

Abstract
Viral encephalitis is a major cause of morbidity and mortality worldwide, yet there is no proven efficacious therapy for most viral infections of the central nervous system (CNS). Many of the viruses that cause encephalitis induce apoptosis and activate c-Jun N-terminal kinase (JNK) following infection. We have previously shown that reovirus infection of epithelial cell lines activates JNK-dependent apoptosis. We now show that reovirus infection resulted in activation of JNK and caspase-3 in the CNS. Treatment of reovirus-infected mice with a cell-permeating peptide that competitively inhibits JNK activity resulted in significantly prolonged survival of intracerebrally infected mice following an otherwise lethal challenge with T3D (100× 50% lethal dose). Protection correlated with reduced CNS injury, reduced neuronal apoptosis, and reduced c-Jun activation without altering the viral titer or viral antigen distribution. Given the efficacy of the inhibitor in protecting mice from viral encephalitis, JNK inhibition represents a promising and novel treatment strategy for viral encephalitis.
Viral encephalitis is a major cause of morbidity and mortality throughout the world. Despite the importance of neurotropic viruses in human disease, effective therapies are available for only a few neurotropic viruses, and even when these infections are optimally treated, residual mortality and neurological sequelae remain considerable. In the case of flavivirus infections, which include Japanese encephalitis virus, the most common cause of viral encephalitis worldwide, and West Nile virus, the most common cause of epidemic encephalitis in the United States, no established treatment exists (34). Similarly, herpes simplex virus (HSV) encephalitis is the most common cause of acute sporadic encephalitis in the Western world (37). While treatment with acyclovir improves outcomes, residual morbidity and mortality remain significant. Novel strategies for treating viral central nervous system (CNS) infections are urgently needed.
Neurotropic viruses, including HSV, flaviviruses, rhabdoviruses, bunyaviruses, and alphaviruses, cause disease by triggering apoptosis in neurons (9, 12, 17, 25, 33, 39). Apoptosis in neurons infected with HSV or West Nile virus is caspase dependent (26, 39), and HSV infection in the CNS is also associated with activation of c-Jun N-terminal kinase (JNK) (26, 40). Moreover, JNK is activated following infection with many different viruses, including echovirus (16), human immunodeficiency virus (21), severe acute respiratory syndrome coronavirus (20), coxsackievirus B3 (19), Sindbis virus (22), and mammalian reovirus (8). Given that JNK is commonly activated following many viral infections and apoptosis is a common mechanism of neuronal cell death following viral infection, we evaluated the role of JNK in virus-induced encephalitis.
Reovirus infection of the mouse CNS is a classic experimental model of viral encephalitis that allows detailed examination of viral pathogenesis in the CNS and in primary cortical neuronal cultures. As with flaviviruses and herpesviruses, reovirus infection of the CNS causes tissue injury and disease by triggering apoptosis (23), resulting in 100% mortality from encephalitis following intracerebral inoculation of >10 PFU of either of the prototypic serotype 3 strains Abney (T3A) and Dearing (T3D) (15). Encephalitis due to reovirus-induced apoptosis is associated with activation of caspase-3, and immunohistochemistry studies have shown that areas of histological damage in the cingulate cortex, hippocampus, and thalamus colocalize with reovirus antigen and caspase-3 activation (28). Our previous studies supported the potential benefit of neuroprotective strategies in the treatment of viral encephalitis by showing that agents such as minocycline can delay disease progression and prolong survival of infected mice (30); however, all treated mice eventually succumbed to infection, emphasizing the need to develop more efficacious treatments. Neuroprotective strategies aimed at blocking JNK and JNK-dependent apoptotic signaling pathways have recently shown great efficacy in reducing injury with experimental models of CNS ischemia (3). We now show that JNK inhibition is neuroprotective with an experimental model of viral encephalitis, the first demonstration of the efficacy of this therapeutic approach for an infectious disease.
We show that JNK and its primary target protein, c-Jun, are activated in reovirus-induced encephalitis. We utilize a cell-permeating peptide (d-stereoisomer c-Jun N-terminal kinase inhibitor 1 [D-JNKI-1]; trade name XG-102) to selectively block JNK activity in reovirus-infected neuronal cultures and in the brains of reovirus-infected mice. We show in a mouse model of viral encephalitis that administration of D-JNKI-1 following intracerebral challenge with an otherwise lethal dose of T3D results in long-term survival of infected mice associated with a significant decrease in CNS apoptosis and tissue injury. These studies indicate that inhibition of JNK activation may be a promising novel therapeutic approach for the treatment of viral CNS infection.
MATERIALS AND METHODS
Cell lines and viruses.
L929 mouse fibroblasts (ATCC CL1) were used for viral titer assays and were maintained in 2 × 199 medium supplemented with 10% heat-inactivated fetal bovine serum and 4 mM l-glutamine for these studies.
Reovirus serotype 3, strains Abney (T3A) and Dearing (T3D), are laboratory stocks which have been plaque purified and passaged (twice) in L929 cells to generate working stocks (36). Virus infections in vitro were performed at a multiplicity of infection of 100 to ensure that 100% of susceptible cells are infected and to maximize the synchrony of viral replication.
Primary cell culture.
Primary neuronal cultures were prepared from the cortices of embryonic day 16 Swiss Webster Hsd:nd4 mice (Harlan-Sprague-Dawley, Indianapolis, IN) as described previously (13). Briefly, fetuses were removed aseptically by cesarean section, brains removed, and cortices dissected. Cortices were dissociated by trituration in neurobasal medium supplemented with 1% fetal bovine serum and l-glutamine (0.6 mM) and viable cells plated at a density of 5 × 104 cells per poly-d-lysine/laminin-coated coverslips (Biocoat; Becton Dickinson, Franklin Lakes, NJ) for immunocytochemistry studies.
Cultures were plated out and maintained in serum-free neurobasal medium supplemented with B-27 nutrient supplement (2% [vol/vol]), antibiotic/antimycotic (penicillin-streptomycin; 1,000 units/ml), and l-glutamine (0.6 mM). All studies were performed on primary neuronal cultures at 7 to 9 days in vitro. All culture media and supplements were purchased from Invitrogen unless stated otherwise.
