

Abstract
Human vaccination with BioThrax™ requires six injections followed by annual boosters. This makes it difficult for the compliance of the immunization program and underscores the need for development of a new and optimized vaccination protocol. Current research aims to demonstrate the proof of concept to develop a needle free mucosal immunization protocol using a murine anthrax model. A/J mice were immunized with BioThrax™ via an intranasal route. Sera, saliva, vaginal, and nasal washes were evaluated for protective antigen (PA) specific antibody responses by ELISA. Antigen-specific, antibody-secreting lymphocytes were measured by ELISPOT. Sera neutralization antibody titers were determined by in vitro anthrax lethal toxin (Letx) neutralization assay. Immunized animals were challenged by a lethal dose of Bacillus anthracis Sterne spores to determine the efficacy of the vaccination. Nasal mucosal immunization with BioThrax™ elicited robust serum and mucosal antibody responses against PA. The antigen specific antibodies neutralized anthrax Letx, as demonstrated by in vitro neutralization assays. Two doses of intranasal BioThrax™ were sufficient to completely protect A/J mice against challenge with 100×LD 50 Bacillus anthracis Sterne spores. The data suggests that intranasal administration may be an effective immunization modality for an improved immunization program against anthrax.
Keywords: Anthrax Vaccine, Bacillus anthracis, Mucosal Immunization, Protective Immunity
1. Introduction
Anthrax is caused by the Gram-positive, spore-forming, rod-shaped bacterium Bacillus anthracis[1]. Intentional dissemination of B. anthracis spores by bioterrorists recently resulted in 11 cases of cutaneous anthrax and 11 cases of inhalational anthrax, including 5 deaths in the US [2, 3]. This bioterror threat underscores the urgent need for the development of an improved immunization program against anthrax.
Protective antigen (PA), one of the anthrax toxin components, has been shown to be an essential component of an anthrax vaccine [4, 5]. Interestingly, researchers have found that anti-PA antibody specific immunity has anti-spore activity and might play a role in impeding the early stages of infection with B. anthracis spores [6]. The current US human anthrax vaccine, BioThrax™ (Anthrax Vaccine Adsorbed/AVA, BioPort Corporation, Lansing, MI), consists of aluminum hydroxide-adsorbed supernatant material from fermentor cultures of a toxigenic, non-encapsulated strain of B. anthracis. It contains primarily protective antigen (PA) and undefined quantities of lethal factor (LF) and edema factor (EF), the other two anthrax toxin components. Human vaccination with AVA requires six immunizations followed by annual boosters [7, 8]. 1% systemic and 3.6% local adverse events in humans occur with vaccine injections [9].
In addition to PA, LF and EF may play an important role in providing immunity [10-14]. Immunization with solely somatic components of B. anthracis, such as surface polysaccharides and cell-associated antigens EA1 and EA2, has not provided protective immunity [15]. However, studies of spore vaccines suggest that some other B. anthracis antigens may contribute in a significant manner to protective immunity [16, 17].
Although neutralizing antibodies to anthrax toxins addresses the toxemia component of anthrax disease, immune responses to additional virulence factors or somatic antigens in the spore and vegetative cell may be critical to the development of complete immunity to anthrax exposure. An improved vaccine should be designed to target both toxemia and septicemia [4, 18]. It has been reported that the BioThrax™ products contain some somatic antigens and are able to evoke host immune responses against the somatic components of B. anthracis[19]. Therefore, BioThrax™ may be able to target both toxemia and septicemia. Thus, BioThrax™ may have better protective efficacy against anthrax in comparison with that of the recombinant PA (rPA-based) vaccine currently under development.
Evidence has shown that mucosal, but not parenteral, immunization induces immune responses in both systemic and secretory immune compartments [20]. Thus, a vaccine administered parenterally, such as the BioThrax™ vaccine, most likely would not induce mucosal immunity against inhalational anthrax. Therefore, we have studied intranasal mucosal immunization with human anthrax vaccine and demonstrated that this immunization method may provide an efficacious and more acceptable vaccine strategy to prevent multi-type anthrax disease states.
2. Materials and methods
2.1. Materials
Protective antigen (PA) and lethal factor (LF) were purchased from List Biological Laboratories, Inc. (Campbell, CA). Biothrax™ vaccine (AVA) was produced by BioPort Corporation (Lansing, MI). B. anthracis Sterne strain spores were from an anthrax spore vaccine, which is a viable suspension of the Sterne strain 34F2 spores in saponin (Colorado Serum Company, Denver, CO). Mouse monocyte macrophage cell line J774A.1 was purchased from the American Type Culture Collection (Manassas, VA).
