

Abstract
Dengue virus envelope protein (E) contains two N-linked glycosylation sites, at Asn-67 and Asn-153. The glycosylation site at position 153 is conserved in most flaviviruses, while the site at position 67 is thought to be unique for dengue viruses. N-linked oligosaccharide side chains on flavivirus E proteins have been associated with viral morphogenesis, infectivity, and tropism. Here, we examined the relevance of each N-linked glycan on dengue virus E protein by removing each site in the context of infectious viral particles. Dengue viruses lacking Asn-67 were able to infect mammalian cells and translate and replicate the viral genome, but production of new infectious particles was abolished. In addition, dengue viruses lacking Asn-153 in the E showed reduced infectivity. In contrast, ablation of one or both glycosylation sites yielded viruses that replicate and propagate in mosquito cells. Furthermore, we found a differential requirement of N-linked glycans for E secretion in mammalian and mosquito cells. While secretion of E lacking Asn-67 was efficient in mosquito cells, secretion of the same protein expressed in mammalian cells was dramatically impaired. Finally, we found that viruses lacking the carbohydrate at position 67 showed reduced infection of immature dendritic cells, suggesting interaction between this glycan and the lectin DC-SIGN. Overall, our data defined different roles for the two glycans present at the E protein during dengue virus infection, highlighting the involvement of distinct host functions from mammalian and mosquito cells during dengue virus propagation.
Dengue virus (DV), a member of the flavivirus genus of the Flaviviridae family, causes the most prevalent arthropod-borne viral disease of humans, which is associated with a large social and economic burden. DV contains a single-stranded RNA genome of positive polarity that is translated in the cytoplasm as a single polyprotein. Processing of the polyprotein by cellular and viral proteases results in three structural and seven nonstructural mature proteins. The structural proteins are the capsid (C), which assembles with the genomic RNA, the glycosylated envelope protein (E), and the precursor of the membrane protein (prM). Flavivirus assembly takes place in the endoplasmic reticulum (ER) (23, 24). The RNA-C complex buds into the ER lumen, acquiring the lipid bilayer, E, and prM. Furin-mediated proteolysis of prM in the trans-Golgi network (40) triggers rearrangement, the homodimerization of E, and the formation of mature viral particles (1).
It is postulated that the interaction of E with a host cell receptor directs DV particles to the endocytic pathway. The acidic environment in the endosome is believed to produce major conformational changes in E that induce fusion of the viral and host cell membranes, which in turn releases the viral genome into the cytoplasm. Several studies indicate that cell surface heparan sulfates (HS) are involved in the attachment of DV to mammalian but not to mosquito cells (5, 9, 13, 14). It was demonstrated that positively charged residues on the surface of E bind to the negatively charged HS (5, 14). Since HS is ubiquitously expressed on many cell types, a more-specific protein receptor is thought to be required to explain the limited cell tropism of DV. Significant effort has been made to identify the cellular receptor for DV. Different cellular proteins have been proposed as possible DV receptors, including heat shock protein 70 (HSP70) and HSP90, GRP78/Bip, CD14, and a 37/67-kDa high-affinity laminin receptor (15, 17, 28, 39, 42). Recently, the dendritic cell (DC) endocytic receptor DC-SIGN (CD209), which is expressed in human skin at the anatomical site of DV exposure, has been identified as a cellular factor required for productive infection of immature DCs (32, 41). Ectopic expression of DC-SIGN renders many poorly susceptible cells infectible by the four DV serotypes (26). However, DC-SIGN is not an authentic entry receptor responsible for DV internalization, but rather acts as a critical attachment factor that concentrates DV particles at the DC surface, enhancing viral entry (26). DC-SIGN is a tetrameric C-type lectin that binds, through its C-terminal carbohydrate recognition domain (CRD), high-mannose N-linked glycans on the surfaces of DV particles (26, 32).
The E protein has two potential N-linked glycosylation sites, at Asn-67 and Asn-153. The glycosylation site at Asn-153 is conserved in most flaviviruses, while glycosylation at Asn-67 is unique for DV (12). Recently, a cryoelectron microscopy reconstruction of DV complexed with the carbohydrate binding domain of DC-SIGN has shown interaction of the lectin with the N-glycan at Asn-67 (36). Previous analysis of the crystal structure of E from DV and other flaviviruses indicated that the carbohydrate moiety at Asn-153 extends across the dimer interface covering the fusion peptide (29, 38). Stabilization of the E dimer by this oligosaccharide is consistent with the properties of DV mutants at position 153, which fuse with target membranes at a higher pH (10, 21). Interestingly, the presence or absence of specific glycosylation at Asn-153 in DV-4 or at the corresponding amino acid in West Nile virus (WNV) E protein (Asn-154) is associated with neuroinvasiveness in mice (4, 19).
Extensive structural analysis supports the notion that sugar molecules on the viral surface are important for DV entry. In addition, other reports have provided evidence that glycosylation of flavivirus structural proteins could have other functions during viral maturation. Specific inhibition of ER α-glycosidases, blocking the trimming of the N-linked oligosaccharides, impaired both the secretion and infectivity of several flaviviruses (6, 45, 46). These inhibitors prevent the removal of the terminal glucose residues on the N-linked glycans. The lack of this modification on the sugar tree could alter the mechanism for controlling proper protein folding mediated by ER chaperones (35, 44). Therefore, it is likely that glycosylation of DV structural proteins plays a role during viral morphogenesis.
In this study, we investigated the relevance of N-glycosylation of DV type 2 (DV-2) E during different stages of viral replication by the removal of individual glycosylation sites in the context of DV infectious clones. We found that both potential sites, Asn-67 and Asn-153, are utilized for glycosylation in mosquito and mammalian cells. Viruses lacking one or both N-glycosylation sites in the E protein replicate in C6/36 mosquito cells. In contrast, specific and differential roles of glycosylation at positions 67 and 153 were observed in mammalian cells. We show here that the glycan at position 153 greatly increased viral specific infectivity per RNA molecule, while glycosylation at Asn-67 was essential for viral assembly or exit. Infection of different mammalian cells with DV-2 carrying the mutation N67Q resulted in one round of viral replication, but viral progeny was undetectable. Furthermore, our data emphasize a role for the glycosylation site at position Asn-67 in DC-SIGN-mediated enhancement of DV entry. Viruses lacking this carbohydrate showed a reduced capacity to infect Raji-DC-SIGN cells as well as monocyte-derived dendritic cells.
MATERIALS AND METHODS
Cells.
Human hepatocytic cells (Huh-7) and monkey kidney cells (Vero) were cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal calf serum (FCS) and 1% penicillin/streptomycin. Baby hamster kidney cells (BHK) were cultured in minimum essential medium alpha supplemented with 10% fetal bovine serum, 100 U/ml penicillin, 100 μg/ml streptomycin. Raji is a nonadherent human B-cell line. Raji cells expressing DC-SIGN were generated by transduction with a retroviral vector as described previously (26). Raji cells were cultured in RPMI 1640 medium supplemented with 10% FCS and 1% penicillin/streptomycin. Aedes albopictus C6/36 cells were cultured at 28°C in Leibovitz's L-15 medium. DCs were generated by differentiation of human monocytes as previously described (25).
Recombinant DV.
DV cDNAs carrying mutations in the E protein were obtained using a modified pD2/IC-30P-A plasmid (20) that includes a unique AflII restriction site just upstream of the polyprotein stop codon (pD2/ICAflll) (3). To facilitate insertion of the mutations, we generated an intermediate plasmid using pGEM-T (Promega), including the fragment obtained by digestion of pD2/ICAflll with SphI-NsiI, to generate pGEMSphI-NsiI. Mutation N153Q in DV-2 E protein was introduced into pGEM.DV-SphI-NsiI by PCR using the primers sense AVG-408 (5′-CGACGTCGCATGCAGTCGGACAAGACACAGGAAAACATGGC-3′) and antisense AVG-410 (5′-GAATACATCCTGTTTTTCTTTC-3′). This PCR fragment was digested by using the unique restriction sites SphI and HpaI and cloned into the pGEM.DV-SphI-NsiI digested with the same restriction enzymes. The resulting plasmid was digested with SphI-NsiI, and the fragment was cloned into pD2/ICAflll to generate the plasmid pD2.N153Q. Mutation N67Q in the E protein was generated by overlapping PCR using the following primers: PCR1 sense AVG-101 (5′-TCCAGACTTTACGAAACACG-3′) and antisense AVG-411 (5′-GCGAGATTCTGTTGTTGTTTGGGTTAGCTTTGC-3′) and PCR2 sense AVG-412 (5′-ATAGAGGCAAAGCTAACCCAAACAACAACAGAAT-3′) and antisense AVG-239 (5′-TCTGTGATGGAACTCTGTGG-3′). The PCR fragment was digested with SacI-SphI and cloned directly into pD2/ICAflII to generate the plasmid pD2.N67Q. To obtain the double mutant N67Q N153Q, the pGEM.DV-SphI-NsiI carrying the mutation N153Q was digested with SphI-NsiI and the resulting fragment was cloned into pD2.N67Q to generate pD2.N67Q-N153Q.