Cortical neurons were characterized both morphologically and by immunocytochemistry using a mouse monoclonal antibody directed against microtubule-associated protein 2 (MAP-2) (1:100; Abcam). Primary neuronal cultures were typically comprised of 90 to 95% neurons.
Immunocytochemistry.
Primary cortical neuron cultures were infected with T3A at a multiplicity of infection of 100 and harvested at 24 and 48 h postinfection using 4% paraformaldehyde for immunocytochemistry analysis as previously described (13, 28). D-JNKI-1 or TAT control peptide was added to the medium 30 min prior to infection at a final concentration of 4 μM based on previously described neuronal toxicity and efficacy studies (4). Dual-label immunocytochemistry staining was performed to identify c-Jun activation using an antibody specific to phosphorylated serine 73 c-Jun (pS73 c-Jun) (cy3) and MAP-2 neuron-specific antibody (fluorescein; Abcam) or monoclonal antibody specific to the σ3 protein of reovirus (fluorescein).
Peptide inhibitors.
D-JNKI-1 (trade name XG-102) and empty TAT control protein were provided by Christophe Bonny through Xigen S.A., Rue des Terreaus 17, CH-1003 Lausanne, Switzerland.
In vivo studies.
Two-day-old Swiss Webster pups were intracerebrally (i.c.) inoculated with 100 or 1,000 PFU of virus (T3A or T3D) in a 10-μl volume as described previously (28). For whole-brain lysates, mice were infected with 1 × 105 PFU of T3A and sacrificed at day 7 postinfection. At 8 (T3D) or 10 (T3A) days after infection with T3 reovirus, mice were sacrificed and brain tissue removed for histological studies or organ lysates. Therapeutic injections were administered at a dose of 11 mg/kg of body weight/day for both D-JNKI-1 and TAT control therapy in a vehicle phosphate-buffered saline (PBS) and a volume of 10 μl by intraperitoneal (i.p.) injection. All T3A infection experiments included treatment with inhibitor 6 h prior to infection, and T3D infection experiments included treatment with inhibitor or control peptide starting 24 h postinfection. Daily injections of D-JNKI-1, control TAT protein, or PBS were continued for 10 days or until mice were moribund and required sacrifice. All experiments performed conformed to local IACUC and national USPHS guidelines on the ethical use of animals and were approved by IACUC.
Western blotting.
Whole-brain lysates were prepared as previously described (13). Lysates were boiled for 5 min and electrophoresed (Hoefer Pharmacia Biotech, San Francisco, CA) in 10% or 12% tricine/polyacrylamide gels at a constant voltage of 70 V through the resolving gel. Proteins were electroblotted onto Hybond-C nitrocellulose membranes (Amersham Biosciences) and immunoblotting performed as described previously (27). Immunoblots were probed with antibodies directed against stress-activated protein kinase/JNK and phosphorylation-specific Thr183/Tyr185 stress-activated protein kinase/JNK (Cell Signaling Technology, Danvers, MA), serine 63 phosphorylation-specific c-Jun (Cell Signaling Technology, Danvers, MA), and actin (Calbiochem, Sunnyvale, CA). The Typhoon 9400 Imager (Amersham, Piscataway, NJ), and associated software was used for densitometry measurements of Western blots.
Histological studies.
Brain tissue was fixed in 10% formalin for 20 h at room temperature. Tissue was transferred to 70% ethanol before paraffin embedding and cutting of sections. Coronal brain sections (4 μm thick) were prepared, deparaffinized in xylene, and rehydrated in consecutive 100%- to 80%-ethanol washes. Antigen retrieval was performed using 10 mM citrate buffer (pH 6.0). Tissue sections were permeabilized in Neuropore (Trevigen, Inc., Gaithersburg, MD) overnight at 4°C and blocked in 10% normal goat serum (in 1× phosphate-buffered saline with 0.3% Triton [PBST]) for 2 h at room temperature. Sections were incubated overnight at 4°C with rabbit polyclonal antibodies directed against pS73 c-Jun or rabbit polyclonal antibodies directed against cleaved caspase-3 (Asp175) per the manufacturer's recommendations (Cell Signaling Technology). Following washes with PBST, sections were incubated with either fluorescein- or cy3-conjugated goat antirabbit antibody (Vector Laboratories; 1:100) diluted in PBST for 2 h at room temperature. For dual-label studies, tissue sections were incubated with a reovirus σ3-specific monoclonal antibody (38) (4F2; 1:100) overnight before washing with PBST, incubation with fluorescein-conjugated goat anti-mouse secondary antibody (Vector Laboratories; 1:100) for 2 h, washing again with PBST, and incubation for 20 min with 1 μg/ml Hoechst 33342 (Invitrogen). Sections were mounted using VectorShield (Vector Laboratories). Immunostaining for digital immunofluorescence was visualized using a Zeiss Axioplan 2 digital deconvolution microscope with a Cooke Sensicam 12-bit camera.
Viral titer assays.
Tissues prepared for determination of viral titer were immediately transferred to 1 ml of sterile PBS upon removal and stored at −80°C. Samples were freeze-thawed on three separate occasions before sonication. Serial dilutions of homogenized brain tissue were prepared in gel saline and viral titer determined by plaque assays as previously described (11).
Statistical analysis.
All statistical analyses were performed using Instat and Prism (GraphPad Software, Inc., San Diego, CA). Statistical comparisons were made using a two-tailed, unpaired t test with Welch's correction. Survival rates were assessed by the log rank test.
RESULTS
C-Jun is phosphorylated in reovirus-infected primary neuronal cultures.
