

Abstract
Middle T antigen (MT) of polyomavirus is known to play an important role in virus-mediated cellular transformation. While MT has been extensively examined in spontaneously immortalized rodent fibroblasts, its interactions with cells of other types and species are less well understood. We have undertaken a cross-species and cross-cell-type comparison of MT-induced transformation in cells with genetically defined backgrounds. We tested the transforming abilities of a panel of MT mutants, Y250F, Y315F, and Y322F, that are selectively mutated in the binding sites for the principal effectors of MT—Src homology 2 domain-containing transforming protein, phosphatidylinositol 3-kinase (PI3K), and phospholipase C-γ, respectively—in fibroblasts and epithelial cells of murine or human origin. We found that the Y315F mutation disabled the ability of MT to induce transformation in all cell types and species tested. While Y315F also failed to activate the PI3K pathway in these cells, genetic evidence has indicated Y315 may make other contributions to transformation. To confirm the role of PI3K, the PIK3CA gene, encoding p110α, the prime effector of PI3K signaling downstream from activated growth factor receptors, was genetically ablated. This abolished the transforming activity of MT, demonstrating the essential role for this PI3K isoform in MT-mediated transformation. The Y250F mutant was able to transform the human, but not the murine, cells that were examined. Interestingly, this mutant fully activates the PI3K pathway in human cells but activated PI3K signaling poorly in the murine cells used in the study. This again points to the importance of PI3K activation for transformation and suggests that the mechanism by which MT activates the PI3K pathway differs in different species.
Polyomavirus (PyV) is a small, double-stranded, closed-circular-DNA virus with an approximately 5-kb genome divided into two roughly equal regions. The late transcripts produce the viral capsid proteins, whereas the early region encodes three so-called tumor (T) antigens (46) that are important for both productive infection and transformation. The 785-amino-acid large T antigen (LT) is a nuclear protein with origin-specific DNA binding properties and an ATPase activity essential for viral replication (46). LT cannot transform cells in culture but has the ability to immortalize primary cells (16, 32). Small t antigen (st) is a 195-amino-acid protein found both in the nucleus and in the cytoplasm (46). It is known to bind and thereby inhibit protein phosphatase 2A (PP2A) (39, 52). Middle T antigen (MT), a 421-amino-acid protein associated with membranes and underlying cytoskeletal elements, has been shown to be essential for transformation of rodent fibroblasts in tissue cultures (20, 34, 47). Mammary epithelial cell-specific expression of MT in transgenic mice results in the induction of multifocal mammary tumors with 100% penetrance (21).
MT functions by providing a platform for the assembly of cellular signaling proteins (11, 26). Like st, it binds to the A and C subunits of PP2A (39, 49), thus replacing the B subunit of PP2A and consequently altering its substrate specificity and localization. Upon association with PP2A, MT recruits the Src family tyrosine kinases c-src, yes, and fyn to the complex, thereby activating their tyrosine kinase activities (1, 7, 9, 10, 18, 23, 30, 31). In turn, MT becomes phosphorylated on three major tyrosine residues, 250, 315, and 322 (24, 41, 42), providing docking sites for the phosphotyrosine binding domain of Src homology 2 domain-containing transforming protein (SHC) (3, 12), the Src homology 2 domain(s) of p85, the adaptor subunit of class IA phosphatidylinositol 3-kinases (PI3Ks), (8, 27, 28, 45, 51), and phospholipase C-γ (PLC-γ) (44), resulting in the activation of the respective downstream signaling cascades. Very recent evidence suggests some additional role for minor tyrosine phosphorylation sites in the MT C-terminal region (6).
Class IA PI3Ks are heterodimeric lipid kinases consisting of a p85 regulatory subunit and a p110 catalytic subunit (reviewed in reference 14). In response to growth factor stimulation and the subsequent activation of receptor tyrosine kinases (RTKs), class IA PI3Ks are recruited to the membrane via interaction of the p85 subunit with phosphotyrosine-containing motifs on the activated receptor. The p110 catalytic subunit of PI3K then catalyzes the phosphorylation of phosphatidylinositol 4,5-bisphosphate to form phosphatidylinositol 3,4,5-trisphosphate. This lipid second messenger in turn activates the serine/threonine kinase AKT and other downstream effectors to regulate multiple cellular functions, including proliferation, survival, and migration. There are three highly homologous p110 catalytic isoforms encoded by three distinct genes, PIK3CA, PIK3CB, and PIK3CD. Earlier studies showed that class IA PI3K was physically and functionally associated with activated RTKs and the transforming activity of MT (27). Recently, cancer genomic sequencing technology has identified that the PIK3CA gene, encoding the p110α isoform, is frequently mutated in a number of the most common forms of cancer, including brain, colon, and breast cancer (40). In addition, recent efforts using gene targeting (15, 54) and chemical inhibitors specific for PI3K isoforms (29) showed that p110α is the primary responsive PI3K for various normal and oncogenic growth factor signals.
The adapter protein SHC serves to couple MT to the Ras/mitogen-activated protein kinase (MAPK) pathway. In transient transfection experiments, MT efficiently activates extracellular signal-regulated kinases (ERKs) (48). However, ERKs have rarely been reported to be phosphorylated on their key activation sites in cells stably expressing MT, perhaps due to the presence of a feedback loop in the MAPK pathway itself or due to signaling from another MT-activated signaling pathway. SHC binds to the so-called NPXY (Asn-Pro-Thr-Tyr) motif in MT, and this binding is interrupted by mutation of the N, P, or Y residue (3, 12, 13). While PLC-γ clearly binds to MT at phosphorylated Tyr-322 (44), previous observations have suggested a minimal role for residue 322 in MT (19).