The cDNA of DV carrying the reporter Renilla luciferase was constructed using the full-length cDNA pD2/ICAflII. A cassette containing the encephalomyocarditis virus internal ribosome entry site (EMCV IRES) fused to the Renilla luciferase was generated by overlapping PCR using the following primers: PCR1 sense AVG-428 (5′-AAGAAGCAGGAGTTCTGTGGTAGTAACCGCGGGCCCGGGATCCG-3′) and antisense AVG-481 (5′-CATAAACTTTCGAAGTCATACCGGTCACCATGGTTGTGGCCATAT-3′) and PCR2 sense AVG-480 (5′ ATATGGCCACAACCATGGTGACCGGTATGACTTCGAAAGTTTATG-3′) and antisense AVG-387 (5-GTTTCATCTTAAGTTTTGCTTTCTATTGTTCATTTTTGAGAACTC-3′). This PCR product was digested with HpaI-AflII and cloned into a previously described plasmid, pGEM-3′UTR-AflII (3), to generate a plasmid containing the EMCV IRES-Renilla-DV-3′ UTR. This plasmid was digested with StuI-AflII, and the DNA fragment was cloned into pD2/ICAflII to obtain the cDNA of DV-Renilla (pDV-R). To generate the plasmid pDV-R.N67Q, pD2.N67Q was digested with SacI-SphI and the fragment containing the N67Q mutation was cloned into pDV-R.
Viral RNA transcription and transfection.
The wild-type (WT) and mutant cDNAs of DV-2 were linearized by digestion with XbaI. Linearized plasmids were phenol-chloroform extracted, ethanol precipitated, and resuspended in RNase-free water at a concentration of 100 ng/μl. In vitro transcriptions by T7 RNA polymerase were performed in a 40-μl reaction mixture using 0.5 μg of DNA template, 2 mM of m7GpppA cap structure analogue, 0.8 mM ATP, and 2 mM of UTP, CTP, and GTP. The transcription reaction mixture was incubated at 37°C for 2 h. The DNA templates were removed from the reaction mixture by DNase I digestion, and the RNAs were purified by using an RNeasy mini kit (QIAGEN Inc.) and quantified spectrophotometrically.
For RNA transfections, mammalian BHK-21 and mosquito C6/36 cells were grown to 60 to 70% confluence in 35-mm culture dishes. Lipofectamine 2000 (Invitrogen) was used to transfect RNA transcripts (300 ng). We prepared complexes of RNA-Lipofectamine 2000 (in μg and μl, respectively) in a 1:6 ratio for both cell lines and followed the transfection procedure according to the manufacturer's instructions. The BHK-21 cells were maintained at 37°C, and the Aedes albopictus C6/36 cells were maintained at 28°C.
To prepare stocks of WT and mutant viruses, the RNAs (2 μg) were transfected into BHK-21 and C6/36 cells. Viruses were harvested 5 to 7 days posttransfection and used to infect fresh cells to obtain higher titers. Viral stocks were quantified by plaque assays in BHK cells. For viruses that did not propagate in BHK cells, the amount of infectious particles was evaluated by fluorescence-activated cell sorter (FACS) analysis 24 h postinfection using different cell lines. In addition, DV RNA levels were measured by fluorogenic reverse transcription-PCR (RT-PCR) using TaqMan technology. DV-specific primers and probes targeted to amplify nucleotides 10419 to 10493 within the viral 3′UTR were used as previously described (2).
Immunofluorescence assay.
BHK-21 and C6/36 cells that had been seeded in a 35-mm tissue culture dish with a 1-cm2 coverslip inside were transfected with 300 ng of RNA or infected with 108 RNA molecules of WT or mutant DV quantified by real-time PCR. At each time point, the coverslips were recovered and a 1:200 dilution of murine hyperimmune ascetic fluid against DV-2 in phosphate-buffered saline (PBS)-0.2% gelatin was used for immunofluorescence (IF) assays, as previously described (2).
One-step growth curves.
Subconfluent BHK cells in a six-well plate were infected with equal amounts of WT DV, N153Q DV, or infectious DV-2 carrying a reporter gene (DV-R) recovered from BHK cells. A multiplicity of infection (MOI) of 0.01 or 0.1 was used in each case as indicated. After a 1-h adsorption period, the cells were washed three times with PBS, and 2 ml of growth media was added. At each time point after infection, cell supernatants were collected and frozen at −70°C. For virus quantification at each time point, the supernatants were serially diluted and a plaque assay was performed on BHK cells.
Western blots and glycosidase treatment.
BHK and C6/36 cells plated in 35-mm tissue culture dishes were infected with WT or mutant DV. The cells were harvested, washed with PBS, and lysed using buffer L (50 mM Tris, 150 mM NaCl, 2 mM EDTA, 1% Triton X-100, 0.5 mM phenylmethylsulfonyl sulfate) at pH 7.5. The lysates were centrifuged at 10,000 × g for 10 min to clear cellular debris and denatured in buffer S (5% sodium dodecyl sulfate [SDS], 50 mM Tris [pH 6.8], 10% glycerol) at 70°C for 10 min. Aliquots from each sample were then treated with 250 U of peptide N-glycosidase F (PNGase F), with 500 U of endoglycosidase H (Endo H), or with buffer control, according to the manufacturer's instructions (New England BioLabs, Inc., Beverly, MA). Digestions were carried out for 4 h at 37°C. Samples were then analyzed under denaturing and nonreducing conditions by 9% SDS-polyacrylamide gel electrophoresis (SDS-PAGE), and Western blotting was performed using the specific anti-E monoclonal antibody (mAb) E18 (1:500) (33). In order to determine the amounts of secreted E protein, the medium of infected BHK cells grown in 100-mm tissue culture dishes was collected at 30, 60, and 90 h postinfection. The infection was normalized by focus forming units in C6/36 cells (MOI of 0.1). Ten milliliters of medium was ultra-centrifuged at 4°C for 3 h at 140,000 × g, and viruses were resuspended in 300 μl of PBS. Thirty microliters of each sample was loaded into a 9% SDS-PAGE gel, and Western blots were performed as described above.
Construction of DV-2 sE expression plasmid.
Coding sequences for the mature forms of DV-2 prM and E proteins were obtained by PCR from the infectious cDNA clones (pD2/ICAflll, pD2.N67Q, and pD2.N153Q) described above. They were used as templates to generate recombinant Semliki Forest virus (SFV) encoding the DV-2 prM- and transmembrane-deleted forms of E as previously described (26, 27). The sequence coding for prM and the ectodomain of DV-2 E (soluble E [sE]; amino acids 115 to 674 in the DV-2 polyprotein) were amplified by PCR (Pwo DNA polymerase; Roche Applied Science) using the sense primer 5′-ATATTGCGCGCATGAAGTGCCTTTTGTACTTAGCCTTTTTATTCATTGGGGTGAATTGCTTCCATTTAACCACACGT-3′ and the antisense primer 5′-ATATTATGCATCTAGGCGGGGGCCACCTGGGAGGTCTCGGTTCCTTTCTTAAACCAGTTGAGCTTCAG-3′. The sense and antisense primers code for the signal peptide of the vesicular stomatitis virus G protein (MKCLLYLAFLFIGVNC) and the 1D4 tag (TETSQVAPA) peptide sequence, respectively. The amplified sequences coding for E were digested with BssHII and NsiI and introduced into the corresponding sites of the pSFV2 vector (22). The recombinant pSFV-E plasmids were sequenced through the prM/sE coding region.
Pulse-chase assays and expression and detection of recombinant sE proteins.