We have previously shown that JNK and the related downstream transcription factor, c-Jun, are activated in reovirus-infected epithelial cell lines and that inhibition of JNK results in a marked reduction in reovirus-induced apoptosis (7, 8). We therefore evaluated the role of JNK in reovirus-induced apoptosis in neurons and the CNS. We used a c-Jun serine 73 phosphorylation (pS73)-specific antibody to examine c-Jun activation in primary neuronal cultures. Infection of primary cortical neurons with T3A resulted in a two- to threefold up-regulation in the percentage of neurons with detectable pS73 c-Jun at 24 h and a sixfold increase at 48 h postinfection compared to results with mock-infected controls (P = 0.01) (Fig. 1A and B). We then used D-JNKI-1, which competitively blocks access of JNK to c-Jun and other substrates by a direct competitive mechanism (1, 2), to inhibit the activation of c-Jun in reovirus-infected neurons. D-JNKI-1 was designed by linking a 10-amino-acid human immunodeficiency virus Tat transporter sequence to a 20-amino-acid JNK binding motif of JNK-interacting protein-1 (3). The d-retroinverso form (D-JNKI-1) has a prolonged half-life in vivo and is particularly important for treating neurons, since it is uniquely resistant to degradation from neuronal proteases (3). The level of specificity of D-JNKI-1 for the JNK binding motif is extremely high based on previous studies showing that it doesn't inhibit other kinases, including extracellular signal-related kinase (ERK), p38, protein kinase C, p34, calcium/calmodulin-dependent protein kinase, and protein kinase A (3). Primary cortical neurons were treated with D-JNKI-1 or the TAT peptide that lacks the associated inhibitory JNK-interacting protein 1 sequence (TAT control peptide) for 30 min followed by infection with T3A. T3A infection of primary cortical neuronal cultures resulted in increased activation of pS73 c-Jun compared to results with mock infection (Fig. 1). Neurons that stained positive for pS73 c-Jun colocalized with cells positive for MAP-2 staining (Fig. 1B) and cells positive for σ3 reovirus antigen (Fig. 1E). T3A-infected neurons treated with D-JNKI-1 exhibited a reduction in activated pS73 c-Jun in MAP-2-positive cells (Fig. 1C) despite the continued presence of σ3 reovirus antigen (Fig. 1F). In order to quantitatively assess this result, treatment groups were counted using a blinded method, and three separate replicates showed that the percentage of cells positive for pS73 c-Jun was significantly decreased in the D-JNKI-1 treated neurons at 48 h postinfection with T3A (13%) compared to results with vehicle-treated (32%) or TAT control peptide-treated (30%), T3A-infected neurons (P = 0.04) (Fig. 1G).
FIG. 1.

Reovirus-induced activation of pS73 c-Jun in primary cortical neuronal culture is inhibited with D-JNKI-1. Representative photographs are shown from 48 h postinfection. Primary MCCs were mock infected, T3A infected, or treated with D-JNKI-1 prior to T3A infection. (A to C) Primary MCCs were colabeled with antibodies to pS73 c-Jun (red) and neuron-specific antibodies to MAP2 (green). (D to F) Primary MCCs were colabeled with antibodies to pS73 c-Jun activation (red) and antibodies to σ3 reovirus antigen (green). (G) Graphic display of the percentage of primary MCCs that stained positive for pS73 c-Jun. Blinded cell counts from three replicates are shown of mock-infected or T3A-infected primary MCCs treated with D-JNKI-1, TAT control, or vehicle (PBS). Vertical bars indicate standard errors of the means. *, P ≤ 0.05. Original magnification of images shown in panel A, ×400.
JNK and c-Jun are activated following reovirus infection of the CNS.
We next examined whether infection with T3 reovirus in an in vivo model of encephalitis (29) resulted in the same degree of JNK and c-Jun activation as was seen in primary neuronal cultures in vitro. In order to evaluate JNK activation in reovirus-induced encephalitis, we infected 2-day-old Swiss Webster mice with T3A at a dose of 1 × 105 PFU by i.c. injection and sacrificed the animals at day 8 postinfection. A high viral challenge was selected in an effort to maximize in vivo cell signaling activation. Western blot analysis of whole-brain lysates showed that Thr183/Tyr185 phosphorylated JNK was increased for T3A-infected mice compared to levels for mock-infected mice in the absence of a change in the levels of total JNK (Fig. 2). Given that c-Jun is an important target of activated JNK, we also looked for the presence of activated c-Jun in mice infected with T3A compared to mock-infected mice at day 8 postinfection using antibodies specific for the serine 63 (pS63) and pS73 phosphorylated forms of c-Jun. A dose of 1 × 105 PFU of T3A was used to maximize cell signal activation for Western blots, and a dose of 1 × 103 PFU was used for histological analysis in an effort to activate signaling events but preserve neuronal architecture. Antibodies to the pS73 and pS63 c-Jun were utilized based on their manufacturer's approved applications for immunofluorescence and immunoblotting, respectively. Immunohistochemical studies demonstrated nuclear localization of pS73 c-Jun in brain tissue from T3A-infected mice, with distribution restricted to regions known to be affected during reovirus-induced encephalitis, including the cingulate cortex, hippocampus, and thalamus (Fig. 3A). Immunohistochemical studies of pS73 c-Jun activation and distribution in T3D-infected mouse brains revealed equivalent results (data not shown). Levels of pS63 c-Jun were dramatically higher in whole-brain lysates from T3A-infected mice than in those from mock-infected control animals (Fig. 3B).
FIG. 2.
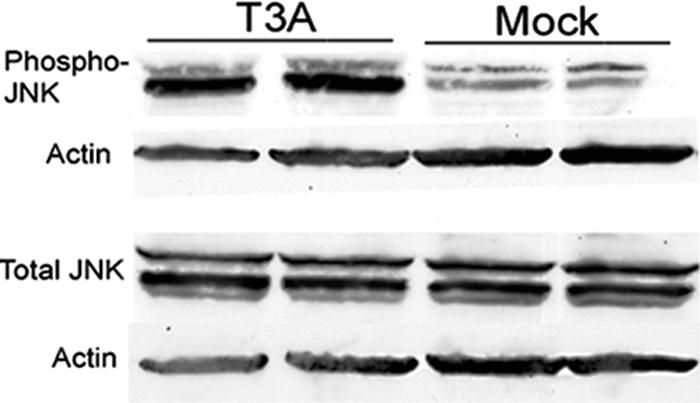
T3A-induced JNK phosphorylation in brains of neonatal mice. Two-day-old Swiss Webster mice were mock or T3A infected and sacrificed when moribund. Tissue was processed for whole-brain lysates and subjected to immunoblotting for evidence of phosphorylated JNK and total JNK. Each band represents an individual.