Studies using MT mutants defective in their binding sites for either SHC or PI3K have indicated that recruitment of both of these signaling proteins is required for efficient transformation under some circumstances. However, conflicting data exist on the importance of each signaling pathway in a given context. For example, substitution of Tyr-315 by phenylalanine at the p85 binding site (Y315F) dramatically reduced polyomavirus-induced transformation in Rat F111 cells (4) and tumorigenesis in mice (17, 45), but expression of the Y315F allele of MT via transfection transforms almost as well as wild-type MT in Rat1 fibroblasts (38). Similarly, mutation of the SHC binding site (Y250F) in MT dramatically reduced transformation in fibroblasts in vitro and tumorigenesis in a mouse mammary tumor virus (MMTV)-MT transgenic model (35, 50). However, the same mutant in the context of PyV showed much more modest and tissue-specific effects on tumorigenesis (2, 53).
In order to examine further the potential differences in the signaling pathways utilized by MT in particular cell types and species, we established well-defined in vitro systems, including (i) human mammary epithelial cells (HMECs), (ii) human mammary fibroblasts (HMFs), (iii) mouse mammary epithelial cells (MMECs), and (iv) mouse embryonic fibroblasts (MEFs), thus enabling us to study the role of MT in fibroblasts versus epithelial cells and mouse versus human cells. We show that wild-type MT induces transformation in all of these cell types when p53 is inactivated by introducing a dominant negative form of p53, p53DD. By use of single binding site-defective MT mutants, we observed that the Y315F mutation blocked MT-induced activation of PI3K signal and transformation in all cell types, regardless of the species. In contrast, MT mutated at the SHC binding site (Y250F) retained transforming activity in human cells but failed to transform both murine fibroblasts and epithelial cells. Notably, the Y250F mutant fully activated PI3K in human cells but poorly engaged in PI3K signaling in murine cells. These data suggest that the mechanism of MT for activating the PI3K signal may differ in the human and murine cells tested in this study. Furthermore, using our recently generated conditional knockout system for the PIK3CA gene, we show that genetic ablation of the p110α isoform of PI3K abolished MT-induced transformation in MEFs, as assayed in both cell culture and xenograft tumor growth in athymic mice, indicating the essential role for p110α in MT-mediated cellular transformation.
MATERIALS AND METHODS
Vectors and retrovirus production.
Wild-type MT and its mutants (Y250F, Y315F, Y322F, and YYY [Y250/315/322F]) were subcloned from their original pLJ vectors into the retrovirus vector pWZL-blasticidin. Retroviruses were produced as previously described (55).
Cell culture.
The culture conditions for HMECs were as described previously (55). The MMEC line HC11 was cultured in RPMI 1640 supplemented with 10% fetal bovine serum (FBS), insulin (5 μg/ml), and epidermal growth factor (5 ng/ml). Both HMFs and MEFs were cultured in Dulbecco's modified Eagle medium (DMEM) supplemented with 10% FBS. Stable cell lines were generated by infection with virus carrying genes encoding p53DD, simian virus 40 (SV40) LT, or various alleles of MT. All infections were carried out in the presence of 4 μg/ml polybrene. After infection, cells were subjected to selection with the appropriate antibiotic (G418, 250 μg/ml; blasticidin, 2.5 μg/ml). Infection frequencies were usually 20 to 40%.
AI growth assays.
For anchorage-independent (AI) growth assays, 5 × 104 (HMEC) or 1 × 105 (HC11, HMF, MEF) cells were seeded per 60-mm plate, with a bottom layer of 0.6% Bacto agar in DMEM and a top layer of 0.3% Bacto agar in the respective growth medium. Fresh growth medium was added after 1.5 weeks. Colony formation was scored after 3 weeks, and only colonies of 0.2 mm in diameter were counted. At least three independent assays were performed in triplicate.
Focus formation assays.
HMFs and MEFs were seeded on 100-mm plates to allow these fibroblasts to reach about 70% to 80% confluence the next day for infection with various retroviruses. At 24 h postinfection, the medium was changed to DMEM supplemented with 2% FBS. Cells were cultured without splitting for 3 weeks, and the growth medium was changed every 3 days. Confluent monolayer cultures with foci were fixed with methanol and subsequently stained with 0.5% crystal violet. The number of foci formed by wild-type MT was defined as 100% and used to normalize between experiments. Three independent assays were performed in duplicate.
Immunoblotting.
Cells were lysed in 20 mM Tris (pH 7.5), 140 mM NaCl, 1 mM EDTA, 1% Nonidet P-40, 10% glycerol, 1 mM sodium vanadate, 0.5 mM dithiothreitol, 1 mM phenylmethylsulfonyl fluoride, 2 μg/ml aprotinin, 2 μg/ml leupeptin, and 2 μg/ml pepstatin. Each lysate containing ∼60 μg of protein was loaded on the gel. Proteins were separated by sodium dodecyl sulfate-10% polyacrylamide gel electrophoresis and transferred to nitrocellulose membranes. These were blocked and incubated with the following primary antibodies: anti-p53 (DO-1 and Ab421; Santa Cruz); anti-MT (18-8) (39); anti-phospho-Akt (Ser473, Thr308); anti-Akt, anti-phospho-ERK, and anti-ERK (all from Cell Signaling Technology); antibodies for c-Myc (sc-764) and p16 (sc-1207) (both from Santa Cruz); and anti-vinculin (Sigma-Aldrich). Infrared-labeled secondary antibodies (Rockland) and the Odyssey System (Licor) were used for detection.
Tumorigenicity assays.
Six- to 8-week old athymic mice (Ncr:Nu/Nu; Taconic Laboratories) were gamma irradiated with a single dose of 400 rads prior to injections. Cells (2 × 106) were resuspended in 50 μl of PBS and 50 μl of Matrigel (Becton Dickinson) and injected subcutaneously into the flanks of mice anesthetized by inhalation of isoflurane. The conditions of the mice and tumor development were monitored daily. Mice were sacrificed when a single tumor reached 1.5 cm in diameter or after 2 months of monitoring. Animal care and protocols were approved by the Institutional Animal Care and Use Committees of Dana-Farber Cancer Institute and Harvard Medical School.
RESULTS
PyV MT complements p53 inactivation to transform telomerase-immortalized human cells.