2.2. Animals, treatments, and sample collection
Female A/J mice at 6 - 8 weeks old were purchased from Jackson Laboratory (Bar Harbor, ME) and housed under BSL2 pathogen-free conditions in the vivarium facility at the University of Rochester Medical Center. The animal research herein reported was conducted in facilities with programs accredited by the Association for the Assessment and Accreditation of Laboratory Animal Care.
Experiment 1
After anesthesia with katamine and xylazine, mice (8 mice/group) were immunized intranasally by instillation with different doses of BioThrax™ (7.5, 15, and 30 μl) in treatment groups or 30 μl physiological saline in control groups. Animals were immunized once (week 0), twice (weeks 0, 3), or three times (weeks 0, 3, and 5). Animal sera were obtained by retro-orbital bleeding at weeks 0, 3, 5, 7 and stored at -20°C until use. Immunized animals were challenged by subcutaneous injection with a dose of approximately 100 times the 50% lethal dose (LD50) [21], 1.85 × 105 spores of B. anthracis Sterne, eight weeks after the first immunization when serum anti-PA antibody level was at its peak time after three time immunizations.
The animals were monitored four times a day for one week after spore challenge and twice a day thereafter. The endpoint for the spore challenge experiment was the occurrence of severe anthrax symptoms or two weeks after spore injection. Any animals that developed severe symptoms of anthrax, such as ruffled coat, hunching, shivering, uncoordinated movements, dehydration, respiratory difficulties, massive edema, and skin lesions, were euthanized promptly during the two weeks.
Experiment 2
Mice were allotted into treatment groups A, B, and C (6 mice/group) and the corresponding control groups A’, B’, and C’ (4 mice/group), respectively. The animals were immunized by the intranasal route with 30 μl of BioThrax™ once (week 0) in group A, twice (weeks 0, 3) in group B, and 3 times (weeks 0, 3, 5) in group C. The control groups were administered 30 μl physiological saline accordingly. Saliva, nasal wash, vaginal wash, and spleen samples were collected at week 3 from groups A and A’, at week 5 from groups B and B’, and at week 7 from groups C and C’. After the animals were injected intraperitoneally with 10 μg carbachol (Sigma, St. Louis, MO) in 0.1 ml 0.9% NaCl solution to stimulate flow, saliva were collected by means of a 200-pld pipettor fitted with a plastic tip [22]. After collection of saliva, the animals were sacrificed. 50 μl 0.9% NaCl solution was instilled into the vaginal and nasal cavities to collect vaginal and nasal wash samples, respectively. Then, spleens were obtained to isolate lymphocytes for ELISPOT assay.
2.3. Quantitative ELISA for measurement of serum antibody concentration
Serum anti-PA IgG, IgG1, IgG2a, and IgA antibody concentrations were determined using ELISA Quantitation Kits (Bethel Lab. Inc., Montgomery TX) with a modified procedure. Briefly, 96-well flat-bottom immuno plates (Nalge Nunc International, Rochester, NY) were coated with 1 μg/well capture antibodies (goat anti-mouse IgG-, or IgG1-, or IgG2a-, or IgA-affinity purified) for standard curve or 100 ng/well PA (List Biological Laboratories, Inc.) for sera samples antibody concentration measurement in 100 μl coating buffer (0.05 M carbonate-bicarbonate buffer, pH 9.6) and incubated overnight at 4°C. The plates were then washed five times with washing buffer (0.05% Tween 20 in PBS) and blocked with 200 μl PBS (pH 7.4) containing 1% bovine serum albumin (BSA) for 1 h at room temperature. After five washes, 100 μL serial dilutions of reference serum containing the given amount of mouse antibodies (for standard curve) or 1:100 dilutions of mouse sera samples in PBS (pH 7.4) containing 0.05% Tween 20 and 1% BSA were added to each well, and this was incubated for 2 h at 37°C. The plates were washed with washing buffer five times and incubated with 100 μl/well of 1:10,000 dilution of goat anti-mouse IgG, IgG1 or IgG2a conjugated to alkaline phosphates for 1 h at room temperature. Unbound antibodies were removed by washing five times with washing buffer, and the bound antibody was detected following incubation with p-nitrophenylphosphate phosphatase substrate system (KPL, Gaithersburg, MD) for 30 min. The reaction was stopped by adding 0.5M EDTA, and the absorbance values were obtained using a Dynatech MR4000 model microplate reader at 405 nm. A standard curve was generated for each set of samples, and serum antibody concentrations were calculated in accordance to the standard curve.
2.4. Measurement of anthrax lethal toxin neutralizing antibody titer
The Letx (LF + PA) neutralization was measured by the ability of sera to neutralize the cytotoxicity of Letx for J774A.1 mouse macrophage cells. This colorimetric toxin neutralization assay (TNA) was performed as described previously, except that pooled sera (2 mice/pool) were used in the assays [14]. The Letx-neutralizing antibody titers of individual serum, calculated by linear regression analysis, were expressed as the reciprocal of the antibody dilution preventing 50% of cell death [23].
