

Abstract
Interaction of the C-terminal domains of Sendai virus (SeV) P and N proteins is crucial for RNA synthesis by correctly positioning the polymerase complex (L+P) onto the nucleocapsid (N/RNA). To better understand this mechanism within the paramyxovirus family, we have studied the complex formed by the SeV C-terminal domains of P (PX) and N (NTAIL) proteins by solution nuclear magnetic resonance spectroscopy. We have characterized SeV NTAIL, which belongs to the class of intrinsically disordered proteins, and precisely defined the binding regions within this latter domain and within PX. SeV NTAIL binds with residues 472 to 493, which have a helical propensity (residues 477 to 491) to the surface created by helices α2 and α3 of PX with a 1:1 stoichiometry, as was also found for measles virus (MV). The binding interface is dominated by charged residues, and the dissociation constant was determined to be 57 ± 18 μM under conditions of the experiment (i.e., in 0.5 M NaCl). We have also shown that the extreme C terminus of SeV NTAIL does not interact with PX, which is in contrast to MV, where a second binding site was identified. In addition, the interaction surfaces of the MV proteins are hydrophobic and a stronger binding constant was found. This gives a good illustration of how selection pressure allowed the C-terminal domains of N and P proteins to evolve concomitantly within this family of viruses in order to lead to protein complexes having the same three-dimensional fold, and thus the same function, but with completely different binding interfaces.
Sendai virus (SeV) was discovered in 1952 (26) and is also referred to as murine parainfluenza virus as it was found to infect the respiratory tract of mice, to cause pneumonia, and to readily spread to uninfected animals. It has recently been shown that SeV was able to replicate in the upper and lower respiratory tracts of chimpanzees and African green monkeys and could, therefore, theoretically also cause zoonotic disease in humans (37). SeV belongs to the paramyxovirus family. This family includes several human pathogens, such as human parainfluenza viruses, measles and mumps viruses, which cause severe respiratory diseases in infants and young children. Despite extensive vaccination campaigns against measles and mumps, the diseases have not been eradicated and outbreaks even occur within vaccinated populations (36). Moreover, no vaccine exists for human parainfluenza viruses. As for other paramyxoviruses, the genome of SeV is a negative-sense single-stranded RNA molecule, approximately 15,000 nucleotides in length. Within the virion, the nucleoprotein (N) packages the genomic RNA into a helical protein-RNA complex termed the nucleocapsid, with a stoichiometry of one N protomer for six nucleotides (6, 13, 24). In the cytoplasm of an infected cell, the viral RNA polymerase uses the nucleocapsid as a template for both the replication and encapsidation of the viral genome, as well as the transcription to messenger RNAs encoding the viral proteins. The viral RNA polymerase has no known homologue in humans and is therefore an interesting target for antiviral treatment. This enzyme is constituted of two proteins, the large (L) protein and the phosphoprotein (P). The L protein contains the polymerase activity as well as the capping and polyadenylation activities (1, 15, 41). The P protein plays a crucial role in the enzyme by positioning L onto the N/RNA template through an interaction with the C-terminal domain of N (14). Without P, L is not functional. The N, P, and L proteins of SeV and measles and mumps viruses are functionally equivalent. However, sequence identity between proteins from these viruses is limited, and the viruses have been placed in different genera (Respirovirus, Morbilivirus, and Rubulavirus, respectively).
The Paramyxoviridae L protein is the least-characterized protein of the replicative complex and is also the biggest protein encoded by the viral genome (∼2,250 amino acids [aa]). Bioinformatic studies showed that six regions of good conservation can be defined within the L protein sequences. These regions have been proposed to be important for the various enzymatic activities necessary for viral transcription and replication (31, 35).