PyV MT was one of the first viral oncogenes to be isolated as an independent genetic element and studied for its transforming potential (47). However, almost all of our understanding of MT is derived from rodent cell systems. To investigate the transforming activity of MT in human cells, we stably introduced MT into a previously characterized telomerase-immortalized HMEC (hTERT-HMEC) line. Like many cell lines originating from breast epithelium, hTERT-HMECs have spontaneously gained elevated levels of Myc expression and lost expression of p16INK4A (55). We found that MT failed to transform the hTERT-HMECs despite the high level of myc and loss of p16INK4A in these cells (Fig. 1A). Previous work with REF52 cells has suggested that MT can induce an ARF/p53-mediated block to cell division (33). When p53 was inactivated by introducing either SV40 LT or a dominant negative allele of p53 (p53DD) into these epithelial cells, MT potently transformed them, as measured by AI growth in soft agar (Fig. 1A). To examine potential cell-type-specific effects of MT, we also analyzed the effect of MT in a telomerase-immortalized human mammary fibroblast (hTERT-HMF) line (55). Like hTERT-HMECs, these hTERT-HMFs have lost p16INK4A expression, but the level of c-myc is undetectable in these cells (Fig. 1B). Again, MT cooperates with p53 inactivation to induce transformation in human fibroblasts (see Fig. 3).
FIG. 1.
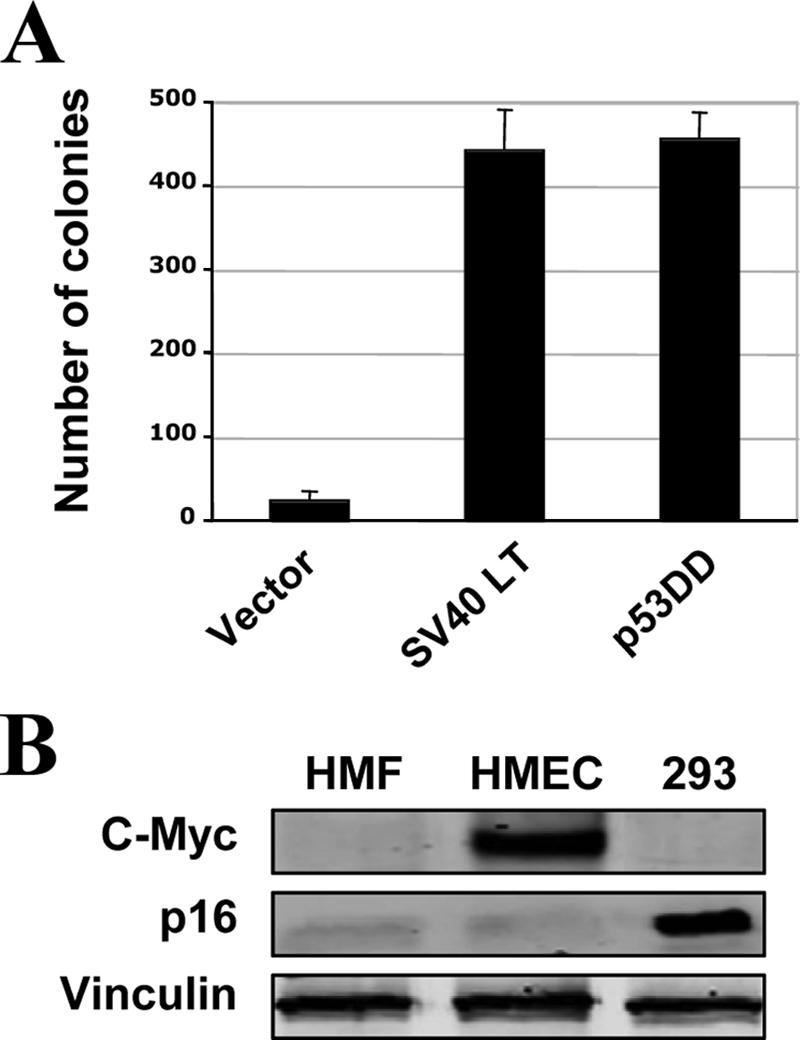
(A) Wild-type MT complements SV40 LT or p53DD to induce colony growth of hTERT-immortalized HMECs in soft agar. Bar graphs reflect averages ± standard deviations for three independent experiments. (B) Western blot analysis of the expression levels of c-Myc and p16INK4A in hTERT-immortalized HMFs. Cell lysates from HMECs immortalized by hTERT, which are known to have high levels of myc and loss of p16INK4A expression, as well as lysates from 293 cells, were used as controls.
FIG. 3.

The PI3K binding site at Y315 is important for MT-mediated transformation in HMFs. (A) Cells were starved, and lysates were prepared for immunoblot analysis as described in the legend to Fig. 2. (B) Soft agar colony growth of HMF-hTERT-p53DD cells expressing various alleles of MT. The number of colonies raised by wild-type MT was defined as 100% and used for normalization. Means ± standard deviations for four experiments are shown in each case. (C) Focus formation analysis of HMF-hTERT-p53DD cells expressing various alleles of MT was carried out as described in Materials and Methods. The number of foci raised by wild-type MT was defined as 100% and used for normalization. Means ± standard deviations for four experiments are shown.
The PI3K binding site is important for MT-mediated transformation in human cells.
MT is known to activate multiple cellar effectors when it is expressed in cells. However, MT-stimulated signaling pathways and their roles in MT-mediated transformation of human cells have not been studied extensively. To dissect the effector pathway(s) required for MT transforming activity in human cells, we individually introduced into HMECs and HMFs expressing both hTERT and p53DD a panel of MT mutants, each carrying a single binding site mutation that selectively blocks the binding of MT to SHC (Y250F), PI3K (Y315F), or PLC-γ (Y322F), as well as a triple mutant (Y250F/Y315F/Y322F [YYY]) that ablated all three binding sites. Two of the major signaling pathways activated by MT are the MAPK and PI3K pathways, while the importance of the PLC-γ pathway has been difficult to detect. To study these signaling pathways in the resultant human cell lines, we starved cells expressing the various MT alleles in basal medium and measured phosphorylation of Akt at both Ser-473 (Fig. 2A and 3A) and Thr-308 (data not shown) or phosphorylation of ERK (Fig. 2A and 3A) as signaling readouts for PI3K or MAPK activation. Notably, MT activated PI3K in both human cell types (Fig. 2A and 3A). The Y315F mutant and the YYY triple mutant failed to stimulate Akt phosphorylation (Fig. 2A and 3A), consistent with the fact that Y315 is required for MT to associate with and activate PI3K. However, we did not observe elevated levels of ERK phosphorylation in MT-expressing cells compared to control cells (Fig. 2A and 3A).