2.5. Determination of antibody forming cells (AFCs) by enzyme-linked immunospot assay (ELISPOT)
ELISPOT was performed as previously described with modifications [20, 24]. Multiscreen-IP plates (Millipore Corp., Billerica, MA) were pre-wetted with 35% ethanol, washed with PBS, and coated with either PA or BSA (Promega, Madison, WI) at 5 μg/ml, and incubated overnight at 4°C. The plates were then decanted and washed with PBS. 200 μl of Rg-10 medium containing RPMI 1640 (with L-glutamine but no phenol red from Invitrogene), antibiotics (100 IU Penicillin, 100 μg/ml Streptomycin, and 0.25 µg/ml Amphotericin B), 10% heat-inactivated FBS (Hyclone, Logan, UT), and 50 µM β-mercaptoethanol were added into each well and incubated at 37°C and 5% CO2for at least 2 h until use. After the control (4 mice/group) or immunized mice (6 mice/group) were sacrificed, their spleens were collected and placed in ice-cold Rg-10 medium. Spleens were mashed through two meshed screens, collected in 15 ml Rg-10, and centrifuged at 1000 RPM for 10 min at room temperature. To lyse the red blood cells, the pellet was resuspended into 1 ml sterile ACK solution (150 mM NH4Cl, 10 mM KHCO3, 0.1 mM Na2 EDTA, pH 7.3). This suspension was raised to 25 ml with Rg-10 and then centrifuged as above. The pellet of lymphocytes was resuspended and centrifuged three more times and finally resuspended into 1.5 ml Rg-10. Cells were counted and tested for viability using the trypan blue exclusion assay. Cells were appropriately diluted so that 5 × 105 cells per well could be added by delivering 100 μl cells/well. Cells were then incubated at 37°C, 5% CO 2 for 5 h, removed, and washed 5 times with PBS + 0.1% Tween (PBST). HRP- conjugated antibodies goat anti mouse IgA and IgG (Zymed/Invitrogen) along with IgG1 and IgG2a (Bethyl Laboratories, Montgomery, Texas) were mixed in PBS + 0.1% Tween + 2% BSA (PBSTB) and filtered with an Acrodisc 25 mm PF filter and added to wells at 1 μg/ml. The plates and antibody were incubated overnight at 4°C. The following morning, the plates were washed 4 times with PBST and then once with PBS. 50 μl HRP substrate TrueBlue™ (KPL, Gaithersburg, MD) was added according to manufacturer’s protocol. The plates were rinsed and allowed to dry for at least one day, read on a CTL Immunospot reader (CTL, Cleveland OH), and analyzed using the Immunospot 3 program (CTL). Each well was run in triplicate, and the negative control (IP plate coated with BSA) was subtracted from the experimental during analysis.
2.6. Statistical analysis
Antibody concentration and ELISPOT data among different groups at different time points were compared and analyzed using the Fisher LSD test, ANOVA; survival analysis was performed by Time Log-Rank Test. In comparing groups, P-values < 0.05 were considered significantly different. All statistical analysis was performed using Statistica 7.1 software from StatSoft, Inc. (Tulsa, OK).
3. Results
3.1. Sera antibody response after intranasal immunization with BioThrax™
Table 1 summarizes serum antibody responses against PA after single and multiple immunizations with different dosages of BioThrax ™. As expected, the anti-PA serum antibody responses appear dose-dependent. A single intranasal immunization with BioThrax ™ even at the lowest dosage tested (7.5 μl) elicited robust anti-PA IgG, IgG1, and IgG2a response 3 weeks after immunization in comparison with the control (P < 0.05). The antibody levels reached their peaks at about 5 weeks after vaccine inoculation. The rise in antigen specific IgG1 and IgG2a after intranasal immunization with BioThrax™ suggested that both Th2 and Th1 immune responses, with predominant Th2 responses against PA were elicited (values of IgG2a/IgG1 < 1.0). The 15 and 30 μl dosage groups produced significantly high anti-PA total IgG responses compared to the 7.5 μl dosage group at week 5. However, no significant difference was found in the anti-PA total IgG responses between the 15 and 30 μl dosage groups (P = 0.079).
Table 1.