SeV P protein (568 aa) is a modular protein with distinct functional domains (39). The N-terminal part of P (PNT) is a chaperone for N and prevents it from binding to nonviral RNA in the infected cell (11). The C-terminal part of P (PCT) is only functional as an oligomer and forms with L the polymerase complex (8). PNT is poorly conserved and unstructured in solution (18, 19), while PCT contains the oligomerization domain (PMD) that folds as a homotetrameric coiled coil (40) containing the L binding region and a C-terminal partially folded domain, PX (residues 474 to 568), identified as the nucleocapsid binding site (33). Interestingly, PX is also expressed as an independent polypeptide in infected cells (10). PX has a C-subdomain (residues 516 to 568) that consists of three α-helices arranged in an antiparallel triple-helical bundle linked to an unfolded flexible N-subdomain (residues 474 to 515) (3) (Fig. 1). The structure of the entire SeV PCT was modeled by combining the independently obtained three-dimensional (3D) structures of its constituent parts with small-angle scattering data on the whole domain (3). All paramyxovirus P proteins are likely to be oligomeric, containing sequence repeats in the central region of the molecule characteristic of helical coiled coils (8). Despite the low sequence conservation, the extreme C terminus of measles virus (MV) P protein (XD, residues 459 to 507) adopts a similar α-helical fold to the SeV PX C-subdomain (16) (Fig. 1). As for the corresponding region of P from mumps virus (residues 343 to 392), spectroscopic studies have shown that it is also predominantly α-helical, but with a lesser degree of tertiary organization (21). Nevertheless, in all three viruses it is this very C-terminal end of P that interacts with the nucleocapsid.
FIG. 1.

Schematic representation of the sequences of the C-terminal domains of N and P proteins used or discussed in this article. (From top to bottom) SeV NTAIL, with the striped region indicating the SeV P protein binding site on NTAIL, as suggested by the work of Çevik et al. (7), and the black regions indicating the two predicted α helices (Jpred program). Two shorter SeV NTAIL constructs were also used: NTAIL(443-524) and NTAIL(443-501). For comparison, the MV NTAIL is shown with the two regions involved in binding to the C-terminal domain of MV P protein (XD) indicated: region 488 to 504, which undergoes induced α-helical folding (16, 22), and the additional site region 517 to 525 found by Bourhis et al. (5). The C-terminal domain of SeV P protein (PX [474 to 568]) and the C-terminal domain of MV protein P (XD [459 to 507]) are also depicted. In the last two domains, the gray regions correspond to the three α helices observed in these domains.
As for other paramyxoviruses, SeV N protein (524 aa) is divided into two regions (27): the well-conserved NCORE (residues 1 to 400) and the hypervariable NTAIL (residues 401 to 524) (Fig. 1). SeV NCORE contains all the regions required for self-assembly and RNA binding, whereas SeV NTAIL is involved in PX binding (9). In all Paramyxoviridae, NTAIL has very few predicted secondary structure elements (18). Using various biochemical and biophysical approaches, it was shown that NTAIL of MV is unstructured in solution (4) but undergoes an induced folding in the presence of the C-terminal domain of MV P (4, 16). Using both solution nuclear magnetic resonance (NMR) spectroscopy and X-ray crystallography, Kingston et al. (22) confirmed that a specific region of MV NTAIL (residues 487 to 503) (Fig. 1) binds as an α-helix to the surface created by the second (α2) and third (α3) helices of MV XD (Fig. 1), in an orientation parallel to helix 3, creating a four-helix bundle. Recently an additional site (residues 517 to 525) within MV NTAIL has been shown to be involved in binding to XD but retains its unfolded character (5). In contrast, it was shown that the nucleocapsid binding domain of the mumps P protein, does not bind to NTAIL but to its structured NCORE domain (21). Altogether these results showed that polymerase binding mechanisms are similar in morbiliviruses (MV) and respiroviruses (SeV) but differ significantly in rubulaviruses (mumps virus). However, some differences also seem to exist between the MV and SeV polymerase binding mechanisms. Çevik et al. (7) have identified, by mutation and deletion, residues in SeV NTAIL (residues 461 to 472) that are essential for the interaction with P, but unlike for measles virus, these residues do not match the main predicted α-helix in SeV NTAIL (Fig. 1). Moreover mutant studies of SeV PX have shown that not only residues in the folded C-subdomain, but also a few residues in the unfolded N-subdomain of PX (absent in the MV study; Fig. 1), are important for the complex formation with N (42).