FIG. 2.

The PI3K binding site at Y315 is important for MT-mediated transformation in HMECs. (A) Cells were starved in basal medium overnight, and lysates were prepared as described in Materials and Methods for immunoblot analysis of p-Akt and p-ERK. The slightly slower-migrating bands (indicated by the arrows) are p-Akt, while the more rapidly migrating species is nonphosphorylated Akt. Expressions of MT and its various mutant alleles are as indicated. (B) Soft agar colony growth of HMEC-hTERT-p53DD cells expressing various alleles of MT. The assay was carried out as described in Materials and Methods. (C) The number of colonies raised by wild-type MT was defined as 100% and used for normalization. Means ± standard deviations for three experiments are shown for each allele.
To test the transforming activities of the MT mutants, we first performed AI growth assays for both HMECs and HMFs. Often, the transformation potential of MT alleles has been assayed by focus formation, but this assay cannot be performed on epithelial cells. AI growth assays were chosen to allow us to test the various types and species of cells with a uniform assay format. The triple mutant YYY abolished the transforming ability of MT in these cells (Fig. 2B and C and 3B) as expected. When we examined mutants in single effector binding sites, we found that the Y315F mutant failed to induce colony growth in soft agar in both human cell types tested, while Y250F and Y322F mutants retained high transforming capacity (Fig. 2B and C and 3B). However, the Y250 site and its attendant SHC binding have previously been shown to be crucial for MT transformation in rodent fibroblasts, as measured by focus formation (3, 12, 13). We also tested the MT mutants in human fibroblasts by a focus formation assay, as is traditionally done with rodent fibroblasts. As shown in Fig. 3C, the MT mutants displayed the same transformation patterns in the HMF focus assay as they had in the AI growth assay.
Both the PI3K and SHC binding sites on MT are important for transformation of murine cells.
To examine species-specific effects of MT on transformation, we included murine cells in our study, namely, the MMEC line HC11 and MEFs. Virtually all murine cell lines suffer some form of p53 inactivation to allow their immortalization (43). One potential source of variation among rodent cell lines stems from the fact that p53 can be inactivated by various means. To be consistent with our previous studies, we again used p53DD in our work with murine cells. Thus, our primary MEFs were prepared from 13.5-day embryos and immortalized by the introduction of p53DD at passage one. The MMEC line HC11 is known to express a mutated p53. By introducing DDp53, we hoped to override the signal of the endogenous protein, thus providing a genetic background with respect to p53 similar to those of the other cell lines.
We stably introduced the panel of MT mutants described above into the two murine cell lines via retrovirus-mediated infection. Cells were starved and lysates were prepared for analysis of cellular signaling as before. Wild-type MT stimulated phosphorylation of Akt but not ERK in these cells, the same signaling pattern obtained from the analysis of MT in human cells (Fig. 4A and 5A). Once again the Y315F mutant failed to elevate the phospho-Akt levels in both HC11 cells and MEFs (Fig. 4A and 5A). Interestingly, the phospho-Akt levels induced by the Y250F mutant were also greatly reduced compared to wild-type MT in murine cells (Fig. 4A and 5A). As we did not observe a significant effect of the Y250F mutant on PI3K signaling in human cells, this finding indicates that the mechanism by which MT activates PI3K may differ in human and murine cells.
FIG. 4.

Both PI3K and SHC binding sites on MT are important for transformation of HC11 MMECs. (A) Cells stably expressing various alleles of MT were starved, and lysates were prepared for Western blot analysis as described in Materials and Methods. Expressions of various MT alleles and phosphorylation of Akt or ERK are indicated. (B) Soft agar colony growth of HC11-p53DD cells expressing various alleles of MT. The number of colonies raised by wild-type MT was defined as 100% and used for normalization. Means ± standard deviations for four experiments are shown.
FIG. 5.

Both PI3K and SHC binding sites on MT are important for transformation of MEFs. (A) Cells stably expressing various alleles of MT were starved, and lysates were prepared for Western blot analysis as described in Materials and Methods. Expressions of various MT alleles and phosphorylation of Akt or ERK are indicated. (B) Soft agar colony growth of MEF-p53DD cells expressing various alleles of MT. The number of colonies raised by wild-type MT was defined as 100% and used for normalization. (C) Focus formation analysis of MEF-p53DD cells expressing various alleles of MT, as described in Materials and Methods. The number of foci raised by wild-type MT was defined as 100% and used for normalization. Means ± standard deviations for four experiments are shown.
We then tested the transforming activities of MT and each mutant in both HC11 cells and MEFs by AI growth assays. Wild-type MT efficiently induced transformation in both murine cell types (Fig. 4B and 5B). As was the case with the human cells, the Y315F mutant was unable to efficiently transform either murine epithelial cells or fibroblasts (Fig. 4B and 5B), providing further evidence for a role of PI3K activation in MT-induced transformation. The Y250F mutant, defective in the SHC binding that has previously been reported to be essential for MT-induced transformation in established murine fibroblast cell lines (3, 12, 13), also failed to transform both murine cell types (Fig. 4B and 5B). As expression of this mutant efficiently induced transformation in both human cell types, the behavior of the Y250F mutation clearly demonstrates that MT-mediated transformation does indeed show species specificity. Expression of the Y322F mutant, which is defective in its binding to PLC-γ, transformed both murine cell types efficiently, showing only a moderate reduction in numbers compared to those elicited by wild-type MT. Once again, focus formation assays were performed for the various mutants with the murine fibroblasts, with results entirely in accord with the AI growth assays (Fig. 5C).