Summary of anti-PA antibody concentrations in sera from immunized A/J mice with BioThrax™
| Anti-PA Antibody | Time | One Immunization | Two Immunizations | Three Immunizations | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| 7.5 μl | 15 μl | 30 μl | 7.5 μl | 15 μl | 30 μl | 7.5 μl | 15 μl | 30 μl | ||
| IgG | 0 wk | 0.06 ± 0.00aX | 0.07 ± 0.01aX | 0.06 ± 0.00aX | 0.06 ± 0.00aX | 0.06 ± 0.01aX | 0.06 ± 0.01aX | 0.05 ± 0.00aX | 0.06 ± 0.01aX | 0.05 ± 0.00aX |
| 3 wk | 3.86 ± 0.30aX | 5.95 ± 0.58aX | 6.61 ± 0.57aX | 4.04 ± 0.50aX | 4.75 ± 0.47aX | 5.64 ± 0.86aX | 3.67 ± 0.41aX | 4.41 ± 0.40aX | 6.15 ± 0.63aX | |
| 5 wk | 5.09 ± 0.55aX | 8.11 ± 0.67bX | 12.24 ± 1.12bX | 12.77 ± 1.38aY | 36 .1 ± 3.18bY | 38.70 ± 3.11bY | 15.23 ± 1.76aY | 33.20 ± 2.42bY | 41.59 ± 2.08cY | |
| 7 wk | 3.87 ± 0.43aX | 4.21 ± 0.44aX | 7.65 ± 1.15aX | 19.15 ± 1.54aY | 37.94 ± 2.85bY | 50.17 ± 2.68cY | 27.14 ± 1.87aZ | 42.63 ± 3.15bZ | 59.22 ± 2.25cZ | |
|
| ||||||||||
| IgG1 | 0 wk | 0.09 ± 0.01aX | 0.09 ± 0.01aX | 0.08 ± 0.01aX | 0.10 ± 0.02aX | 0.08 ± 0.01aX | 0.08 ± 0.01aX | 0.08 ± 0.01aX | 0.08 ± 0.01aX | 0.08 ± 0.01aX |
| 3 wk | 3.31 ± 0.53aX | 5.52 ± 0.36aX | 6.12 ± 0.74aX | 3.62 ± 0.40aX | 3.92 ± 0.45aX | 4.99 ± 0.65aX | 3.73 ± 0.37aX | 4.04 ± 0.49aX | 3.73 ± 1.00aX | |
| 5 wk | 4.48 ± 0.37aX | 6.81 ± 0.28aX | 10.49 ± 0.74aX | 11.01 ± 1.03aY | 27.13 ± 1.71bY | 30.38 ± 1.61cY | 10.78 ± 1.26aY | 27.0 ± 2.58bY | 29.59 ± 1.33bY | |
| 7 wk | 3.67 ± 0.40abX | 3.28 ± 0.36bX | 6.61 ± 1.00aX | 16.26 ± 1.21aY | 28.58 ± 1.87bY | 36.38 ± 2.75cY | 21.55 ± 1.65aZ | 33.2 ± 2.52bZ | 49.55 ± 1.75cZ | |
|
| ||||||||||
| IgG2a | 0 wk | 0.08 ± 0.01aX | 0.07 ± 0.01aX | 0.08 ± 0.02aX | 0.08 ± 0.01aX | 0.08 ± 0.01aX | 0.07 ± 0.01aX | 0.09 ± 0.02aX | 0.08 ± 0.01aX | 0.07 ± 0.01aX |
| 3 wk | 0.43 ± 0.04aX | 0.66 ± 0.06aX | 1.18 ± 0.25aX | 0.40 ± 0.05aX | 0.68 ± 0.08aX | 0.90 ± 0.12aX | 0.72 ± 0.18aX | 0.95 ± 0.15aX | 0.87 ± 0.13aX | |
| 5 wk | 0.68 ± 0.13aXY | 0.61 ± 0.08aX | 1.48 ± 0.23aX | 0.35 ± 0.04aX | 2.53 ± 0.40bY | 5.92 ± 0.82cY | 1.30 ± 0.15aY | 3.33 ± 0.34aY | 6.02 ± 0.58bY | |
| 7 wk | 0.23 ± 0.02aX | 0.47 ± 0.06aX | 0.98 ± 0.21aX | 0.35 ± 0.05aX | 3.86 ± 0.45bY | 6.46 ± 0.76cY | 2.36 ± 0.30aY | 5.45 ± 0.62bZ | 8.88 ± 0.88cZ | |
NOTE: Antibody responses were measured by ELISA after 1 (week 0), 2 (weeks 0, 3), and 3 (weeks 0, 3, 5) immunization with a dosage of 7.5, 15, or 30 μl of BioThrax™ . Antibody concentrations are expressed as Mean ± SE, mg/ml (n = 8).Values in columns without the same letters (a, b, c) differ significantly between different dosages in the same immunization schedule (P < 0.05). Values in columns without same capital letters (X,Y,Z) differ significantly between different immunization schedules in the same dosage (P < 0.05). Data were analyzed using Fisher LSD test, and ANOVA by STATISTIA 7.1 software (StatSoft, Inc., Tulsa, OK)
In order to show that booster immunization could enhance the anti-PA antibody responses, we also immunized animals 2 and 3 times (Table 1). The first booster immunization with all three dosages significantly enhanced anti-PA IgG, IgG1 and IgG2a responses at week 5 compared to a single immunization (P < 0.05); the second booster further significantly enhanced anti-PA IgG, IgG1, and IgG2a immune responses at week 7 (P < 0.05).