FIG. 3.

T3A-induced activation of c-Jun in brains of neonatal mice. Two-day-old Swiss Webster mice were mock or T3A infected and sacrificed when moribund. (A) Histological tissue sections from T3A-infected mice were evaluated using fluorescence immunohistochemistry for pS73 c-Jun (green) in the cingulate cortex, hippocampus, and thalamus. Images (original magnification, ×400) provide representative observations of the 12 animals sampled per group. (B) Whole-brain lysate samples were evaluated for evidence of activated (pS63) c-Jun. Each band represents an individual. Images are representative of eight animals sampled per group.
D-JNKI-1 protects against virus-induced encephalitis.
In order to evaluate the importance of JNK and c-Jun activation during viral encephalitis, we next sought to inhibit JNK prior to infection. Six hours following treatment with D-JNKI-1 by i.p. injection (11 mg/kg) or PBS vehicle control (10 μl), 2-day-old mice were infected with a 10× 50% lethal dose (LD50) (100 PFU) of T3A by i.c. injection. Dosing of D-JNKI-1 was based on prior dosing and efficacy experiments (2, 3). Mice subsequently received daily i.p. treatment with D-JNKI-1 (11 mg/kg/day) or vehicle control for a total of 10 days postinfection. Mock-infected litters received identical dosing of D-JNKI-1. Mice were sacrificed at days 8 to 10 based on the presence of clinical illness in the concurrent vehicle control mice. Histology sections were first analyzed by hematoxylin-and-eosin staining and scored blind for CNS injury based on a previously published neuropathological injury scoring system (14, 35). Inhibition of JNK with D-JNKI-1 significantly reduced virus-induced brain injury in T3A-infected mice compared to results with untreated mice in the cingulate cortex (P = 0.005), hippocampus (P = 0.02), and thalamus (P = 0.006) (Fig. 4A and B).
FIG. 4.

D-JNKI-1 treatment protects from T3A-induced CNS injury. Mice were treated with D-JNKI-1 or vehicle control, followed by viral challenge with T3A. (A) Representative brain tissue samples were stained with hematoxylin and eosin in order to evaluate for evidence of histological damage. Samples represent four replicates of the experiment consisting of six mice per treatment group per replicate. Original magnification of images shown in panel A,×200. (B) Blind neuropathology scoring of histologic sections show significantly decreased injury in the D-JNKI-1-treated mice compared to untreated, T3A-infected mice in areas typical of reovirus histologic injury: the cingulate cortex (*, P = 0.005), hippocampus (*, P = 0.02), and thalamus (*, P = 0.006). Histopathology scoring: 0, no lesions; 1, <10% of total section shows CNS injury; 2, 10 to 40%; 3, 40 to 75%; 4, >75%.
Given the degree of neuroprotection seen in T3A reovirus-infected mice treated with D-JNKI-1, we evaluated survival of D-JNKI-1-treated mice compared to that of mock-infected mice. Mice were treated with D-JNKI-1 (11 mg/kg/day i.p.) and challenged 6 h later with T3A i.c. (10× LD50, 100 PFU). Mice then received D-JNKI-1 for 10 days postinfection (11 mg/kg/day i.p.). D-JNKI-1-treated, T3A-infected mice had a significantly prolonged survival (mean day of death [MDD]) compared to T3A-infected, vehicle-treated controls (14 ± 1.9 [n = 6] versus 10.6 ± 0.6 [n = 6], respectively; P = 0.006) (Fig. 5A). Since the D-JNKI-1-treated, infected mice exhibited prolonged survival but still died, we examined pathological samples from moribund mice for the cause of death. Pathological examination of moribund mice suggested that death was not due to CNS injury, which was mild or absent, but instead resulted from myocarditis (data not shown), a well-recognized consequence of murine T3A systemic infection (32). In an effort to isolate the therapeutic benefits of reduction in CNS injury in the absence of concomitant myocardial injury, mice were challenged with reovirus T3D. This strain of virus produces encephalitis identical to that with T3A but is not associated with cardiac injury (10, 32). We also modified our experimental protocol to determine whether initiation of JNK-inhibition after a viral challenge would be as efficacious as treatment prior to a viral challenge.
FIG. 5.

D-JNKI-1 treatment results in significantly prolonged survival for both T3A- and T3D-infected mice. (A) Mice were treated with D-JNKI-1, challenged with T3A 6 h later, and sacrificed when moribund. T3A-infected, untreated mice had an MDD of 10.6 (standard deviation [SD], ±0.6), and T3A-infected, D-JNKI-1 treated mice had an MDD of 14 (SD, ±1.9; P = 0.006). (B) Mice were challenged with T3D and treated 24 h later with vehicle control, TAT control peptide, or D-JNKI-1. Survival studies revealed 38% long-term survival in the T3D-infected, D-JNKI-1-treated group. Of the individuals that died, the MDD was prolonged for T3D-infected, D-JNKI-1-treated mice compared to T3D-infected, untreated mice (13.2; SD, ±1.42; and 9.8; SD, ±2.3 respectively; P < 0.00001). Survival of TAT peptide-treated T3D-infected mice did not significantly differ from that of vehicle control, T3D-infected mice (data not shown).
Two-day-old mice were infected with T3D (1 × 103 PFU i.c.; 100× LD50), followed 24 h later by treatment with D-JNKI-1 (11 mg/kg/day i.p. for 10 days), TAT control peptide (11 mg/kg/day i.p. for 10 days), or vehicle (10 μl PBS i.p. for 10 days). All untreated, T3D-infected mice died [n = 26]. By contrast, 38% (10 of 26) of T3D-infected mice treated with D-JNKI-1 survived the infection (Fig. 5B). For T3D-infected mice suffering from fatal disease despite D-JNKI-1 treatment, survival was significantly prolonged compared to that of vehicle and TAT controls (MDD, 13.2 [±1.4] versus 9.8 [±2.3] days postinfection; P < 0.00001).