Recombinant SFV-E particles were produced by electroporation of in vitro-synthesized vector RNA into BHK cells (27). sE proteins were produced by infection of BHK cells with recombinant SFV-E particles (MOI, 50) as previously described (26, 27). For the pulse-chase assays, the cells were starved at 7 h postinfection for 45 min in serum and methionine/cysteine-free DMEM (ICN Biomedicals) prior to the addition of 200 μCi·ml−1 [35S]cysteine and [35S]methionine (Pro-Mix 35S; Amersham Biosciences). The pulse times were 15 min and 25 min for BHK and C6/36 cells, respectively. Subsequently, the cells were extensively washed and the synthesis of sE proteins was continued to 7 h in serum-free DMEM. The cells were lysed in buffer A (20 mM Tris [pH ∼7.5], 50 mM NaCl, 2 mM EDTA, 1% Triton X-100, supplemented with a protease inhibitor cocktail [Roche]). The supernatants and lysed cells were clarified prior to protein immunoprecipitation by using mAb 1D4 (20 μg·ml−1) that recognizes the C9-tag peptide sequence (University of British Columbia, Vancouver, British Columbia, Canada) and analyzed by SDS-PAGE (Nupage Novex N,N-methylenebisacrylamide-Tris gels; Invitrogen) as previously described (26). The gels were fixed, dried, and exposed onto Biomax MR1 film (Kodak). 35S-radiolabeled sE proteins were quantified by ImageJ software. The percentage of secreted sE protein was calculated as follows: % secreted sE protein = 100·[(RUtx− − sRUt0)/(cRUt0 − RUBG)], where t0 is the beginning of the kinetics; RUtx is the secreted sE protein in relative units at a given time point of the kinetics; sRUt0 is the secreted sE protein in relative units at t0; cRUt0 is the sE protein in the cells in relative units at t0; and RUBG is the background in relative units, corresponding to a negative control (empty well).
For standard protein production, the cells' media were replaced by 0% FCS Glasgow minimal essential medium at 7 h postinfection and the synthesis of sE proteins was continued to 16 h postinfection. The cells were lysed in buffer A. The supernatant and lysed cells were clarified, and the proteins were then analyzed by Western blotting as previously described (26).
Flow cytometry assays.
Cells (105) were exposed to DV particles for 2 h at 37°C in FCS-free RPMI 1640 supplemented with 0.2% bovine serum albumin, 1% penicillin/streptomycin. The cells were subsequently washed with complete RPMI 1640 to remove excess virus and incubated at 37°C. Intracellular viral antigens were stained as previously described (26). Briefly, infected cells were fixed and permeabilized with cytofix and cytoperm buffers according to the manufacturer's instructions (Pharmingen). The permeabilized cells were stained with the DV E mAb 4G2 (10 μg·ml−1; LGC Promochem) for 45 min at 4°C prior to washing and addition of the secondary phycoerythrin-conjugated anti-mouse mAb (R0480; 1/500) for 35 min at 8°C. The stained cells were subsequently washed and analyzed by FACS (FACSCalibur, BD Biosciences). Data were processed with CellQuest software (BD Biosciences). For the binding experiments, Vero and BHK cells (2 × 105) were incubated with equivalent genome copy numbers of WT DV-2 and the N67Q mutant (50 genome equivalents of DV/cell, determined by quantitative PCR) for 2 h at 4°C in RMPI medium supplemented with 1% bovine serum albumin and 10 mM HEPES. The cells were washed three times with cold PBS and fixed in 4% paraformaldehyde in PBS for 30 min. The protocol was followed as described above for FACS analysis. The data are representative of two independent experiments.
RESULTS
Replication of DV lacking N-glycosylation of the E protein in mosquito cells.
To determine the role of DV E glycosylation during viral replication, we generated recombinant viruses with specific mutations in each of the potential N-glycosylation sites, Asn-67 and Asn-153. These glycosylation sites were removed by changing the asparagine into glutamine in a cDNA clone of DV-2 that was used to produce full-length mutated RNAs. The WT and mutated RNAs carrying the N67Q, N153Q, or N67Q+N153Q substitutions were transfected into C6/36 mosquito cells. Viral replication was analyzed by IF using antibodies against DV-2. Seven days after transfection with WT and mutant RNAs, about 50% of the monolayer was IF positive (Fig. 1A). To confirm that the mutations introduced into the viral RNA were maintained in the replicating viruses, the RNA was purified from viral particles and used in RT-PCR and sequencing analysis. We observed that the three recombinant viruses conserved the original mutations (data not shown).
FIG. 1.
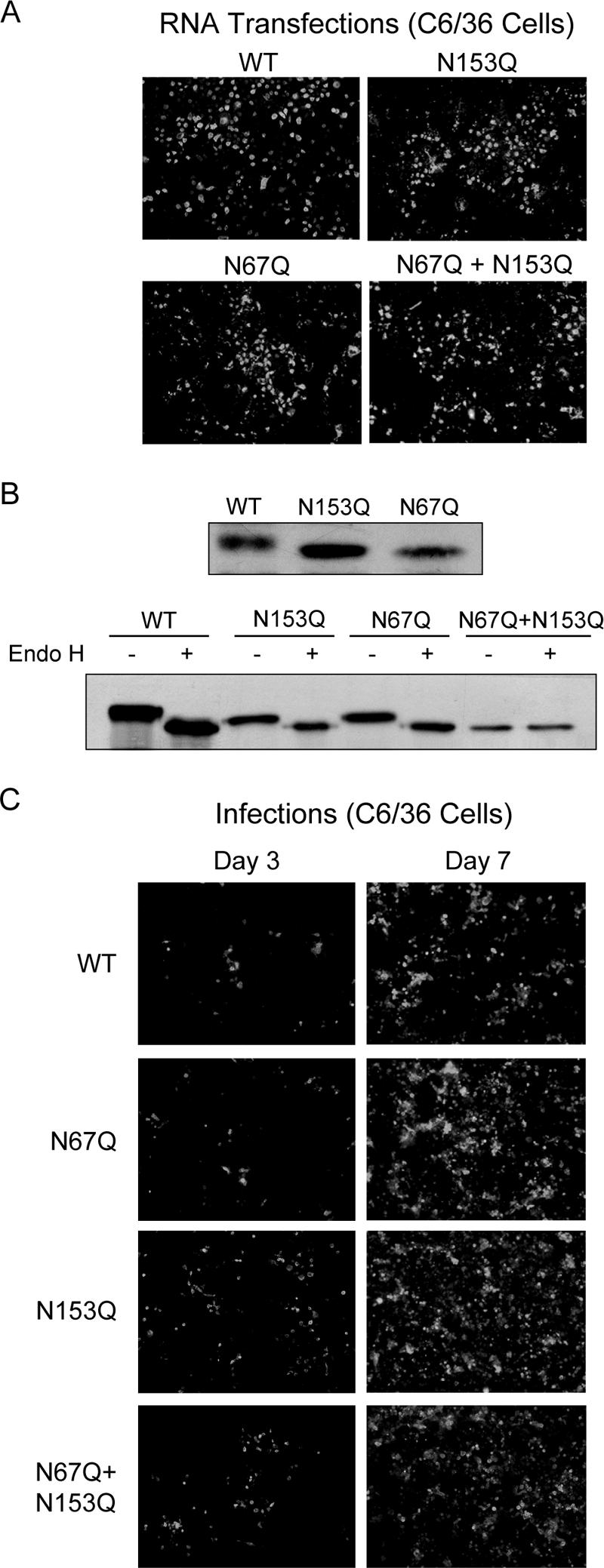
Replication of DV glycosylation mutants in mosquito cells. (A) IF assays of C6/36 cells transfected with DV RNAs, WT and mutated in the glycosylation site at Asn-67 (N67Q), Asn-153 (N153Q), and in both sites (N67Q+N153Q). The IF analysis was performed at 7 days after RNA transfection using polyclonal antibodies against DV-2. Images were taken at ×200 magnification. (B) Western blots of DV WT E and glycosylation mutants from C6/36 cell lysates at 7 days postinfection. The lysates were treated with Endo H (+) or buffer control (−). (C) IF analysis of C6/36 cells at 3 and 7 days postinfection with WT DV and the N67Q, N153Q, and N67Q+N153Q glycosylation mutants. Infection was normalized based on the amount of viral RNA in the inoculum.
To analyze whether the two glycosylation sites were utilized in mosquito cells, lysates from infected cells were prepared and analyzed by Western blotting. The mobility of the mutated E proteins (N67Q and N153Q) was faster than that of the WT E, suggesting that both asparagines were glycosylated (Fig. 1B). To confirm this observation, we examined the Endo H sensitivities of E proteins obtained from cells infected with WT and mutated viruses. This treatment resulted in a noticeable mobility shift of the three proteins, WT, N67Q, and N153Q, with the larger change observed for the WT protein, which presumably carries two glycan molecules. In agreement with this observation, the N67Q+N153Q double-mutant E protein was insensitive to Endo H treatment (Fig. 1B).