The p110α isoform of PI3K is essential for MT-mediated cellular transformation.
All of our experiments using MT mutants defective in single binding sites for cellular effectors point to the idea that the interaction of MT with PI3K is important for MT transforming activity in mammalian cells. However, it is still possible that another unidentified signaling molecule(s) utilizes the same binding site as PI3K-p85 to mediate MT transformation. The PI3K binding site on the platelet-derived growth factor receptor, for example, can interact with Nck (36). For MT itself, genetic evidence strongly suggests pY315 may generate signals other than from PI3K (22). Such observations make the use of simple direct interpretations of Y315F to connect PI3K signaling to transformation inappropriate. A direct confirmation of the role of PI3K in MT transformation is required. To do this, we tested the transforming activity of MT in p110α-null MEFs prepared from mice that we had recently generated that allowed conditional knockout of the PIK3CA gene (54). MEFs deficient for p110α were generated from control MEFs containing the homozygous floxed allele via Cre-mediated deletion and immortalized by introducing p53DD, as described previously (54). Control MEFs and p110α-null MEFs were then infected with retrovirus expressing wild-type MT. Notably, ablation of p110α completely blocked phospho-Akt and focus formation induced by MT (Fig. 6A and B). This is consistent with our previous observations that p110α-deficient cells are resistant to transformation by mutationally activated RTKs, further supporting the notion that MT is a viral analogue of an activated receptor.
FIG. 6.

The p110α isoform of PI3K is required for MT-mediated cellular transformation. (A) Both p110α+/+ and p110α−/− MEFs were infected with a control empty-vector virus or viruses encoding wild-type MT, as indicated. Focus formation assays were carried out as described in Materials and Methods. Foci were scored when wild-type MEFs had been cultured for 3 weeks and p110α knockout (−/−) cells had been cultured for 5 weeks. The bar graph reflects means ± standard deviations for three experiments. (B) Wild-type MT was stably introduced into both p53DD-immortalized wild-type MEFs (+/+) and p110α-null MEFs (−/−). (C) The indicated cells were injected subcutaneously into nude mice as described in Materials and Methods. Tumors were scored at 3 to 6 weeks postinjection.
To gain further insight into the effect of p110α ablation on tumorigenesis induced by MT, we injected p53DD-immortalized control MEFs or p110α knockout MEFs expressing MT into nude mice subcutaneously and scored the ability of the injected cells to form tumors. While control MEFs expressing MT formed palpable tumors in nude mice within 2 weeks that reached ∼6 mm in diameter in 4 weeks (six of six mice), MT expression in p110α knockout MEFs failed to promote tumor formation in animal hosts over a period of 6 weeks (zero of six mice) (Fig. 6C). Taken together, our data demonstrate that the transforming activity of MT depends on PI3K activation and that the p110α isoform of MT is essential for MT-mediated transformation in mammalian cells.
DISCUSSION
The present work points to the overarching importance of PI3K pathway activation in MT-mediated transformation in the human and mouse fibroblasts and mammary epithelial cells that were examined. However, the means used by MT for activation of the PI3K pathway differs between mice and humans. For the first time, we were able to use MEFs derived from knockout mice to further pin down the involvement of PI3K isoforms, finding a specific requirement for p110α in MT transformation. This result eliminates one nagging issue in MT study. Almost all previous ideas concerning the role of PI3K in MT transformation were based on the use of PI3K binding site mutants in MT. That site is likely to have other, non-PI3K functions (22). We can now point with confidence to the role of PI3K in MT transformation. Other MT effectors were either species specific, as was the case for SHC in murine cells, or relatively unimportant in any situation, as was the case for PLC-γ.
We aimed to gain insights into the individual importance of the intracellular signaling pathways activated by PyV MT in cellular transformation via a cross-species and cross-cell-type study. While the interaction of MT with rodent fibroblasts has previously been studied, we have compared and contrasted mouse and human cells of two distinct cell lineages. Single binding site mutations of Y250, Y315, and Y322 were used to block the binding of SHC, p85, or PLC-γ to MT. The most prominent effects were observed for the p85 binding site mutant, Y315F. This mutant was transformation defective in both cell types and both species, as measured by AI growth and focus formation assays. Western blot analysis, using antibodies to phospho-Akt, revealed that the PI3K pathway was strongly activated in all cell types expressing wild-type MT, while expression of the Y315F mutant failed to activate the PI3K pathway in all cell lines tested. This is consistent with the generally held hypothesis that PI3K pathway activation is essential for MT-mediated transformation. Interestingly, the transforming potential of the SHC binding site mutant (Y250F), previously reported to be essential for MT to transform established fibroblast cell lines (35, 37), did vary with the species examined. While this mutant was unable to induce transformation in either murine fibroblasts or epithelial cells, in agreement with published data for rodent fibroblasts, expression of Y250F was fully transforming in both human epithelial cells and fibroblasts. Western blot analyses of the activation of the PI3K pathway in these Y250F cell lines revealed significantly reduced phospho-Akt levels in both murine cell lines, whereas the human cell lines expressing the Y250F mutant showed phospho-Akt levels similar to those of their wild-type MT counterparts. These data suggest that there may be differences in the mechanism of PI3K activation between human and murine cells, particularly in the importance of secondary binding sites for p85. It has previously been shown that while Y315 is the primary binding site for p85, Y250 can also play a role in PI3K binding of MT via a known interaction of Grb2/Gab2 (a PI3K adapter) with SHC (37). For the moment, we can hypothesize that another adapter with a binding site on MT different from Y250 plays the role of Gab2 in human cells, but it will clearly require further work to test this possibility. The fact that the Y250F mutation reduces PI3K activation in murine cells but not human cells and reduces transformation in murine but not human cells should not be interpreted as suggesting that PI3K binding/activation is the only essential role of Y250 in murine cells. SHC binding via Y250 is known to be important in transforming rodent fibroblasts. Notably, the P248H mutation (22), which lies in the NPXY motif in MT, also blocks SHC binding and abolishes MT-mediated transformation in our MEF cells (but not in HMECs) without altering PI3K activation (unpublished results).
