoto J, Yamada T, Takata Y, Markham GD, Takusagawa F. Crystal structure of the S-adenosylmethionine synthetase ternary complex: a novel catalytic mechanism of S-adenosylmethionine synthesis from ATP and Met. Biochemistry. 2004;43:1821–1831. doi: 10.1021/bi035611t. [DOI] [PubMed] [Google Scholar]
- 15.Taylor WR. A deeply knotted protein structure and how it might fold. Nature. 2000;406:916–919. doi: 10.1038/35022623. [DOI] [PubMed] [Google Scholar]
- 16.Mallam AL, Jackson SE. Folding studies on a knotted protein. J. Mol. Biol. 2005;346:1409–1421. doi: 10.1016/j.jmb.2004.12.055. [DOI] [PubMed] [Google Scholar]
- 17.Taylor WR, Xiao B, Gamblin SJ, Lin K. A knot or not a knot? SETting the record ‘straight’ on proteins. Comput. Biol. Chem. 2003;27:11–15. doi: 10.1016/s1476-9271(02)00099-3. [DOI] [PubMed] [Google Scholar]
- 18.Jacobs SA, Harp JM, Devarakonda S, Kim Y, Rastinejad F, Khorasanizadeh S. The active site of the SET domain is constructed on a knot. Nat. Struct. Biol. 2002;9:833–838. doi: 10.1038/nsb861. [DOI] [PubMed] [Google Scholar]
- 19.Lua RC, Grosberg AY. Statistics of knots, geometry of conformations, and evolution of proteins. PLoS Comput. Biol. 2006;2:e45. doi: 10.1371/journal.pcbi.0020045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lickorish WBR. An Introduction to Knot Theory. New York: Springer; 1997. [Google Scholar]
- 21.Hartshorn MJ. AstexViewer: a visualisation aid for structure-based drug design. J. Comput. Aided Mol. Des. 2002;16:871–881. doi: 10.1023/a:1023813504011. [DOI] [PubMed] [Google Scholar]
- 22.Noguchi T, Akiyama Y. PDB-REPRDB: a database of representative protein chains from the Protein Data Bank (PDB) in 2003. Nucleic Acids Res. 2003;31:492–493. doi: 10.1093/nar/gkg022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Van Petegem F, Clark KA, Chatelain FC, Minor DL., Jr Structure of a complex between a voltage-gated calcium channel beta-subunit and an alpha-subunit domain. Nature. 2004;429:671–675. doi: 10.1038/nature02588. [DOI] [PMC free article] [PubMed] [Google Scholar]