Treatment with D-JNKI-1 decreases activated c-Jun and caspase-3.
We next evaluated mice infected with T3A and T3D and treated with D-JNKI-1 for evidence of c-Jun activation in order to understand whether D-JNKI-1 prevented c-Jun phosphorylation and subsequent activation. Our prior in vitro studies with nonneuronal cells had suggested that both reovirus-induced JNK and ERK activation were involved in c-Jun phosphorylation, and while inhibitors of either kinase reduced c-Jun phosphorylation, complete inhibition of c-Jun phosphorylation required inhibition of both kinases (8). By contrast, our studies with primary neuron cultures (see above) suggested that treatment with D-JNKI-1 resulted in near-complete abrogation of reovirus-induced c-Jun phosphorylation without concomitant ERK inhibition. Brain sections were analyzed from mice treated with D-JNKI-1 (11 mg/kg/day for 10 days), followed 6 h later by infection with T3A (1 × 102 PFU i.c.) and mice infected with T3D (1 × 103 PFU) and treated 24 h later with D-JNKI-1 (11 mg/kg/day for 10 days). These mice were sacrificed at days 8 (T3D) and 10 (T3A), and tissue sections were analyzed with dual-label fluorescence immunohistochemistry using an antibody to pS73 c-Jun and a monoclonal σ3-specific antireovirus antibody or antibody to MAP-2. Whole-brain lysate was also prepared in order to evaluate pS63 c-Jun activity by Western blot analysis. In the absence of D-JNKI-1, T3A- and T3D-infected mice exhibited up-regulation and nuclear localization of phosphorylated c-Jun in the areas of reovirus antigen and histological damage within the cingulate cortex, hippocampus, and thalamus. pS73 c-Jun typically colocalized with σ3 reovirus antigen-positive neurons and MAP-2-positive cells in untreated, infected mice (Fig. 6A). Following treatment with D-JNKI-1, virus-induced c-Jun phosphorylation was greatly diminished despite the continued presence of detectable viral antigen in these same regions of the brain (Fig. 6A). Diminished levels of pS63 c-Jun were confirmed by Western blot analysis of whole-brain lysates on three replicates per treatment group as represented by densitometry studies (Fig. 6B).
FIG. 6.

D-JNKI-1 treatment of T3A- or T3D-infected mice decreases activation of c-Jun in vivo. (A) Representative immunohistochemical staining of brain sections of mock-, T3A-, or T3D-infected mice treated with vehicle (PBS), TAT control peptide, or D-JNKI-1 (DJNKI). Successive sections were stained with antibodies to pS73 c-Jun (PcJun) (green) and colabeled with either antibody to reovirus σ3 protein (red) or antibody to MAP2 neuron-specific antigen (red). Images are provided from the cingulate cortex (T3A) or the hippocampus (T3D); original magnification, ×400. (B) Representative immunoblots of whole-brain lysate are shown following mock or T3A infection and treatment with vehicle (PBS) or D-JNKI-1. Blots were probed for pS63 c-Jun and actin. Densitometry studies represent values obtained from individual blots from three mice per treatment group normalized to corresponding actin densitometry values. Vertical bars indicate standard errors of the means.
In order to evaluate the ability of D-JNKI-1 treatment to reduce neuronal apoptosis following T3 infection, we examined adjacent tissue sections from the same animals described above using dual-label fluorescence immunohistochemistry with a cleavage-specific antibody to caspase-3 and a reovirus σ3 protein-specific antibody. In mice infected with T3A or T3D and treated with PBS or TAT control peptide, cleaved (activated) caspase-3 exhibited colocalization with reovirus antigen. Representative samples from T3D-infected mice are provided (Fig. 7). Following treatment with D-JNKI-1, there was a marked reduction in activated or cleaved caspase-3 despite the continued presence of reovirus antigen (Fig. 7). Diminished activated caspase-3 was seen throughout the areas of typical reovirus-induced injury and apoptosis.
FIG. 7.

D-JNKI-1 treatment of T3D-infected mice decreases cleaved caspase-3. Adjacent tissue sections from mock- or T3D-infected mice treated with TAT control peptide or D-JNKI-1 were evaluated with dual-label fluorescence immunohistochemistry using antibody to σ3 reovirus protein (Reo Ag) (green) and cleavage specific (activated) caspase-3 (Casp) (red). Representative images are shown (original magnification, ×400) from the hippocampus of the T3D-infected groups. T3A data are not shown.
D-JNKI-1 does not alter viral titer.
Our studies indicated that treatment with D-JNKI-1 dramatically reduced reovirus-induced CNS injury and apoptosis and was associated with prolonged and enhanced survival of infected mice. However, immunohistochemistry studies (see above) of viral antigen using reovirus σ3 protein-specific antibodies suggested that the number of viral antigen-immunoreactive cells might be modestly decreased in D-JNKI-1-treated mice. In order to evaluate the effect of D-JNKI-1 therapy on viral replication, we evaluated viral titers from mice infected with T3A or T3D and treated with D-JNKI-1. Mice were treated with D-JNKI-1 (11 mg/kg/day i.p. for 10 days), challenged with T3A (1 × 102 PFU i.c.) 6 h later, and sacrificed at day 10 postinfection for brain titer assay. T3D-infected mice (1 × 103 PFU i.c.) were treated with D-JNKI-1 (11 mg/kg/day i.p. for 10 days) beginning 24 h postinfection and sacrificed at day 8 postinfection for brain titer assay. Viral titers were not significantly different between D-JNKI-1-treated mice and vehicle controls following either T3A or T3D infection (P = 0.8 or 0.5, respectively) (Fig. 8).
FIG. 8.

D-JNKI-1 treatment does not alter viral growth in the CNS. Two-day-old Swiss Webster mice were infected with T3A or T3D and treated with vehicle control or D-JNKI-1. Six hours following D-JNKI-1 or vehicle control treatment, mice were challenged with T3A and sacrificed on day 10 postinfection. Mice infected with T3D were treated with D-JNKI-1 or vehicle control 24 h postinfection and were sacrificed at day 8 postinfection. Each treatment group represents three replicates with six mice. Vertical bars indicate standard errors of the means.