Viral stocks of the recombinant viruses lacking each glycosylation site were generated and quantified by real-time RT-PCR. Equivalent numbers of genome copies were used to infect fresh cells, and IF analysis was performed as a function of time. The results indicate that recombinant viruses lacking one or both glycosylation sites replicate and propagate in C6/36 cells (Fig. 1C).
Requirement of DV E glycosylation for efficient replication in mammalian cells.
To investigate the requirement of E glycosylation during DV infection in mammalian cells, we transfected the RNAs corresponding to the WT virus and N67Q, N153Q, and N67Q+N153Q mutants into BHK cells. The accumulation of viral proteins was determined by IF at day 5 after transfection. At this time, almost the complete monolayer was viral-antigen positive for the WT and the recombinant N153Q, while the N67Q and N67Q N153Q mutants did not replicate (Fig. 2A).
FIG. 2.

Replication of DV glycosylation mutants in BHK cells. (A) IF assays of BHK cells transfected with DV WT RNA and mutants in the glycosylation site at Asn-67 (N67Q), Asn-153 (N153Q), and in both sites (N67Q+N153Q). The IF analysis was performed at 5 days after RNA transfection using polyclonal antibodies against DV-2. Images were taken at ×200 magnification. (B) IF analysis of BHK cells at 1, 2, and 3 days postinfection with WT DV and the N67Q, N153Q, and N67Q+N153Q glycosylation mutants. Infection was normalized based on the amount of viral RNA in the inoculum. (C) Production of viral particles from BHK cells infected with WT, N67Q, N153Q, and N67Q N153Q viruses was determined by quantification of viral RNA using fluorogenic RT-PCR. RNA copy numbers per ml are shown at different times postinfection. Error bars indicate standard errors of the means. (D) Secretion of E protein from cells infected with WT virus and E glycosylation mutants. The media of infected BHK cells were used for Western blot analysis. Samples of the WT and N67Q and N153Q mutant viruses collected at different times postinfection as indicated above the gel were analyzed.
To characterize the defect of viruses carrying the mutation N67Q in mammalian cells, we used viral stocks produced in C6/36 cells. We infected BHK cells with viruses lacking each of the glycosylation sites obtained in mosquito cells and monitored replication by IF as a function of time. Interestingly, when equivalent numbers of genome copies, determined by real-time RT-PCR, were used for infection, DV proteins were readily detected at day 1 in cells infected with the WT and the N67Q mutant (Fig. 2B). The positive signal for DV antigens observed for the N67Q mutant was maintained for two days but then decreased as a function of time, while the positive signal in cells infected with the WT virus increased, reaching the complete monolayer at day 3. The N153Q virus was delayed in replication; however, at day 3 the IF signal was near the WT level. The N67Q+N153Q double-mutant virus showed delayed infection and, like the N67Q mutant, failed to propagate (Fig. 2B). To exclude a possible temperature-dependent phenotype for the N67Q mutant, BHK cells were infected and grown at 32°C. In these conditions, results similar to the ones observed at 37°C were obtained (data not shown).
To analyze whether viral particles of the N67Q and N67Q+N153Q mutants were released from infected BHK cells, we harvested the supernatants at different times postinfection and quantified the viral RNA by real-time RT-PCR. No viral RNA was detected in the supernatants of cells infected with the two mutants lacking the Asn-67 (Fig. 2C), indicating that viral particle assembly or release was impaired. In contrast, RNA from the N153Q mutant was readily detected; however, the amount of viral RNA quantified for this virus was about two orders of magnitude lower than for the WT. Furthermore, we analyzed the presence of the E protein in the medium by Western blot analysis as a function of time after infection with the WT virus and the N67Q and N153Q mutants. As shown in Fig. 2D, the E protein was accumulated in the medium of cells infected with the WT virus but undetectable after infection with the N67Q mutant. In addition, consistent with the delayed replication of the N153Q mutant observed in the IF analysis, accumulation of E in the medium of cells infected with the N153Q mutant was delayed and reduced in comparison to the accumulation of the WT virus.
Ablation of Asn-153 of the E protein reduces DV infectivity.
Utilization of Asn-153 for N-glycosylation in mammalian cells was determined by Western blot analysis. The E protein obtained from BHK cells infected with N153Q virus showed a faster mobility than that from the WT protein (Fig. 3A). In addition, treatment with PNGase F resulted in a mobility change of both the WT and the N153Q E proteins, suggesting the utilization of both potential glycosylation sites, Asn-67 and Asn-153, in mammalian cells (Fig. 3A).
FIG. 3.

Decreased infectivity of DV particles bearing the N153Q mutation in the E protein. (A) Western blots of WT DV E and the N153Q glycosylation mutant from BHK cell lysates at 3 days postinfection. The lysates were treated with PNGase F (+) or buffer control (−). (B) The top panel shows a representative image comparing the plaque phenotype of WT DV with that of the N153Q mutant virus. The plot shows one-step growth curves of WT DV and the N153Q mutant in BHK cells. The cells were infected at an MOI of 0.01, and titers were determined at each time point by plaque assay. Error bars indicate standard errors of the means. At 2 and 4 days, as indicated by the arrows, the supernatants of infected cells were used to extract and quantify the viral RNA by fluorogenic RT-PCR. Values are means ± standard errors. (C) The PFU/RNA ratios obtained in supernatants at 2 and 4 days postinfection are shown for the WT and N153Q mutant viruses. (D) Comparison of stability of WT and N153Q DV. Viral stocks were incubated at 37°C, aliquots were removed at different times, and the amounts of infectious particles were determined by plaque assay in BHK cells. Error bars indicate standard errors of the means.
To determine the replication defect of the N153Q mutant DV, we analyzed first whether the mutant viral particles were less infectious or if they were inefficiently released from infected cells. To this end, we infected BHK cells with the same MOI for WT DV and the N153Q mutant, and the infectivity-to-viral-RNA ratio was analyzed in the supernatants of infected cells. Supernatants were harvested from WT- and N153Q DV-infected BHK cells and centrifuged to remove cell debris, and the amount of infectious particles was determined by plaque assay. In parallel, viral RNA was extracted and quantified by real-time RT-PCR (Fig. 3B). The N153Q virus produced smaller plaques than the WT virus (Fig. 3B). The differences between WT and N153Q DV in the ratio of viral infectivity to genome copies were 10- and 17-fold at 2 and 4 days postinfection, respectively. At day 2 postinfection, the amount of viral particles produced by the N153Q mutant was similar to that for the WT virus, but the infectivity of the mutant particles was 10-fold lower (Fig. 3C). These results suggest that, even though large amounts of viral particles were released from cells infected with N153Q mutant DV, their infectivity was greatly reduced in comparison to that of the WT virus. One possibility is that the particles carrying the mutated E display lower stability. In order to compare the stability of N153Q and WT viral particles, we performed a heat inactivation experiment. We incubated both viral stocks at 37°C and determined infectivity as a function of time (Fig. 3D). Similar reductions in the viral titers were observed, suggesting comparable stability of WT and N153Q particles in these experimental conditions.
Replication defects of DV lacking glycosylation at Asn-67.
The N67Q mutant DV was able to enter BHK cells, but viral particles were not produced (Fig. 2). To better examine the requirement of glycosylation at Asn-67 during DV replication, we followed infection of BHK cells by FACS analysis. Cells were infected with WT and N67Q mutant DV, with the virus inoculum normalized based on the amount of infectious particles, and the percentages of cells producing viral proteins were determined at different times postinfection by FACS analysis. The numbers of cells positive for the DV antigen were similar for the WT and the N67Q virus at 18 and 24 h postinfection (Fig. 4A). However, after 24 h, the percentage of N67Q DV-infected cells greatly decreased, while the number of cells infected with the WT DV continued to grow. This observation suggests an essential role of glycosylation at Asn-67 during DV propagation. Similar defects were observed in HeLa, Huh-7, and A549 cells infected with the N67Q virus (data not shown).
FIG. 4.

Replication of DV-2 bearing the N67Q mutation in different mammalian cells. (A) BHK cells were infected with equal amounts of WT DV and N67Q DV infectious particles. Cells were harvested at 18, 24, 48, and 72 h postinfection, and viral infectivity was quantified by FACS analysis using mAb 4G2. Values are the means ± standard errors of triplicate experiments. (B) Binding of WT DV and the N67Q mutant. FACS analyses detecting cell-associated viral antigens in BHK and Vero cells are shown. The cells were incubated with medium as control (gray) or WT or N67Q DV corresponding to 50 genome equivalents of DV/cell (black), kept at 4°C, and analyzed after 2 h. The numbers in parentheses represent the means of the fluorescence intensities. The data are representative of two independent experiments. (C) Infection of BHK and Vero cells with WT DV and the N67Q mutant. The cells were infected with equivalent genome copy numbers of WT DV and the N67Q mutant (determined by quantitative PCR). The cells were stained for intracellular E protein production 24 h postinfection and analyzed by FACS. The data are representative of three independent experiments.