Although SHC binding is important for MT-mediated transformation of rodent cells, activation of the MAPK pathway is rarely, if ever, seen in cell lines stably expressing wild-type MT, even though transient expression of MT can indeed activate the MAPK pathway via SHC (48) and MAPK activation is seen in MT-induced tumors in vivo (5). It has always been presumed that some feedback loop or cross-talk from another pathway was inhibiting MAPK activation. Our experiments with human cells give a hint as to one possible mechanism underlying this inhibition. In murine cells, we can see clearly that phospho-ERK levels are elevated in cells expressing the Y315F mutant but not in wild-type MT (Fig. 4A and 5A). This suggests that cross-talk from the PI3K pathway may contribute to the silencing of MAPK signals.
For the PLC-γ binding mutant (Y322F), a slight reduction in the number of colonies and foci formed, 10 to 30% compared to the wild-type MT-expressing cells, was observed in all cell lines examined in this study. However, both epithelial cells and fibroblasts of murine or human origin were still efficiently transformed. It is likely that under more restrictive conditions, e.g., serum deprivation, these slight effects would be more prominent (44).
In all past discussions of MT transformation, the term PI3K was used to nonspecifically refer to the aggregate of class IA PI3K activities. We were able to begin to examine the role of specific PI3K isoforms by construction of a conditional p110α knockout mouse, enabling us to look at wild-type MT transformation in a PI3K knockout background. While previous work had shown that MMTV-MT-derived mammary tumor cells expressing a dominant negative p85 subunit undergo elevated levels of apoptosis (50) and that the expression of a constitutively activated AKT dramatically accelerates oncogenesis and reduces apoptosis, as observed in MMTV-Y315/322F-MT cells (25), our demonstration of a requirement for the p110α subunit in MT transformation finally establishes the essential role for the PI3K pathway in MT-mediated transformation. These data are also in nice accord with our previous studies on growth factor receptors in cells lacking p110α and the finding that ablation of p110α greatly diminishes transformation by oncogenic alleles of growth factor receptors (54). The failure of MT to transform the p110α knockout cells, which still express p110β, emphasizes that the two PI3K isoforms are not functionally equivalent. Future work will be needed to elucidate key differences between the two.
Acknowledgments
This work was supported by grants from the NIH (PO1-CA50661, T.M.R and B.S.S; PO1-CA089021, T.M.R; CA30002, T.M.R.; CA34722, B.S.S), by a Claudia Barr Award (J.J.Z), by DoD BC051565 (J.J.Z), by a Friends Woman's Cancer Program Award (J.J.Z.), and by the German Research Foundation (postdoctoral fellowship to T.U.).
In compliance with Harvard Medical School guidelines, we disclose a consulting relationship with Novartis Pharmaceuticals, Inc. (T.M.R.).
Footnotes
Published ahead of print on 18 April 2007.
REFERENCES
- 1.Bolen, J. B., C. J. Thiele, M. A. Israel, W. Yonemoto, L. A. Lipsich, and J. S. Brugge. 1984. Enhancement of cellular src gene product associated tyrosyl kinase activity following polyomavirus infection and transformation. Cell 38:767-777. [DOI] [PubMed] [Google Scholar]
- 2.Bronson, R., C. Dawe, J. Carroll, and T. Benjamin. 1997. Tumor induction by a transformation-defective polyomavirus mutant blocked in signaling through Shc. Proc. Natl. Acad. Sci. USA 94:7954-7958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Campbell, K. S., E. Ogris, B. Burke, W. Su, K. R. Auger, B. J. Druker, B. S. Schaffhausen, T. M. Roberts, and D. C. Pallas. 1994. Polyoma middle tumor antigen interacts with SHC protein via the NPTY (Asn-Pro-Thr-Tyr) motif in middle tumor antigen. Proc. Natl. Acad. Sci. USA 91:6344-6348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Carmichael, G., B. S. Schaffhausen, G. Mandel, T. J. Liang, and T. L. Benjamin. 1984. Transformation by polyomavirus is drastically reduced by substitution of phenylalanine for tyrosine at residue 315 of middle-sized tumor antigen. Proc. Natl. Acad. Sci. USA 81:679-683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cecena, G., F. Wen, R. D. Cardiff, and R. G. Oshima. 2006. Differential sensitivity of mouse epithelial tissues to the polyomavirus middle T oncogene. Am. J. Pathol. 168:310-320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chen, L., X. Wang, and M. M. Fluck. 2006. Independent contributions of polyomavirus middle T and small T to the regulation of early and late gene expression and DNA replication. J. Virol. 80:7295-7307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cheng, S. H., R. Harvey, P. C. Espino, K. Semba, T. Yamamoto, K. Toyoshima, and A. E. Smith. 1988. Peptide antibodies to the human c-fyn gene product demonstrate pp59c-fyn is capable of complex formation with the middle-T antigen of polyomavirus. EMBO J. 7:3845-3855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Courtneidge, S. A., and A. Heber. 1987. An 81 kd protein complexed with middle T antigen and pp60c-src: a possible phosphatidylinositol kinase. Cell 50:1031-1037. [DOI] [PubMed] [Google Scholar]
- 9.Courtneidge, S. A., and A. E. Smith. 1983. Polyomavirus transforming protein associates with the product of the c-src cellular gene. Nature 303:435-439. [DOI] [PubMed] [Google Scholar]