3.2. Anti-PA IgA in mucosal secretions
To determine if intranasal mucosal immunization with BioThrax™ is able to elicit antigen-specific mucosal immunity, we measured secretory anti-PA antibody levels in saliva, nasal, and vaginal washes from immunized animals. Anti-PA IgA concentrations in mucosal secretions after 1-, 2-, and 3-time immunization with a dosage of 30 μl vaccine are shown in Fig. 1. Significant mucosal anti-PA IgA responses after a single intranasal immunization with BioThrax™ were observed (week 3), and booster immunizations (week 5 and 7) further significantly enhanced mucosal IgA responses (P < 0.05).
Fig.1. Anti-PA IgA concentration in saliva, nasal and, vagina washes.

Mice were immunized intranasally with 30 μl of BioThrax™ once (week 0), twice (weeks 0, 3), or three times (weeks 0, 3, 5). Saliva, nasal wash, and vaginal wash samples were collected at week 3, 5, and 7 to determine anti-PA IgA concentration by quantitative ELISA. (BioThrax™, n = 6; control, n = 4)
3.3. Antigen specific antibody secreting cells (ASC) by ELISPOT assay
A significant increase in the number of IgG, IgG1, and IgG2a antigen specific antibody secreting lymphocytes was measured upon stimulation with PA (Fig. 2), consistent with the ELISA data shown in Table 1. A high percentage of PA-specific IgA secreting cells were also identified, indicating that mucosal IgA responses were elicited by intranasal immunization. The number of PA-specific, antibody-secreting cells increased when booster immunizations were given.
Fig. 2. Determination of PA specific antibody forming cells by ELISPOT assay.
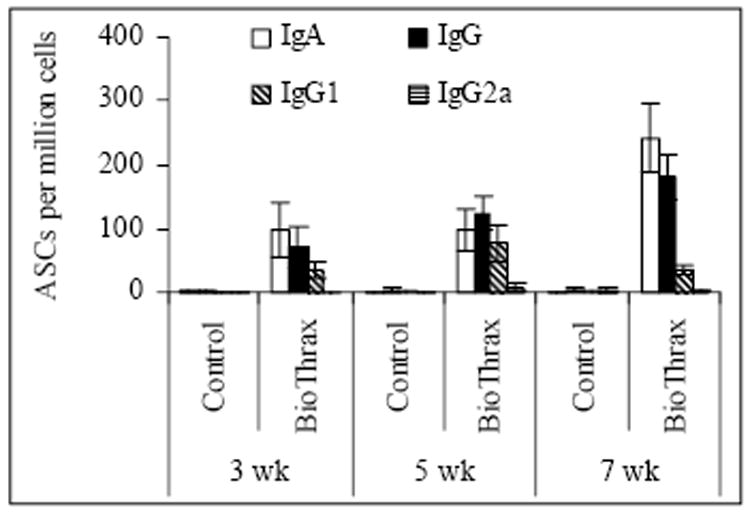
After mice were immunized intranasally with 30 μl of BioThrax™ once (week 0), twice (weeks 0, 3), or three times (weeks 0, 3, 5), they were sacrificed for collection of spleen tissue samples for ELISPOT assay. (BioThrax™, n = 6; control, n = 4)
3.4. In vitro protection against anthrax lethal toxin
Anti-PA antibodies in the sera were capable of neutralizing anthrax Letx using an in vitro protection assay. As shown in figure 3 (insert part), a single intranasal immunization with a dosage of 7.5, 15, or 30 μl of BioThrax™ resulted in Letx neutralization antibody titers (geomean) up to 11, 32, and 160, respectively. A dose-dependent toxin neutralization antibody response was observed, and booster immunization further significantly enhanced Letx neutralization antibody titers to 38, 1280, and 1522, respectively (P < 0.05), as did a second booster (Fig. 3).
Fig. 3. Neutralization of anthrax lethal toxin by sera from mice immunized with BioThrax™.