Normalization of the viral input by the amount of infectious particles as described in the previous experiments could mask differences in viral entry. Therefore, to analyze a possible role of the glycan at Asn-67 during entry, we normalized the inoculum by the amount of viral RNA. In this case, BHK and Vero cells were incubated with equivalent genome copy numbers of WT and N67Q mutant DV. The viral antigen was quantified by FACS analysis either after 2 h of incubation at 4°C to assess binding or after 24 h at 37°C to evaluate viral entry and antigen production inside the cell. The results shown in Fig. 4B indicate similar binding of the WT and the N67Q mutant to BHK and Vero cells. In addition, the quantification of viral antigens at 24 h shows that, even though the N67Q mutant virus was able to infect both cell types, the specific infectivity was two- to threefold lower than that observed for the WT virus (Fig. 4C).
Secretion defect of DV E glycoprotein lacking glycosylation at Asn-67.
In order to determine whether the synthesis or secretion of the E in mosquito and mammalian cells was affected by the presence or absence of glycosylation, we examined the accumulation and secretion of WT, N67Q, and N153Q E proteins in both C6/36 and BHK cells. To this end, the coding sequence for prM and the extracellular domain of E, corresponding to amino acids 115 to 674 of the viral polyprotein, was produced using an SFV expression system as previously described (27). The presence of sE in total cell extracts and supernatants was evaluated by Western blotting, using mAb 1D4 directed against the C9 tag peptide introduced into the carboxy-terminal domain of sE (Fig. 5A). In C6/36 cells, both WT sE and the glycosylation mutants were efficiently produced and secreted (Fig. 5A, left panel). In contrast, while the E proteins were detected in the cytoplasm of BHK cells, N67Q E secretion was severely impaired, as indicated by the lack of sE in the supernatant (Fig. 5A, right panel). Secretion of the N153Q mutant was only slightly reduced in comparison to secretion of the WT protein (Fig. 5A, right panel).
FIG. 5.

Secretion of DV-2 E glycosylation mutants expressed in mosquito and mammalian cells. Soluble forms of WT DV-2 E protein and glycosylation mutants were expressed using the SFV vector either in C6/36 cells or BHK cells. (A) Western blots of WT DV-2 E protein and glycosylation mutants 1 day postinfection with SFV vector in cell extracts (C) and in supernatants (S), as indicated above each gel. The extracts and supernatants of uninfected cells (UI) were used as negative controls. The E glycoproteins were detected by using mAb 1D4 directed against the C9 tag peptide introduced into the C terminus of E. Molecular markers are shown on the right. (B) Kinetics of processing and secretion of DV-2 E protein in BHK and C6/36 cells. The WT E and glycosylation mutants were pulse-labeled with [35S]Cys/Met. Cytoplasmic extracts and supernatants were subjected to immunoprecipitation using mAb 1D4 at 1, 3, 5, and 7 h postinfection with SFV. Radiolabeled E proteins immunoprecipitated from cell extracts or from the medium, as indicated above each gel, are shown for each virus. Molecular markers are shown on the left. (C) Quantification of the secreted E proteins shown in panel B. The amount of WT E and the N67Q and N153Q E mutants was determined as a function of time using ImageJ software. These data are representative of two independent experiments.
To further analyze the secretion defect of the N67Q mutant, we performed pulse-chase labeling experiments. The secretion kinetics of WT and mutated E proteins were followed in both mosquito and mammalian cells (Fig. 5B). In C6/36 cells, WT, N67Q, and N153Q E proteins were detected in the supernatants of infected cells after 1 h of chase, with a significant increase at 7 h. In BHK cells, WT and N153Q E proteins were efficiently produced and secreted after 3 h of chase. In contrast, the secretion of N67Q mutant E was dramatically reduced in comparison to the secretion of the WT control and the N153Q E (Fig. 5B, right panel). Quantification of immunoprecipitated proteins at different times after the chase in BHK cells indicated that the secretion of the N67Q mutant E was reduced by about 90% relative to that of the WT protein (Fig. 5C). Although the folding/trafficking of the soluble form of E and the membrane-bound protein could be influenced by different factors, in these experiments it was possible to compare the secretion of the same protein, glycosylated or not, in the two cell types. Taken together, the results suggest that the N-linked glycosylation site at position 67 is required for the efficient secretion of DV-2 E in mammalian cells, but not in mosquito cells.
Reporter virus confirms a propagation defect of DV lacking Asn-67 of the E protein.
To further confirm the replication defect of N67Q mutant DV, we used an infectious DV-2 carrying a reporter gene, DV-R. In this construct, a Renilla luciferase coding sequence was introduced preceding the viral 3′ UTR under the translation control of the EMCV IRES. Transfection of the DV-R RNA into BHK cells resulted in productive viral replication. A viral RNA 10% larger than the WT genome was efficiently replicated and encapsidated, and viral stocks on the order of 1 × 106 viral particles per ml were obtained. One-step growth curves comparing the replication of the WT and DV-R showed slight lower replication of the virus carrying the luciferase gene; the titers at 24 and 48 h were about fivefold lower than the WT titers (Fig. 6A)
FIG. 6.

Reporter DV as a tool to study viral replication. (A) Replication of reporter virus DV-R in BHK cells. The top panel shows a schematic representation of the DV-R genome. The boxes denote the coding sequences of the bicistronic construct, including the viral sequences in the first cistron and the Renilla luciferase (Ren) coding sequence in the second cistron. The bottom panel shows a one-step growth curve comparing WT DV and DV-R. BHK cells were infected at an MOI of 0.1. Viral titers were determined by plaque assays at different times. (B) Time courses of luciferase activity determined in cytoplasmic extracts of DV-R-infected cells in the presence or absence of inhibitors heparin (20 μg/ml) (Hep.) and adenosine analogue (25 μM) (RdRp Inh.). (C) Time courses of luciferase activity determined in cytoplasmic extracts of BHK cells infected with WT DV-R and the N67Q glycosylation mutant. Error bars indicate standard errors of the means.
Upon infection of BHK cells with DV-R, an increase of luciferase activity was observed. The kinetics was similar to the one previously reported for a WNV reporter construct (37). Known inhibitors that interfere with viral entry and RNA synthesis were used to characterize different steps of viral replication by using the reporter activity. The addition of the entry inhibitor, heparin, at the time of infection rendered background levels of luciferase (Fig. 6B). In addition, treatment with an adenosine analogue, an inhibitor of the viral polymerase, used at concentrations that abolished DV-2 replication, showed an effect on luciferase activity after 12 h, suggesting that after this time, newly synthesized RNA is used to produce the reporter protein. With this inhibitor there is a pronounced drop in luciferase activity after 30 h, reflecting complete inhibition of RNA amplification. Therefore, DV-R provides a useful investigational tool for analyzing different steps of the viral life cycle, including morphogenesis, using a sensitive reporter assay.
In order to use DV-R to study the defect of the mutant lacking glycosylation at Asn-67, this mutation was introduced in the cDNA of the DV-R construct and viral particles were produced in C6/36 cells by transfection of the in vitro-transcribed RNA. It is important to mention that replication of DV-R cannot be monitored by reporter activity in C6/36 cells because the EMCV IRES does not function in insect cells. Infection of BHK cells with WT DV-R and the N67Q mutant, normalized by genome copy number, showed similar luciferase levels for the two viruses until 48 h. However, the reporter signal for the mutant virus decreased dramatically after 60 h in comparison to the signal for the WT (Fig. 6C). These results indicate that viral translation and RNA synthesis were not significantly affected by the lack of Asn-67, while the viral propagation of this glycosylation mutant was impaired.
Glycosylation at position 67 of DV E protein enhances viral entry in DC-SIGN-expressing cells.
A recent cryoelectron microscopy study indicated that the CRD domain of DC-SIGN interacts preferentially with the N-linked glycans present at position 67 on DV-2 particles (36). To study whether the glycosylation at Asn-67 was required for DC-SIGN-mediated enhancement of DV infection, Raji, Raji-DC-SIGN, and immature DCs were infected with WT and N67Q DV. The intracellular viral antigen production was quantified by FACS analysis 24 h postinfection using mAb 4G2 against DV E protein.