- 10.Courtneidge, S. A., and A. E. Smith. 1984. The complex of polyomavirus middle-T antigen and pp60c-src. EMBO J. 3:585-591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dilworth, S. M. 2002. Polyomavirus middle T antigen and its role in identifying cancer-related molecules. Nat. Rev. Cancer 2:951-956. [DOI] [PubMed] [Google Scholar]
- 12.Dilworth, S. M., C. E. Brewster, M. D. Jones, L. Lanfrancone, G. Pelicci, and P. G. Pelicci. 1994. Transformation by polyomavirus middle T-antigen involves the binding and tyrosine phosphorylation of Shc. Nature 367:87-90. [DOI] [PubMed] [Google Scholar]
- 13.Druker, B. J., L. E. Ling, B. Cohen, T. M. Roberts, and B. S. Schaffhausen. 1990. A completely transformation-defective point mutant of polyomavirus middle T antigen which retains full associated phosphatidylinositol kinase activity. J. Virol. 64:4454-4461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Engelman, J. A., J. Luo, and L. C. Cantley. 2006. The evolution of phosphatidylinositol 3-kinases as regulators of growth and metabolism. Nat. Rev. Genet. 7:606-619. [DOI] [PubMed] [Google Scholar]
- 15.Foukas, L. C., M. Claret, W. Pearce, K. Okkenhaug, S. Meek, E. Peskett, S. Sancho, A. J. Smith, D. J. Withers, and B. Vanhaesebroeck. 2006. Critical role for the p110alpha phosphoinositide-3-OH kinase in growth and metabolic regulation. Nature 441:366-370. [DOI] [PubMed] [Google Scholar]
- 16.Freund, R., R. T. Bronson, and T. L. Benjamin. 1992. Separation of immortalization from tumor induction with polyoma large T mutants that fail to bind the retinoblastoma gene product. Oncogene 7:1979-1987. [PubMed] [Google Scholar]
- 17.Freund, R., C. J. Dawe, J. P. Carroll, and T. L. Benjamin. 1992. Changes in frequency, morphology, and behavior of tumors induced in mice by a polyomavirus mutant with a specifically altered oncogene. Am. J. Pathol. 141:1409-1425. [PMC free article] [PubMed] [Google Scholar]
- 18.Glover, H. R., C. E. Brewster, and S. M. Dilworth. 1999. Association between src-kinases and the polyomavirus oncogene middle T-antigen requires PP2A and a specific sequence motif. Oncogene 18:4364-4370. [DOI] [PubMed] [Google Scholar]
- 19.Gorga, F. R., C. E. Riney, and T. L. Benjamin. 1990. Inositol triphosphate levels in cells expressing wild-type and mutant polyomavirus middle T antigens: evidence for activation of phospholipase C via activation of pp60c-src. J. Virol. 64:105-112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Griffin, B. E., Y. Ito, U. Novak, N. Spurr, S. Dilworth, N. Smolar, R. Pollack, K. Smith, and D. B. Rifkin. 1980. Early mutants of polyomavirus(dl8 and dl23) with altered transformation properties: is polyomavirus middle T antigen a transforming gene product? Cold Spring Harbor Symp. Quant. Biol. 44:271-282. [DOI] [PubMed] [Google Scholar]
- 21.Guy, C. T., R. D. Cardiff, and W. J. Muller. 1992. Induction of mammary tumors by expression of polyomavirus middle T oncogene: a transgenic mouse model for metastatic disease. Mol. Cell. Biol. 12:954-961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hong, Y. K., A. Mikami, B. Schaffhausen, T. Jun, and T. M. Roberts. 2003. A new class of mutations reveals a novel function for the original phosphatidylinositol 3-kinase binding site. Proc. Natl. Acad. Sci. USA 100:9434-9439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Horak, I. D., T. Kawakami, F. Gregory, K. C. Robbins, and J. B. Bolen. 1989. Association of p60fyn with middle tumor antigen in murine polyomavirus-transformed rat cells. J. Virol. 63:2343-2347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hunter, T., M. A. Hutchinson, and W. Eckhart. 1984. Polyoma middle-sized T antigen can be phosphorylated on tyrosine at multiple sites in vitro. EMBO J. 3:73-79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hutchinson, J., J. Jin, R. D. Cardiff, J. R. Woodgett, and W. J. Muller. 2001. Activation of Akt (protein kinase B) in mammary epithelium provides a critical cell survival signal required for tumor progression. Mol. Cell. Biol. 21:2203-2212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ichaso, N., and S. M. Dilworth. 2001. Cell transformation by the middle T-antigen of polyomavirus. Oncogene 20:7908-7916. [DOI] [PubMed] [Google Scholar]
- 27.Kaplan, D. R., M. Whitman, B. Schaffhausen, D. C. Pallas, M. White, L. Cantley, and T. M. Roberts. 1987. Common elements in growth factor stimulation and oncogenic transformation: 85 kd phosphoprotein and phosphatidylinositol kinase activity. Cell 50:1021-1029. [DOI] [PubMed] [Google Scholar]
- 28.Kaplan, D. R., M. Whitman, B. Schaffhausen, L. Raptis, R. L. Garcea, D. Pallas, T. M. Roberts, and L. Cantley. 1986. Phosphatidylinositol metabolism and polyoma-mediated transformation. Proc. Natl. Acad. Sci. USA 83:3624-3628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Knight, Z. A., B. Gonzalez, M. E. Feldman, E. R. Zunder, D. D. Goldenberg, O. Williams, R. Loewith, D. Stokoe, A. Balla, B. Toth, T. Balla, W. A. Weiss, R. L. Williams, and K. M. Shokat. 2006. A pharmacological map of the PI3-K family defines a role for p110alpha in insulin signaling. Cell 125:733-747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kornbluth, S., M. Sudol, and H. Hanafusa. 1987. Association of the polyomavirus middle-T antigen with c-yes protein. Nature (London) 325:171-173. [DOI] [PubMed] [Google Scholar]
- 31.Kypta, R. M., A. Hemming, and S. A. Courtneidge. 1988. Identification and characterization of p59fyn (a src-like protein tyrosine kinase) in normal and polyomavirus transformed cells. EMBO J. 7:3837-3844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Larose, A., N. Dyson, M. Sullivan, E. Harlow, and M. Bastin. 1991. Polyomavirus large T mutants affected in retinoblastoma protein binding are defective in immortalization. J. Virol. 65:2308-2313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lomax, M., and M. Fried. 2001. Polyomavirus disrupts ARF signaling to p53. Oncogene 20:4951-4960. [DOI] [PubMed] [Google Scholar]