Serial 2-fold dilutions of sera were pre-incubated with recombinant LF and PA, and then added to J774A.1 mouse macrophage cells. The cell viability was measured by colorimetric toxin neutralization assay (TNA). The neutralization titers were expressed as the reciprocal of antibody dilution preventing 50% of cell death. (n = 4, 2 mice/sera pool, 4 pools)
3.5. Protective immunity elicited by intranasal immunization with BioThrax™
A single intranasal immunization with 7.5, 15, and 30 μl BioThrax™ partially protected mice against the anthrax spore challenge (Fig. 4A), with protection rates of 50%, 50%, and 62%; one additional booster immunization resulted in 100% protection with the 15 μl and 30 μl immunization dosages, while one booster immunization with 7.5 μl vaccine raised the protection rate from 50% to 62% (Fig. 4B). A second booster immunization in 7.5 μl immunization dosage group increased the protection rate from 62% to 85% (figure 4C).
Fig. 4. Protection against B. anthracis spore challenge in immunized mice .
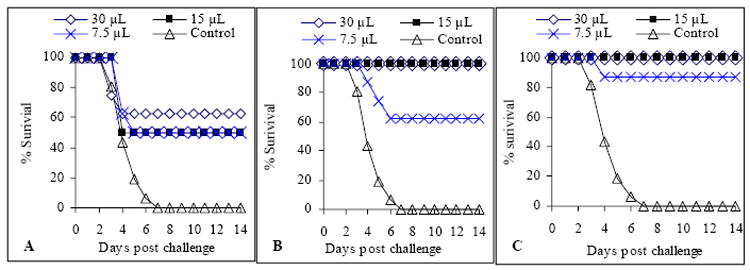
Mice were immunized with BioThrax™ once (week 0, Panel A), twice (weeks 0, 3, Panel B), or three times (weeks 0, 3, 5, Panel C), and then challenged with 100 × LD50 B. anthracis Sterne spore in week 8. Mice in control groups were injected with physiological saline three times (weeks 0, 3, 5). (n = 8)
4. Discussion
We have shown that an intranasal immunization with BioThrax™ is capable of eliciting significant Th2 and Th1 immune responses, with predominant Th2 responses, against PA and that the anti-PA antibodies are capable of neutralizing the toxicity of Letx. Intranasal mucosal immunization with BioThrax™ elicited mucosal secretory IgA responses as well. This resulted in host protective immunity against B. anthracis spore challenge. The protective immune response appeared to be dose-dependent and boostable. This is the first report to show the protective efficacy of needle-free intranasal mucosal immunization with 1-3 doses of human anthrax vaccine.
B. anthracis spores may invade the host via the mucosal route and cause inhalational anthrax which is the major threat when B. anthracis spores are used as a biologic weapon. Previous studies suggest that the upper respiratory tract and other mucosal tissues are affected by inhalational anthrax [2, 25-27]. Therefore, host immunity at the mucosal sites of entry may be effective for protection against inhalational anthrax. Previous studies by Welkos et al. have demonstrated that anti-PA antibodies stimulate spore uptake and interfere with germination in macrophage culture and animal models [6, 28]. This suggests a potentially protective role of anti-PA antibodies at mucosal surfaces against anthrax spore infection. Mucosal, but not parenteral, immunization with PA induces immune responses in both systemic and secretory immune compartments [20]. Our study demonstrates that intranasal immunizations with BioThrax™ elicit robust mucosal and systemic antibody responses against PA. However, more experiments are needed to determine if intranasal immunization with BioThrax™ can provide protection against inhalational anthrax.
The US Department of Health and Human Services has stocked millions of doses of BioThrax™ for emergency preparedness against bioterrorism. In addition, the US Department of Defense has a mandatory program to immunize active duty service personnel against anthrax. The current immunization program with BioThrax™ requires multiple injections by trained personnel. Therefore, considerable medical resources need to be devoted to the immunization program, especially in the case of a mass immunization against potential bioterrorism attack. This makes it difficult for the compliance of the immunization program and underscores the need for development of alternative and optimized vaccination protocols. Recently, Kenny and colleagues demonstrated that a needle-free transcutaneous immunization (TCI) with recombinant PA plus the heat-labile toxin of Escherichia coli protects mice against inhalational anthrax [29]. Current study has shown that a mucosal immunization protocol provides an alternative for immunization against anthrax. Since the anthrax vaccine can be reformulated into a nasal spray or skin patch, self-administration of the vaccine becomes possible. These will be simple and more comfortable ways for administration of the vaccine
The research reported here is a proof of concept that an alternative human immunization strategy against anthrax is possible. Our next steps will be to study the duration of the protective immunity elicited by mucosal immunization in comparison with injection and assess antibody responses in bronchoalveoar lavages (BALs). We will also determine if intranasal immunization with BioThrax™ can protect against inhalational anthrax using guinea pig and nonhuman primate models. In addition, potential side effects of intranasal immunization with BioThrax™ will also need to be addressed. If further studies in animal models are encouraging, we will pursue a clinical trial to determine if humans can be immunized with BioThrax™ by needle-free intranasal mucosal administration.