DISCUSSION
These studies establish that treatment of mice with a cell-permeating peptide that selectively inhibits JNK activation can dramatically prolong and enhance survival of mice in an experimental model of viral encephalitis. Some available antiviral therapies, exemplified by interferon treatment for hepatitis C infection in the liver, are designed to augment specific components of the host's innate immune response (18). Other types of antiviral therapy restrict specific stages in the viral replication cycle, including attachment, uncoating, viral genome replication, or processing of essential proteins. To our knowledge, this is the first time that a targeted cellular therapy designed to interfere with virus-induced activation of an apoptosis-related mitogen-activated protein kinase signaling cascade has been shown to prevent virus-induced apoptosis and tissue injury in vivo, leading to prolonged survival of infected mice. These results are particularly significant, since the JNK inhibitor employed in these studies is currently being evaluated in human clinical trials for treatment of neurologic disease.
Our previous studies with epithelial cell lines have suggested that JNK activation is important in virus-induced apoptosis (8) and that inhibition of JNK with a small peptide inhibitor that prevents phosphorylation results in diminished apoptosis and decreased mitochondrial release of proapoptotic proteins (7). While these studies suggested that c-Jun activation is dependent on both ERK and JNK activation (7), our current results suggest that JNK inhibition with D-JNKI-1 in T3A-infected neurons was greatly diminished with JNK inhibition alone. This may be attributable to the use of the primary neuronal cell types and in vivo brain tissue used in this study or the mechanism of action of D-JNKI-1 compared to that of inhibitors used in prior experiments that prevent phosphorylation of JNK.
Also, we now show that JNK inhibition can reduce apoptosis and tissue injury in vivo in a murine model of encephalitis. In our initial studies with T3A infection following D-JNKI-1 therapy, the inhibitor significantly protected these mice from virus-induced CNS injury, resulting in prolonged survival, although the mice eventually succumbed to infection. This result is similar to that found in our prior studies utilizing the neuroprotective agent minocycline (30). Our postmortem evaluation of T3A-infected, D-JNKI-1-treated mice suggested that our failure to see enhanced survival in the D-JNKI-1-treated mice may be the result of an organ-specific action of this drug, since mice appeared to be dying of myocarditis despite the dramatic reduction in CNS injury. Organ-specific effects of inhibiting reovirus-induced signaling pathways have previously been observed in mice lacking the p50 component of NF-κB (24). Interestingly, these mice showed the same pattern of injury following reovirus infection in the CNS, with dramatically reduced CNS injury but severe myocardial injury, compared to syngeneic controls (24). In an effort to evaluate the neuroprotective efficacy of JNK inhibition without the confounding variable of cardiac injury, we used a reovirus prototype strain (T3D) that induces encephalitis but not myocarditis (10, 32).
A possible role of apoptosis induction in viral pathogenesis is to facilitate viral release from infected cells and subsequent spread in the host. Consequently, inhibition of apoptosis may inhibit or impede viral replication. Viral titers and distribution of viral antigen did not differ significantly between D-JNKI-1-treated and untreated mice; however, immunohistochemistry studies suggested that levels of viral antigen might be lower in D-JNKI-1-treated mice. While this was not supported by subsequent viral titers, these viral titers were assayed at days 8 and 10 postinfection, and further studies of viral kinetics throughout the treatment with D-JNKI-1 will be needed in order to better understand the possible impact of D-JNKI-1 on viral kinetics. The unchanged titers in D-JNKI-1-treated mice suggest that injury from viral infection does not result merely from viral replication but from the associated activation of a variety of host-cell signaling events that ultimately lead to cell dysfunction or death. It may also indicate that effects on virus-induced host cell injury can be dissociated from effects of viral replication, and these two components may be independent and presumably synergistic targets for antiviral therapy. Further studies of the viral dynamics in relation to apoptosis inhibition need to be completed in order to fully characterize the relationship between viral replication and induction of cell death.
Our studies suggest that inhibition of JNK activation using D-JNKI-1 may be a viable option for therapy in virus-induced encephalitis. This type of therapy would presumably be most efficacious for those infections in which JNK is activated and in which this activation is deleterious to host cell survival. While our treatment model resulted in survival of only 38% of the T3D-infected mice, it is likely that this result can be optimized further. We inoculated virus directly into the brain instead of allowing for spread from a distant site of inoculation as would occur during a natural infection. The challenge doses employed were substantial, 100 times the intracerebral LD50, which is significantly greater than would likely occur during natural infection.
While JNK is an important mitogen-activated protein kinase instrumental in multiple cellular processes, including neuronal differentiation and gene expression (5, 6), targeted inhibition of JNK over a short period of time is unlikely to have severe long-term side effects (3) and was not toxic in either our uninfected controls or the T3D-infected, D-JNKI-1-treated mice that survived. Moreover, recent studies of D-JNKI-1 with rats and monkeys have shown no evidence of toxicity, and phase I clinical trials for D-JNKI-1 therapy for stroke patients are currently in progress (www.xigenpharma.com). Targeted inhibition of JNK during neuroinvasive viral infection may also provide a neuroprotective effect that could reduce infection-associated morbidity, such as neuropsychiatric changes, seizures, paralysis, weakness, Parkinsonism, or other movement disorders (31, 37).
Based on prior studies of the specificity of D-JNKI-1 for JNK (3) and our data showing greatly diminished c-Jun and caspase-3 activation following D-JNKI-1 therapy, it is reasonable to conclude that D-JNKI-1 is inhibiting JNK activation and therefore inhibiting apoptosis. Further studies with this model need to be completed in order to characterize the specific role of JNK in apoptosis. This peptide inhibitor may have greater efficacy in vivo by inhibiting a process that the virus actively induces following infection, thereby reducing or slowing the ability of the virus to spread to neighboring cells. This time delay in the optimal viral kinetics of replication or viral spread may allow an organized cell-mediated immune response that is able to control the viral infection, resulting in long-term survival. Since these studies were performed with neonatal mice, in which the host immune response to pathogens is incompletely developed, therapy with this peptide with an adult in vivo model of encephalitis may provide more-striking benefits due to an intact immune response. Using targeted inhibitors of cell signaling proteins, such as D-JNKI-1, as therapy may also be synergistic with specific antiviral therapy or immunotherapy aimed at reducing viral replication, resulting in improved therapeutic efficacy.