- 34.Magnusson, G., M. G. Nilsson, S. M. Dilworth, and N. Smolar. 1981. Characterization of polyoma mutants with altered middle and large T-antigens. J. Virol. 39:673-683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Markland, W., B. A. Oostra, R. Harvey, A. F. Markham, W. H. Colledge, and A. E. Smith. 1986. Site-directed mutagenesis of polyomavirus middle-T antigen sequences encoding tyrosine 315 and tyrosine 250. J. Virol. 59:384-391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nishimura, R., W. Li, A. Kashishian, A. Mondino, M. Zhou, J. Cooper, and J. Schlessinger. 1993. Two signaling molecules share a phosphotyrosine-containing binding site in the platelet-derived growth factor receptor. Mol. Cell. Biol. 13:6889-6896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ong, S. H., S. Dilworth, I. Hauck-Schmalenberger, T. Pawson, and F. Kiefer. 2001. ShcA and Grb2 mediate polyoma middle T antigen-induced endothelial transformation and Gab1 tyrosine phosphorylation. EMBO J. 20:6327-6336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Oostra, B. A., R. Harvey, B. K. Ely, A. F. Markham, and A. E. Smith. 1983. Transforming activity of polyomavirus middle-T antigen probed by site-directed mutagenesis. Nature 304:456-459. [DOI] [PubMed] [Google Scholar]
- 39.Pallas, D. C., L. K. Shahrik, B. L. Martin, S. Jaspers, T. B. Miller, D. L. Brautigan, and T. M. Roberts. 1990. Polyoma small and middle T antigens and SV40 small T antigen form stable complexes with protein phosphatase 2A. Cell 60:167-176. [DOI] [PubMed] [Google Scholar]
- 40.Samuels, Y., Z. Wang, A. Bardelli, N. Silliman, J. Ptak, S. Szabo, H. Yan, A. Gazdar, S. M. Powell, G. J. Riggins, J. K. Willson, S. Markowitz, K. W. Kinzler, B. Vogelstein, and V. E. Velculescu. 2004. High frequency of mutations of the PIK3CA gene in human cancers. Science 304:554. [DOI] [PubMed] [Google Scholar]
- 41.Schaffhausen, B., and T. L. Benjamin. 1981. Comparison of phosphorylation of two polyomavirus middle T antigens in vivo and in vitro. J. Virol. 40:184-196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Schaffhausen, B. S., and T. L. Benjamin. 1979. Phosphorylation of polyoma T antigens. Cell 18:935-946. [DOI] [PubMed] [Google Scholar]
- 43.Sherr, C. J., and R. A. DePinho. 2000. Cellular senescence: mitotic clock or culture shock? Cell 102:407-410. [DOI] [PubMed] [Google Scholar]
- 44.Su, W., W. Liu, B. S. Schaffhausen, and T. M. Roberts. 1995. Association of polyomavirus middle tumor antigen with phospholipase C-gamma 1. J. Biol. Chem. 270:12331-12334. [DOI] [PubMed] [Google Scholar]
- 45.Talmage, D. A., R. Freund, A. T. Young, J. Dahl, C. J. Dawe, and T. L. Benjamin. 1989. Phosphorylation of middle T by pp60c-src: a switch for binding of phosphatidylinositol 3-kinase and optimal tumorigenesis. Cell 59:55-65. [DOI] [PubMed] [Google Scholar]
- 46.Tooze, J. E. 1980. DNA tumor viruses. The molecular biology of tumor viruses, vol. 1 and 2. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- 47.Treisman, R., U. Novak, J. Favaloro, and R. Kamen. 1981. Transformation of rat cells by an altered polyomavirus genome expressing only the middle T protein. Nature (London) 292:595-600. [DOI] [PubMed] [Google Scholar]
- 48.Urich, M., M. Y. el Shemerly, D. Besser, Y. Nagamine, and K. Ballmer-Hofer. 1995. Activation and nuclear translocation of mitogen-activated protein kinases by polyomavirus middle-T or serum depend on phosphatidylinositol 3-kinase. J. Biol. Chem. 270:29286-29292. [DOI] [PubMed] [Google Scholar]
- 49.Walter, G., R. Ruediger, C. Slaughter, and M. Mumby. 1990. Association of protein phosphatase 2A with polyomavirus medium tumor antigen. Proc. Natl. Acad. Sci. USA 87:2521-2525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Webster, M. A., J. N. Hutchinson, M. J. Rauh, S. K. Muthuswamy, M. Anton, C. G. Tortorice, R. D. Cardiff, F. L. Graham, J. A. Hassell, and W. J. Muller. 1998. Requirement for both Shc and phosphatidylinositol 3′ kinase signaling pathways in polyomavirus middle T-mediated mammary tumorigenesis. Mol. Cell. Biol. 18:2344-2359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Whitman, M., D. R. Kaplan, B. Schaffhausen, L. Cantley, and T. M. Roberts. 1985. Association of phosphatidylinositol kinase activity with polyoma middle-T competent for transformation. Nature 315:239-242. [DOI] [PubMed] [Google Scholar]
- 52.Yang, S. I., R. L. Lickteig, R. Estes, K. Rundell, G. Walter, and M. C. Mumby. 1991. Control of protein phosphatase 2A by simian virus 40 small-t antigen. Mol. Cell. Biol. 11:1988-1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yi, X., J. Peterson, and R. Freund. 1997. Transformation and tumorigenic properties of a mutant polyomavirus containing a middle T antigen defective in Shc binding. J. Virol. 71:6279-6286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhao, J. J., H. Cheng, S. Jia, L. Wang, O. V. Gjoerup, A. Mikami, and T. M. Roberts. 2006. The p110α isoform of PI3K is essential for proper growth factor signaling and oncogenic transformation. Proc. Natl. Acad. Sci. USA. [DOI] [PMC free article] [PubMed]
- 55.Zhao, J. J., O. V. Gjoerup, R. R. Subramanian, Y. Cheng, W. Chen, T. M. Roberts, and W. C. Hahn. 2003. Human mammary epithelial cell transformation through the activation of phosphatidylinositol 3-kinase. Cancer Cell 3:483-495. [DOI] [PubMed] [Google Scholar]