Parental Raji cells were poorly susceptible to DV infection. However, DV infectivity is greatly increased by exogenously expressing DC-SIGN in this cell line. While 95% of the Raji cells expressing DC-SIGN were infected by DV, only 0.4% of the parental Raji cells were infected (see WT in Fig. 7A).
FIG. 7.

Glycosylation at Asn-67 enhances DV infection of DC-SIGN-expressing cells. (A) Raji and Raji-DC-SIGN cells were infected with equal amounts of WT and N67Q DV infectious particles. The cells were stained for intracellular E protein production 24 h postinfection and analyzed by FACS as described above. The data are representative of three independent experiments. (B) Immature DCs were infected with WT and N67Q DV at different MOIs, stained for intracellular E antigen production, and analyzed by FACS 24 h postinfection. (C) Time courses of replication of WT DV-2 and the N67Q and N153Q mutants in Raji cells expressing DC-SIGN. The cells were infected with equivalent amounts of WT, N67Q, and N153Q DV infectious particles. Viral infection was quantified by FACS analysis at 18, 24, 48, and 72 h postinfection. Values are the means of duplicate experiments. These data are representative of three independent experiments.
To analyze the contribution of glycosylation at Asn-67 on DC-SIGN-dependent enhancement of infection of DV, we compared the infectivities of WT and N67Q viruses in Raji-DC-SIGN cells, normalizing the viral input by the infectivity in the parental Raji cells. The viral dilution that produced similar infection for each virus in Raji cells was then used to infect DC-SIGN-expressing Raji cells. Interestingly, we observed that while the WT virus infected 95.9% of the DC-SIGN-expressing cells, the N67Q virus exhibited a reduced infectivity (23.7%, Fig. 7A). Similar results were obtained with Raji cells expressing the DC-SIGN homologue L-SIGN (data not shown).
To investigate the capacities of WT and N67Q viruses to infect immature DCs, we generated monocyte-derived DCs and infected them with dilutions of the viral stocks normalized by their infectivities in Raji cells (note the low MOI numbers that represent the low infectivities in the parental Raji cells). As indicated in Fig. 7B, about 95 and 90% of monocyte-derived DCs were productively infected by WT DV at MOIs of 0.5 and 0.05, respectively. In contrast, exposure of DCs to N67Q DV particles resulted in infection of 40% and 25% of cells at MOIs of 0.5 and 0.05, respectively. At low virus concentrations, we observed that monocyte-derived DCs were almost uninfected by N67Q viruses, whereas the WT virus still infected 40% of the cells (Fig. 7B), indicating that a glycan at position 67 in DV enhances infection of immature DCs.
Furthermore, we determined the amounts of Raji-DC-SIGN cells infected with WT, N67Q, and N153Q viruses as a function of time postinfection. The results indicated that both glycosylation mutants showed a decreased infectivity at 24 h after infection in comparison to the infectivity of the WT control (Fig. 7C). However, after 40 h of infection, while the amount of cells infected with the mutant N153Q was similar to the amount infected with the WT virus, the amount of cells infected with the mutant N67Q virus declined, in agreement with the propagation defect observed for this mutant in other mammalian cells. Similar results were obtained with Raji cells expressing L-SIGN (data not shown).
DISCUSSION
In this study, we demonstrate a critical role of N-glycosylation of DV E protein during viral particle formation in mammalian cells. DV naturally cycles between mosquito and human hosts. Although we found that both potential glycosylation sites of DV-2 E protein are utilized in mammalian and mosquito cells, glycosylation of E was not essential for viral replication in insect cells and DV stocks lacking glycosylation in the E protein could be obtained from C6/36 cells.
We have constructed a sensitive DV reporter, DV-R, that allows the discrimination of each step of viral replication, including viral morphogenesis (Fig. 6). The elimination of Asn-67 in the E protein in the context of infectious DV or DV-R resulted in a dramatic decrease of viral particle assembly/release in mammalian cells. In addition, E protein was undetectable in the media of BHK cells infected with the N67Q mutant virus (Fig. 2D). This phenotype could be due to alteration in the folding of E, assembly, or particle release. The replication defect appears to result from the lack of the N-linked glycan rather than the introduction of deleterious amino acid substitutions, because the E protein was fully functional in mosquito cells. All DV glycosylation mutants propagated in mosquito cells (Fig. 1). We provided evidence that the differential requirements of E glycosylation for DV replication in mammalian and mosquito cells can be due to differing E secretion in the two host cells (Fig. 5). It is known that protein folding is dependent on temperature. Therefore, one possible explanation for this host-dependent phenotypic difference could be the ability of mosquito cells to properly fold and secrete the E at the low temperature used to grow C6/36 cells (28°C), while mammalian cells grown at 37°C are unable to secrete this protein. To analyze this possibility, we infected C6/36 and BHK cells, both grown at 32°C. The viruses lacking the glycosylation sites replicated efficiently at 32°C in C6/36 cells, while the same viruses were unable to propagate in BHK cells at the same temperature (data not shown). These results indicate that proper folding of E protein lacking a glycan at position 67 in mosquito cells is not a temperature-dependent phenomenon. Instead, it could be due to different protein folding pathways used by the two host cells. Insect and mammalian cells are known to express different metabolic enzymes involved in processing N-glycans (43). N-linked carbohydrates play important roles in protein folding by serving as ligands of the ER chaperones calnexin and calreticulin. In addition, Erp57, a member of the PDI family, catalyzes correct disulfide bond formation specifically on glycoproteins bound to calnexin and calreticulin (34, 47). Erp57 could be involved in folding of the glycosylated E protein, because it contains 12 cysteines that form six highly conserved disulfide bonds. In light of our results, it is likely that insect cells provide a glycosylation-independent pathway for E protein secretion, while in mammalian cells, host factors would be necessary to specifically recognize the glycan at Asn-67 for proper E-protein folding.
It has been unclear whether DV-2 and DV-4 E proteins were glycosylated at position 153 (18). Using DV-2 16681, our results indicate that Asn-153 is glycosylated in both mammalian and mosquito cells (Fig. 1 and 3). It has been previously shown that serial passages of DV-3 in mosquito cells consistently selected a change in Thr155, resulting in the loss of the glycosylation site at position 153 (21). These results suggested that the sugar moiety at this position might not be advantageous for virus replication in insect cells, which is in agreement with our observations with the N153Q DV that replicated efficiently in C6/36 cells (Fig. 1). In contrast, a deleterious effect on viral infectivity of the N153Q mutant was observed in BHK cells. The amounts of viral particles produced 2 days after BHK cells were infected with similar MOIs of WT and N153Q DV were about two- to threefold higher for the WT DV; however, the infectivity of the released particles containing viral RNA was greatly decreased for the mutant virus (Fig. 3). These results suggest that the glycan at position 153 modestly increases viral particle production but dramatically increases viral infectivity in mammalian cells. In fact, adapted DV mutants previously selected to fuse at a higher pH resulted in the loss of glycosylation at Asn-153, providing evidence for a role for this glycan in the fusion process (10). According to the structure determined for DV and other flaviviruses, this carbohydrate moiety covers the fusion peptide (30, 31, 38). Therefore, the lower infectivity observed for N153Q DV could be explained by a possible role of this glycan in modulating the fusion process.
It has recently been shown for WNV that glycosylation at Asn-154 (corresponding to Asn-153 in DV) plays a role in both viral assembly and infectivity. However, in contrast to our observations using DV, the presence of the N-linked carbohydrate at amino acid 154 had a deleterious effect on viral infectivity. The glycan at Asn-154 decreased WNV infectivity in BHK cells and dramatically reduced its infectivity in C6/36 cells (11). Together, the results indicate that the glycans at position 153 or 154 play different roles in DV and WNV, respectively, and also highlight a host-dependent function. Furthermore, it has been reported that tick-borne encephalitis virus E protein lacking the glycosylation site at position 154 showed a reduced E secretion (24), similar to the phenotype observed here for DV lacking the Asn-67, suggesting analogous roles of carbohydrates present at different positions on the E proteins of these flaviviruses.