Acknowledgments
We express our thanks to Jennifer Smith Leser and Marta Lishnevsky for technical support.
This work was supported by NIH T32AI07537, 5R01NS050138, and 1R01NS051403 and VA Merit funding.
Footnotes
Published ahead of print on 2 May 2007.
REFERENCES
- 1.Barr, R. K., T. S. Kendrick, and M. A. Bogoyevitch. 2002. Identification of the critical features of a small peptide inhibitor of JNK activity. J. Biol. Chem. 277:10987-10997. [DOI] [PubMed] [Google Scholar]
- 2.Bonny, C., A. Oberson, S. Negri, C. Sauser, and D. F. Schorderet. 2001. Cell-permeable peptide inhibitors of JNK: novel blockers of beta-cell death. Diabetes 50:77-82. [DOI] [PubMed] [Google Scholar]
- 3.Borsello, T., P. G. Clarke, L. Hirt, A. Vercelli, M. Repici, D. F. Schorderet, J. Bogousslavsky, and C. Bonny. 2003. A peptide inhibitor of c-Jun N-terminal kinase protects against excitotoxicity and cerebral ischemia. Nat. Med. 9:1180-1186. [DOI] [PubMed] [Google Scholar]
- 4.Borsello, T., K. Croquelois, J. P. Hornung, and P. G. Clarke. 2003. N-methyl-d-aspartate-triggered neuronal death in organotypic hippocampal cultures is endocytic, autophagic and mediated by the c-Jun N-terminal kinase pathway. Eur. J. Neurosci. 18:473-485. [DOI] [PubMed] [Google Scholar]
- 5.Chang, L., Y. Jones, M. H. Ellisman, L. S. Goldstein, and M. Karin. 2003. JNK1 is required for maintenance of neuronal microtubules and controls phosphorylation of microtubule-associated proteins. Dev. Cell 4:521-533. [DOI] [PubMed] [Google Scholar]
- 6.Chang, L., and M. Karin. 2001. Mammalian MAP kinase signalling cascades. Nature 410:37-40. [DOI] [PubMed] [Google Scholar]
- 7.Clarke, P., S. M. Meintzer, Y. Wang, L. A. Moffitt, S. M. Richardson-Burns, G. L. Johnson, and K. L. Tyler. 2004. JNK regulates the release of proapoptotic mitochondrial factors in reovirus-infected cells. J. Virol. 78:13132-13138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Clarke, P., S. M. Meintzer, C. Widmann, G. L. Johnson, and K. L. Tyler. 2001. Reovirus infection activates JNK and the JNK-dependent transcription factor c-Jun. J. Virol. 75:11275-11283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Debiasi, R. L., B. K. Kleinschmidt-Demasters, S. Richardson-Burns, and K. L. Tyler. 2002. Central nervous system apoptosis in human herpes simplex virus and cytomegalovirus encephalitis. J. Infect. Dis. 186:1547-1557. [DOI] [PubMed] [Google Scholar]
- 10.Debiasi, R. L., B. A. Robinson, B. Sherry, R. Bouchard, R. D. Brown, M. Rizeq, C. Long, and K. L. Tyler. 2004. Caspase inhibition protects against reovirus-induced myocardial injury in vitro and in vivo. J. Virol. 78:11040-11050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Debiasi, R. L., M. K. Squier, B. Pike, M. Wynes, T. S. Dermody, J. J. Cohen, and K. L. Tyler. 1999. Reovirus-induced apoptosis is preceded by increased cellular calpain activity and is blocked by calpain inhibitors. J. Virol. 73:695-701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fu, Z. F., and A. C. Jackson. 2005. Neuronal dysfunction and death in rabies virus infection. J. Neurovirol. 11:101-106. [DOI] [PubMed] [Google Scholar]
- 13.Goody, R. J., C. C. Hoyt, and K. L. Tyler. 2005. Reovirus infection of the CNS enhances iNOS expression in areas of virus-induced injury. Exp. Neurol. 195:379-390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hoyt, C. C., S. M. Richardson-Burns, R. J. Goody, B. A. Robinson, R. L. DeBiasi, and K. L. Tyler. 2005. Nonstructural protein σ1s is a determinant of reovirus virulence and influences the kinetics and severity of apoptosis induction in the heart and central nervous system. J. Virol. 79:2743-2753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hrdy, D. B., D. H. Rubin, and B. N. Fields. 1982. Molecular basis of reovirus neurovirulence: role of the M2 gene in avirulence. Proc. Natl. Acad. Sci. USA 79:1298-1302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Huttunen, P., T. Hyypia, P. Vihinen, L. Nissinen, and J. Heino. 1998. Echovirus 1 infection induces both stress- and growth-activated mitogen-activated protein kinase pathways and regulates the transcription of cellular immediate-early genes. Virology 250:85-93. [DOI] [PubMed] [Google Scholar]
- 17.Jan, J. T., and D. E. Griffin. 1999. Induction of apoptosis by Sindbis virus occurs at cell entry and does not require virus replication. J. Virol. 73:10296-10302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kamal, S. M., A. E. Fouly, R. R. Kamel, B. Hockenjos, A. Al Tawil, K. E. Khalifa, Q. He, M. J. Koziel, K. M. El Naggar, J. Rasenack, and N. H. Afdhal. 2006. Peginterferon alfa-2b therapy in acute hepatitis C: impact of onset of therapy on sustained virologic response. Gastroenterology 130:632-638. [DOI] [PubMed] [Google Scholar]