Acknowledgments
This work was supported by the US Public Service research grant R03AI053598 (M. Z.) from the National Institute of Allergy and Infectious Diseases. M. E. P. was supported by National Institutes of Health contract VTEU N01-AI-25460. The authors would like to thank Eric D. Hesek for performing ELISPOT assay. We are grateful to Mary C. Zeng for careful editing of the manuscript. We also appreciate the support from John J. Treanor, Barbara H. Iglewski, Stephen Dewhurst, and Robert C. Rose. The US Department of Defense provided BioThrax™ (AVA) for our research.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Mock M, Fouet A. Anthrax. Annu Rev Microbiol. 2001;55:647–71. doi: 10.1146/annurev.micro.55.1.647. [DOI] [PubMed] [Google Scholar]
- 2.Guarner J, Jernigan JA, Shieh WJ, Tatti K, Flannagan LM, Stephens DS, et al. Pathology and pathogenesis of bioterrorism-related inhalational anthrax. Am J Pathol. 2003;163(2):701–9. doi: 10.1016/S0002-9440(10)63697-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Shieh WJ, Guarner J, Paddock C, Greer P, Tatti K, Fischer M, et al. The critical role of pathology in the investigation of bioterrorism-related cutaneous anthrax. Am J Pathol. 2003;163(5):1901–10. doi: 10.1016/S0002-9440(10)63548-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Brey RN. Molecular basis for improved anthrax vaccines. Adv Drug Deliv Rev. 2005;57(9):1266–92. doi: 10.1016/j.addr.2005.01.028. Epub 2005 Apr 21. [DOI] [PubMed] [Google Scholar]
- 5.Mahlandt BG, Klein F, Lincoln RE, Haines BW, Jones WI, Jr, Friedman RH. Immunologic studies of anthrax. IV. Evaluation of the immunogenicity of three components of anthrax toxin. J Immunol. 1966;96(4):727–33. doi: 10.21236/ad0640357. [DOI] [PubMed] [Google Scholar]
- 6.Welkos S, Little S, Friedlander A, Fritz D, Fellows P. The role of antibodies to Bacillus anthracis and anthrax toxin components in inhibiting the early stages of infection by anthrax spores. Microbiology. 2001;147(Pt 6):1677–85. doi: 10.1099/00221287-147-6-1677. [DOI] [PubMed] [Google Scholar]
- 7.Anthrax Vaccine Adsorbed (BioThrax™) Product Insert . Lansing, Michigan: Bioport Corporation; 2002. January 31, [Google Scholar]
- 8.Friedlander AM, Pittman PR, Parker GW. Anthrax vaccine: evidence for safety and efficacy against inhalational anthrax. Jama. 1999;282(22):2104–6. doi: 10.1001/jama.282.22.2104. [DOI] [PubMed] [Google Scholar]
- 9.Pittman PR, Gibbs PH, Cannon TL, Friedlander AM. Anthrax vaccine: short-term safety experience in humans. Vaccine. 2001;20(56):972–8. doi: 10.1016/s0264-410x(01)00387-5. [DOI] [PubMed] [Google Scholar]
- 10.Pezard C, Weber M, Sirard JC, Berche P, Mock M. Protective immunity induced by Bacillus anthracis toxin-deficient strains. Infect Immun. 1995;63(4):1369–72. doi: 10.1128/iai.63.4.1369-1372.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Little SF. Anthrax vaccines: a development update. BioDrugs. 2005;19(4):233–45. doi: 10.2165/00063030-200519040-00003. [DOI] [PubMed] [Google Scholar]
- 12.Price BM, Liner AL, Park S, Leppla SH, Mateczun A, Galloway DR. Protection against anthrax lethal toxin challenge by genetic immunization with a plasmid encoding the lethal factor protein. Infect Immun. 2001;69(7):4509–15. doi: 10.1128/IAI.69.7.4509-4515.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Galloway D, Liner A, Legutki J, Mateczun A, Barnewall R, Estep J. Genetic immunization against anthrax. Vaccine. 2004;22(1314):1604–8. doi: 10.1016/j.vaccine.2003.09.043. [DOI] [PubMed] [Google Scholar]
- 14.Zeng M, Xu Q, Hesek ED, Pichichero ME. N-fragment of edema factor as a candidate antigen for immunization against anthrax. Vaccine. 2006 Jan 30;24(5):662–70. doi: 10.1016/j.vaccine.2005.08.056. [Epub 2005 Aug 26] [DOI] [PubMed] [Google Scholar]