We observed that the N-linked glycan at position 67 also plays a role in DC-SIGN-mediated enhancement of DV entry (Fig. 7). DV lacking Asn-67 exhibits a reduced capacity to infect immature DCs and Raji-DC-SIGN cells. In keeping with this finding, it has recently been shown that DC-SIGN CRD interacts with N-linked glycans at position 67 on DV-2 particles (36). Furthermore, the introduction of a glycosylation site at position 67 into WNV E protein has been shown to enhance DC-SIGN utilization (7). Reporter virus particles pseudotyped with WNV E protein bearing N-linked glycans at position 67 infected DC-SIGN-expressing cells as efficiently as reporter virus particles bearing the DV-E protein (7). We found that the lack of Asn-67 did not abolish DV infection of DCs or DC-SIGN-expressing Raji cells, suggesting the contribution of other carbohydrate moieties to DC-SIGN-mediated enhancement of infection. DV glycosylation mutants lacking Asn-153 also showed a reduced infectivity in Raji-DCs (Fig. 7C). However, it is unclear whether the glycan at position 153 directly contributes to DC-SIGN-dependent entry or if the lower infectivity of this glycosylation mutant was due to a fusion defect as previously reported (10, 21). Interestingly, using WNV, it has been demonstrated that the presence of a single glycosylation site on either prM or E glycoproteins was sufficient to allow DC-SIGN- or L-SIGN-mediated infection (8), suggesting the presence of immature prM in the viral particle and a possible role of this glycoprotein during WNV entry. Clearly, further studies are necessary to define whether other glycans present at the surface of DV particles, besides the one at Asn-67, are required for DC-SIGN binding and infection of DCs.
Previous reports have shown that the iminoglycans castanospermine (CSP) and deoxinojirimycin, inhibitors of α-glucosidases, decrease DV replication by altering the folding of the prM and E glycoproteins in mouse neuroblastoma cells (6). The decreased secretion of E observed in the presence of CSP suggests a misfolded state of the protein in treated cells (6). The phenotype previously observed with CSP resembles the one observed in the present work with the DV mutant lacking glycosylation at Asn-67 of E. It was also reported that the four serotypes of DV are inhibited by CSP, while yellow fever virus and WNV are partially or almost completely resistant, respectively, to the effects of the drug (45). A similar difference in sensitivity to the effect of deoxinojirimycin was observed in a separate study in which DV was compared to Japanese encephalitis virus (46). The molecular basis for the differential susceptibilities of flaviviruses to α-glucosidase inhibitors remains unclear. Considering the absolute requirement of glycosylation at Asn-67 observed during DV morphogenesis (Fig. 2, 4, and 6), and knowing that DV is the only flavivirus bearing this glycosylation site, a defect in processing the glycan at Asn-67 could be the explanation for the differing susceptibilities to the inhibitors previously observed between DV and other flaviviruses. The presence of a glycosylation site at Asn-67 is absolutely conserved in all DV isolates. Interestingly, a mouse-adapted strain of DV-1 was reported to lack the Thr at position 69, resulting in the loss of the glycosylation site (16).
Taken together, these results define an essential role of E glycosylation at Asn-67 during DV exit from mammalian cells. This finding highlights the specific requirements of host factors for DV protein folding and secretion. We expect that understanding molecular details of DV replication will provide new targets for antiviral strategies.
Acknowledgments
The authors thank Laura Talarico, Fernando Arenzana-Seisdedos, Félix Rey, Laura Burleigh, and members of A. V. Gamarnik's laboratory for helpful discussions. We also thank Michael Diamond and Wen Chang for antibodies against E protein and Richard Kinney for the DV cDNA infectious clone.
This work was supported by grants from HHMI, DENFRAME, PICT-2003, and ICGEB-OPS-RELAB to A.V.G. and the Institut National de la Santé et Recherche Médicale (INSERM) and Pediatric Dengue Vaccine Initiative (PDVI) to A.A. J.A.M. was funded by CONICET and P.-Y.L. by PDVI fellowships.
Footnotes
Published ahead of print on 25 April 2007.
REFERENCES
- 1.Allison, S. L., Y. J. Tao, G. O'Riordain, C. W. Mandl, S. C. Harrison, and F. X. Heinz. 2003. Two distinct size classes of immature and mature subviral particles from tick-borne encephalitis virus. J. Virol. 77:11357-11366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Alvarez, D. E., A. L. De Lella Ezcurra, S. Fucito, and A. V. Gamarnik. 2005. Role of RNA structures present at the 3′UTR of dengue virus on translation, RNA synthesis, and viral replication. Virology 339:200-212. [DOI] [PubMed] [Google Scholar]
- 3.Alvarez, D. E., M. F. Lodeiro, S. J. Luduena, L. I. Pietrasanta, and A. V. Gamarnik. 2005. Long-range RNA-RNA interactions circularize the dengue virus genome. J. Virol. 79:6631-6643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Beasley, D. W., M. C. Whiteman, S. Zhang, C. Y. Huang, B. S. Schneider, D. R. Smith, G. D. Gromowski, S. Higgs, R. M. Kinney, and A. D. Barrett. 2005. Envelope protein glycosylation status influences mouse neuroinvasion phenotype of genetic lineage 1 West Nile virus strains. J. Virol. 79:8339-8347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chen, Y., T. Maguire, R. E. Hileman, J. R. Fromm, J. D. Esko, R. J. Linhardt, and R. M. Marks. 1997. Dengue virus infectivity depends on envelope protein binding to target cell heparan sulfate. Nat. Med. 3:866-871. [DOI] [PubMed] [Google Scholar]
- 6.Courageot, M. P., M. P. Frenkiel, C. D. Dos Santos, V. Deubel, and P. Despres. 2000. Alpha-glucosidase inhibitors reduce dengue virus production by affecting the initial steps of virion morphogenesis in the endoplasmic reticulum. J. Virol. 74:564-572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Davis, C. W., L. M. Mattei, H. Y. Nguyen, C. Ansarah-Sobrinho, R. W. Doms, and T. C. Pierson. 2006. The location of asparagine-linked glycans on West Nile virions controls their interactions with CD209 (dendritic cell-specific ICAM-3 grabbing nonintegrin). J. Biol. Chem. 281:37183-37194. [DOI] [PubMed] [Google Scholar]
- 8.Davis, C. W., H. Y. Nguyen, S. L. Hanna, M. D. Sanchez, R. W. Doms, and T. C. Pierson. 2006. West Nile virus discriminates between DC-SIGN and DC-SIGNR for cellular attachment and infection. J. Virol. 80:1290-1301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Germi, R., J. M. Crance, D. Garin, J. Guimet, H. Lortat-Jacob, R. W. Ruigrok, J. P. Zarski, and E. Drouet. 2002. Heparan sulfate-mediated binding of infectious dengue virus type 2 and yellow fever virus. Virology 292:162-168. [DOI] [PubMed] [Google Scholar]
- 10.Guirakhoo, F., A. R. Hunt, J. G. Lewis, and J. T. Roehrig. 1993. Selection and partial characterization of dengue 2 virus mutants that induce fusion at elevated pH. Virology 194:219-223. [DOI] [PubMed] [Google Scholar]
- 11.Hanna, S. L., T. C. Pierson, M. D. Sanchez, A. A. Ahmed, M. M. Murtadha, and R. W. Doms. 2005. N-linked glycosylation of West Nile virus envelope proteins influences particle assembly and infectivity. J. Virol. 79:13262-13274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Heinz, F. X., and S. L. Allison. 2003. Flavivirus structure and membrane fusion. Adv. Virus Res. 59:63-97. [DOI] [PubMed] [Google Scholar]
- 13.Hilgard, P., and R. Stockert. 2000. Heparan sulfate proteoglycans initiate dengue virus infection of hepatocytes. Hepatology 32:1069-1077. [DOI] [PubMed] [Google Scholar]
- 14.Hung, J. J., M. T. Hsieh, M. J. Young, C. L. Kao, C. C. King, and W. Chang. 2004. An external loop region of domain III of dengue virus type 2 envelope protein is involved in serotype-specific binding to mosquito but not mammalian cells. J. Virol. 78:378-388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hung, S. L., P. L. Lee, H. W. Chen, L. K. Chen, C. L. Kao, and C. C. King. 1999. Analysis of the steps involved in dengue virus entry into host cells. Virology 257:156-167. [DOI] [PubMed] [Google Scholar]