- 19.Kim, S. M., J. H. Park, S. K. Chung, J. Y. Kim, H. Y. Hwang, K. C. Chung, I. Jo, S. I. Park, and J. H. Nam. 2004. Coxsackievirus B3 infection induces cyr61 activation via JNK to mediate cell death. J. Virol. 78:13479-13488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kopecky-Bromberg, S. A., L. Martinez-Sobrido, and P. Palese. 2006. 7a protein of severe acute respiratory syndrome coronavirus inhibits cellular protein synthesis and activates p38 mitogen-activated protein kinase. J. Virol. 80:785-793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kumar, A., S. K. Manna, S. Dhawan, and B. B. Aggarwal. 1998. HIV-Tat protein activates c-Jun N-terminal kinase and activator protein-1. J. Immunol. 161:776-781. [PubMed] [Google Scholar]
- 22.Nakatsue, T., I. Katoh, S. Nakamura, Y. Takahashi, Y. Ikawa, and Y. Yoshinaka. 1998. Acute infection of Sindbis virus induces phosphorylation and intracellular translocation of small heat shock protein HSP27 and activation of p38 MAP kinase signaling pathway. Biochem. Biophys. Res. Commun. 253:59-64. [DOI] [PubMed] [Google Scholar]
- 23.Oberhaus, S. M., R. L. Smith, G. H. Clayton, T. S. Dermody, and K. L. Tyler. 1997. Reovirus infection and tissue injury in the mouse central nervous system are associated with apoptosis. J. Virol. 71:2100-2106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.O'Donnell, S. M., M. W. Hansberger, J. L. Connolly, J. D. Chappell, M. J. Watson, J. M. Pierce, J. D. Wetzel, W. Han, E. S. Barton, J. C. Forrest, T. Valyi-Nagy, F. E. Yull, T. S. Blackwell, J. N. Rottman, B. Sherry, and T. S. Dermody. 2005. Organ-specific roles for transcription factor NF-kappaB in reovirus-induced apoptosis and disease. J. Clin. Investig. 115:2341-2350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pekosz, A., J. Phillips, D. Pleasure, D. Merry, and F. Gonzalez-Scarano. 1996. Induction of apoptosis by La Crosse virus infection and role of neuronal differentiation and human bcl-2 expression in its prevention. J. Virol. 70:5329-5335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Perkins, D., K. A. Gyure, E. F. Pereira, and L. Aurelian. 2003. Herpes simplex virus type 1-induced encephalitis has an apoptotic component associated with activation of c-Jun N-terminal kinase. J. Neurovirol. 9:101-111. [DOI] [PubMed] [Google Scholar]
- 27.Poggioli, G. J., C. Keefer, J. L. Connolly, T. S. Dermody, and K. L. Tyler. 2000. Reovirus-induced G2/M cell cycle arrest requires σ1s and occurs in the absence of apoptosis. J. Virol. 74:9562-9570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Richardson-Burns, S. M., D. J. Kominsky, and K. L. Tyler. 2002. Reovirus-induced neuronal apoptosis is mediated by caspase 3 and is associated with the activation of death receptors. J. Neurovirol. 8:365-380. [DOI] [PubMed] [Google Scholar]
- 29.Richardson-Burns, S. M., and K. L. Tyler. 2004. Regional differences in viral growth and central nervous system injury correlate with apoptosis. J. Virol. 78:5466-5475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Richardson-Burns, S. M., and K. L. Tyler. 2005. Minocycline delays disease onset and mortality in reovirus encephalitis. Exp. Neurol. 192:331-339. [DOI] [PubMed] [Google Scholar]
- 31.Sejvar, J. J., M. B. Haddad, B. C. Tierney, G. L. Campbell, A. A. Marfin, J. A. Van Gerpen, A. Fleischauer, A. A. Leis, D. S. Stokic, and L. R. Petersen. 2003. Neurologic manifestations and outcome of West Nile virus infection. JAMA 290:511-515. [DOI] [PubMed] [Google Scholar]
- 32.Sherry, B. 1998. Pathogenesis of reovirus myocarditis. Curr. Top. Microbiol. Immunol. 233:51-66. [DOI] [PubMed] [Google Scholar]
- 33.Shrestha, B., D. Gottlieb, and M. S. Diamond. 2003. Infection and injury of neurons by West Nile encephalitis virus. J. Virol. 77:13203-13213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Solomon, T. 2004. Flavivirus encephalitis. N. Engl. J. Med. 351:370-378. [DOI] [PubMed] [Google Scholar]
- 35.Thoresen, M., K. Haaland, E. M. Loberg, A. Whitelaw, F. Apricena, E. Hanko, and P. A. Steen. 1996. A piglet survival model of posthypoxic encephalopathy. Pediatr. Res. 40:738-748. [DOI] [PubMed] [Google Scholar]
- 36.Tyler, K. L. 1998. Pathogenesis of reovirus infections of the central nervous system. Curr. Top. Microbiol. Immunol. 233:93-124. [DOI] [PubMed] [Google Scholar]
- 37.Tyler, K. L. 2004. Herpes simplex virus infections of the central nervous system: encephalitis and meningitis, including Mollaret's. Herpes 11(Suppl. 2):57A-64A. [PubMed] [Google Scholar]
- 38.Virgin, H. W., M. A. Mann, B. N. Fields, and K. L. Tyler. 1991. Monoclonal antibodies to reovirus reveal structure/function relationships between capsid proteins and genetics of susceptibility to antibody action. J. Virol. 65:6772-6781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yang, J. S., M. P. Ramanathan, K. Muthumani, A. Y. Choo, S. H. Jin, Q. C. Yu, D. S. Hwang, D. K. Choo, M. D. Lee, K. Dang, W. Tang, J. J. Kim, and D. B. Weiner. 2002. Induction of inflammation by West Nile virus capsid through the caspase-9 apoptotic pathway. Emerg. Infect. Dis. 8:1379-1384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zachos, G., B. Clements, and J. Conner. 1999. Herpes simplex virus type 1 infection stimulates p38/c-Jun N-terminal mitogen-activated protein kinase pathways and activates transcription factor AP-1. J. Biol. Chem. 274:5097-5103. [DOI] [PubMed] [Google Scholar]