- 15.Ezzell JW, Jr, Abshire TG. Immunological analysis of cell-associated antigens of Bacillus anthracis. Infect Immun. 1988;56(2):349–56. doi: 10.1128/iai.56.2.349-356.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cohen S, Mendelson I, Altboum Z, Kobiler D, Elhanany E, Bino T, et al. Attenuated nontoxinogenic and nonencapsulated recombinant Bacillus anthracis spore vaccines protect against anthrax. Infect Immun. 2000;68(8):4549–58. doi: 10.1128/iai.68.8.4549-4558.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Brossier F, Mock M. Toxins of Bacillus anthracis. Toxicon. 2001;39(11):1747–55. doi: 10.1016/s0041-0101(01)00161-1. [DOI] [PubMed] [Google Scholar]
- 18.Wang JY, Roehrl MH. Anthrax vaccine design: strategies to achieve comprehensive protection against spore, bacillus, and toxin. Med Immunol. 2005;4(1):4. doi: 10.1186/1476-9433-4-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kudva IT, Griffin RW, Garren JM, Calderwood SB, John M. Identification of a protein subset of the anthrax spore immunome in humans immunized with the anthrax vaccine adsorbed preparation. Infect Immun. 2005;73(9):5685–96. doi: 10.1128/IAI.73.9.5685-5696.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Boyaka PN, Tafaro A, Fischer R, Leppla SH, Fujihashi K, McGhee JR. Effective mucosal immunity to anthrax: neutralizing antibodies and Th cell responses following nasal immunization with protective antigen. J Immunol. 2003;170(11):5636–43. doi: 10.4049/jimmunol.170.11.5636. [DOI] [PubMed] [Google Scholar]
- 21.Welkos SL, Keener TJ, Gibbs PH. Differences in susceptibility of inbred mice to Bacillus anthracis. Infect Immun. 1986;51(3):795–800. doi: 10.1128/iai.51.3.795-800.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Russell MW, Wu HY. Distribution, persistence, and recall of serum and salivary antibody responses to peroral immunization with protein antigen I/II of Streptococcus mutans coupled to the cholera toxin B subunit. Infect Immun. 1991;59(11):4061–70. doi: 10.1128/iai.59.11.4061-4070.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pitt ML, Little SF, Ivins BE, Fellows P, Barth J, Hewetson J, et al. In vitro correlate of immunity in a rabbit model of inhalational anthrax. Vaccine. 2001;19(32):4768–73. doi: 10.1016/s0264-410x(01)00234-1. [DOI] [PubMed] [Google Scholar]
- 24.Hotomi M, Saito T, Yamanaka N. Specific mucosal immunity and enhanced nasopharyngeal clearance of nontypeable Haemophilus influenzae after intranasal immunization with outer membrane protein P6 and cholera toxin. Vaccine. 16(20):1998. 1950–6. doi: 10.1016/s0264-410x(98)00122-4. [DOI] [PubMed] [Google Scholar]
- 25.Jernigan JA, Stephens DS, Ashford DA, Omenaca C, Topiel MS, Galbraith M, et al. Bioterrorism-related inhalational anthrax: the first 10 cases reported in the United States. Emerg Infect Dis. 2001;7(6):933–44. doi: 10.3201/eid0706.010604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Abramova FA, Grinberg LM, Yampolskaya OV, Walker DH. Pathology of inhalational anthrax in 42 cases from the Sverdlovsk outbreak of 1979. Proc Natl Acad Sci U S A. 1993;90(6):2291–4. doi: 10.1073/pnas.90.6.2291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Vasconcelos D, Barnewall R, Babin M, Hunt R, Estep J, Nielsen C, et al. Pathology of inhalation anthrax in cynomolgus monkeys (Macaca fascicularis) Lab Invest. 2003;83(8):1201–9. doi: 10.1097/01.lab.0000080599.43791.01. [DOI] [PubMed] [Google Scholar]
- 28.Cote CK, Rossi CA, Kang AS, Morrow PR, Lee JS, Welkos SL. The detection of protective antigen (PA) associated with spores of Bacillus anthracis and the effects of anti-PA antibodies on spore germination and macrophage interactions. Microb Pathog. 2005;38(56):209–25. doi: 10.1016/j.micpath.2005.02.001. Epub 2005 Apr 22. [DOI] [PubMed] [Google Scholar]
- 29.Kenney RT, Yu J, Guebre-Xabier M, Frech SA, Lambert A, Heller BA, et al. Induction of Protective Immunity against Lethal Anthrax Challenge with a Patch. J Infect Dis. 2004;190(4):774–82. doi: 10.1086/422694. Epub 2004 Jul 13. [DOI] [PubMed] [Google Scholar]