- 16.Ishak, H., T. Takegami, K. Kamimura, and H. Funada. 2001. Comparative sequences of two type 1 dengue virus strains possessing different growth characteristics in vitro. Microbiol. Immunol. 45:327-331. [DOI] [PubMed] [Google Scholar]
- 17.Jindadamrongwech, S., C. Thepparit, and D. R. Smith. 2004. Identification of GRP 78 (BiP) as a liver cell expressed receptor element for dengue virus serotype 2. Arch. Virol. 149:915-927. [DOI] [PubMed] [Google Scholar]
- 18.Johnson, A. J., F. Guirakhoo, and J. T. Roehrig. 1994. The envelope glycoproteins of dengue 1 and dengue 2 viruses grown in mosquito cells differ in their utilization of potential glycosylation sites. Virology 203:241-249. [DOI] [PubMed] [Google Scholar]
- 19.Kawano, H., V. Rostapshov, L. Rosen, and C. J. Lai. 1993. Genetic determinants of dengue type 4 virus neurovirulence for mice. J. Virol. 67:6567-6575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kinney, R. M., S. Butrapet, G. J. Chang, K. R. Tsuchiya, J. T. Roehrig, N. Bhamarapravati, and D. J. Gubler. 1997. Construction of infectious cDNA clones for dengue 2 virus: strain 16681 and its attenuated vaccine derivative, strain PDK-53. Virology 230:300-308. [DOI] [PubMed] [Google Scholar]
- 21.Lee, E., R. C. Weir, and L. Dalgarno. 1997. Changes in the dengue virus major envelope protein on passaging and their localization on the three-dimensional structure of the protein. Virology 232:281-290. [DOI] [PubMed] [Google Scholar]
- 22.Liljestrom, P., and H. Garoff. 1991. A new generation of animal cell expression vectors based on the Semliki Forest virus replicon. Bio/Technology 9:1356-1361. [DOI] [PubMed] [Google Scholar]
- 23.Lorenz, I. C., S. L. Allison, F. X. Heinz, and A. Helenius. 2002. Folding and dimerization of tick-borne encephalitis virus envelope proteins prM and E in the endoplasmic reticulum. J. Virol. 76:5480-5491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lorenz, I. C., J. Kartenbeck, A. Mezzacasa, S. L. Allison, F. X. Heinz, and A. Helenius. 2003. Intracellular assembly and secretion of recombinant subviral particles from tick-borne encephalitis virus. J. Virol. 77:4370-4382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lozach, P. Y., A. Amara, B. Bartosch, J. L. Virelizier, F. Arenzana-Seisdedos, F. L. Cosset, and R. Altmeyer. 2004. C-type lectins L-SIGN and DC-SIGN capture and transmit infectious hepatitis C virus pseudotype particles. J. Biol. Chem. 279:32035-32045. [DOI] [PubMed] [Google Scholar]
- 26.Lozach, P. Y., L. Burleigh, I. Staropoli, E. Navarro-Sanchez, J. Harriague, J. L. Virelizier, F. A. Rey, P. Despres, F. Arenzana-Seisdedos, and A. Amara. 2005. Dendritic cell-specific intercellular adhesion molecule 3-grabbing non-integrin (DC-SIGN)-mediated enhancement of dengue virus infection is independent of DC-SIGN internalization signals. J. Biol. Chem. 280:23698-23708. [DOI] [PubMed] [Google Scholar]
- 27.Lozach, P. Y., H. Lortat-Jacob, A. de Lacroix de Lavalette, I. Staropoli, S. Foung, A. Amara, C. Houles, F. Fieschi, O. Schwartz, J. L. Virelizier, F. Arenzana-Seisdedos, and R. Altmeyer. 2003. DC-SIGN and L-SIGN are high affinity binding receptors for hepatitis C virus glycoprotein E2. J. Biol. Chem. 278:20358-20366. [DOI] [PubMed] [Google Scholar]
- 28.Martinez-Barragan, J. J., and R. M. del Angel. 2001. Identification of a putative coreceptor on Vero cells that participates in dengue 4 virus infection. J. Virol. 75:7818-7827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Modis, Y., S. Ogata, D. Clements, and S. C. Harrison. 2003. A ligand-binding pocket in the dengue virus envelope glycoprotein. Proc. Natl. Acad. Sci. USA 100:6986-6991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Modis, Y., S. Ogata, D. Clements, and S. C. Harrison. 2004. Structure of the dengue virus envelope protein after membrane fusion. Nature 427:313-319. [DOI] [PubMed] [Google Scholar]
- 31.Mukhopadhyay, S., B. S. Kim, P. R. Chipman, M. G. Rossmann, and R. J. Kuhn. 2003. Structure of West Nile virus. Science 302:248. [DOI] [PubMed] [Google Scholar]
- 32.Navarro-Sanchez, E., R. Altmeyer, A. Amara, O. Schwartz, F. Fieschi, J. L. Virelizier, F. Arenzana-Seisdedos, and P. Despres. 2003. Dendritic-cell-specific ICAM3-grabbing non-integrin is essential for the productive infection of human dendritic cells by mosquito-cell-derived dengue viruses. EMBO Rep. 4:723-728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Oliphant, T., M. Engle, G. E. Nybakken, C. Doane, S. Johnson, L. Huang, S. Gorlatov, E. Mehlhop, A. Marri, K. M. Chung, G. D. Ebel, L. D. Kramer, D. H. Fremont, and M. S. Diamond. 2005. Development of a humanized monoclonal antibody with therapeutic potential against West Nile virus. Nat. Med. 11:522-530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Oliver, J. D., H. L. Roderick, D. H. Llewellyn, and S. High. 1999. ERp57 functions as a subunit of specific complexes formed with the ER lectins calreticulin and calnexin. Mol. Biol. Cell 10:2573-2582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Parodi, A. J. 2000. Protein glucosylation and its role in protein folding. Annu. Rev. Biochem. 69:69-93. [DOI] [PubMed] [Google Scholar]
- 36.Pokidysheva, E., Y. Zhang, A. J. Battisti, C. M. Bator-Kelly, P. R. Chipman, C. Xiao, G. G. Gregorio, W. A. Hendrickson, R. J. Kuhn, and M. G. Rossmann. 2006. Cryo-EM reconstruction of dengue virus in complex with the carbohydrate recognition domain of DC-SIGN. Cell 124:485-493. [DOI] [PubMed] [Google Scholar]
- 37.Puig-Basagoiti, F., T. S. Deas, P. Ren, M. Tilgner, D. M. Ferguson, and P. Y. Shi. 2005. High-throughput assays using a luciferase-expressing replicon, virus-like particles, and full-length virus for West Nile virus drug discovery. Antimicrob. Agents Chemother. 49:4980-4988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rey, F. A., F. X. Heinz, C. Mandl, C. Kunz, and S. C. Harrison. 1995. The envelope glycoprotein from tick-borne encephalitis virus at 2 A resolution. Nature 375:291-298. [DOI] [PubMed] [Google Scholar]
- 39.Salas-Benito, J. S., and R. M. del Angel. 1997. Identification of two surface proteins from C6/36 cells that bind dengue type 4 virus. J. Virol. 71:7246-7252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Stadler, K., S. L. Allison, J. Schalich, and F. X. Heinz. 1997. Proteolytic activation of tick-borne encephalitis virus by furin. J. Virol. 71:8475-8481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tassaneetrithep, B., T. H. Burgess, A. Granelli-Piperno, C. Trumpfheller, J. Finke, W. Sun, M. A. Eller, K. Pattanapanyasat, S. Sarasombath, D. L. Birx, R. M. Steinman, S. Schlesinger, and M. A. Marovich. 2003. DC-SIGN (CD209) mediates dengue virus infection of human dendritic cells. J. Exp. Med. 197:823-829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Thepparit, C., and D. R. Smith. 2004. Serotype-specific entry of dengue virus into liver cells: identification of the 37-kilodalton/67-kilodalton high-affinity laminin receptor as a dengue virus serotype 1 receptor. J. Virol. 78:12647-12656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tomiya, N., S. Narang, Y. C. Lee, and M. J. Betenbaugh. 2004. Comparing N-glycan processing in mammalian cell lines to native and engineered lepidopteran insect cell lines. Glycoconj. J. 21:343-360. [DOI] [PubMed] [Google Scholar]
- 44.Trombetta, E. S., and A. Helenius. 1998. Lectins as chaperones in glycoprotein folding. Curr. Opin. Struct. Biol. 8:587-592. [DOI] [PubMed] [Google Scholar]
- 45.Whitby, K., T. C. Pierson, B. Geiss, K. Lane, M. Engle, Y. Zhou, R. W. Doms, and M. S. Diamond. 2005. Castanospermine, a potent inhibitor of dengue virus infection in vitro and in vivo. J. Virol. 79:8698-8706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wu, S. F., C. J. Lee, C. L. Liao, R. A. Dwek, N. Zitzmann, and Y. L. Lin. 2002. Antiviral effects of an iminosugar derivative on flavivirus infections. J. Virol. 76:3596-3604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zapun, A., N. J. Darby, D. C. Tessier, M. Michalak, J. J. Bergeron, and D. Y. Thomas. 1998. Enhanced catalysis of ribonuclease B folding by the interaction of calnexin or calreticulin with ERp57. J. Biol. Chem. 273:6009-6012. [DOI] [PubMed] [Google Scholar]