The aim of this work is to study in detail the interaction between SeV PX and NTAIL and to unequivocally define the precise regions within PX and NTAIL that mediate association of the polymerase with the nucleocapsid in SeV and to compare these results with the MV XD-NTAIL complex (22). For this, various SeV NTAIL constructs were created by deleting two regions within the entire NTAIL domain (Fig. 1). These NTAIL constructs were then characterized and used for binding assays involving PX using mainly NMR spectroscopy. The results show that PX and NTAIL bind through an interaction between residues 472 to 493 of NTAIL and helices α2 and α3 of PX. The NTAIL extreme C terminus was shown not to interact with PX, in contrast to what was found for MV (5).
While on a global level the mechanisms of transcription and replication in SeV and measles and mumps viruses are very similar, on a molecular level some differences exist which highlight the evolutional flexibility of viruses. Knowledge about these differences in viral working strategy is important for finding ways of attacking these viruses.
MATERIALS AND METHODS
Expression and purification. (i) SeV C-terminal domain of P protein (X protein).
The SeV X protein (PX) spanning aa 474 to 568 of the P protein (Swiss-Prot accession no. P04859) was overproduced with an amino-terminal hexa-His tag followed by a factor Xa cleavage site and purified as described previously (28, 39).
(ii) SeV NTAIL, NTAIL(443-524), and NTAIL(443-501).
A synthetic gene coding for the Harris strain of SeV NTAIL (having the same amino acid sequence as the NTAIL Fushimi strain Q07097, apart from a single mutation at position 410 [E410K]) was produced by GENEART GmbH (Regensburg, Germany) with its DNA sequence adapted to the Escherichia coli codon usage. This gene was cloned via the NdeI and XhoI restriction sites into the pET-TEV vector that is a modified pET28a (Novagen) in which the thrombin cleavage site (CTGGTGCCGCGCGGCAGC) was replaced with a TEV cleavage site (GAAAACCTGTATTTTCAGGGC) and the T7 tag (ATGACTGGTGGACAGCAAATGGGTCGC) was deleted. The expressed protein (NTAIL) consisted of an amino-terminal hexa-His tag followed by a TEV cleavage site. After TEV cleavage, the NTAIL construct contained three additional residues (GHM) at the N terminus. The two shorter constructs, NTAIL(443-524) and NTAIL(443-501), were obtained by subcloning the corresponding region into an expression vector giving rise to a protein having an amino-terminal nona-His tag followed by a TEV cleavage site (RoBioMol cloning platform; IBS, Grenoble, France). The three constructs were then produced in E. coli BL21(DE3) (Novagen) at 37°C after induction with 1 mM isopropyl-β-d-thiogalactopyranoside (IPTG).
For 15N or 15N/13C isotopic labeling, E. coli BL21(DE3) freshly transformed with the appropriate plasmid was grown in 2 liters of M9 minimal medium containing, respectively, 1 g/liter 15NH4Cl, 1 g/liter 15NH4Cl, and 2 g/liter [13C]glucose as sole nitrogen and carbon sources.
The same protocol was used to purify NTAIL, NTAIL(443-524), and NTAIL(443-501). The cell pellet containing the recombinant protein was resuspended in 30 ml Tris-HCl buffer (50 mM [pH 8]) and 500 mM NaCl supplemented with 300 μl of protease inhibitor cocktail (stock solution, 50×) (Complete; Boehringer Mannheim). Cells were disrupted by sonication, and the soluble extract was then recovered after centrifugation at 45,000 × g for 45 min at 4°C. The supernatant was injected onto an immobilized metal affinity chromatography (IMAC)-Ni column (5 cm by 1.5 cm) (QIAGEN), previously equilibrated in Tris-HCl buffer (50 mM [pH 8])-500 mM NaCl, containing 5 mM imidazole. A step gradient of imidazole (5, 20, and 350 mM) was used for elution (25 ml for the 5 and 20 mM fractions and 12 ml for the 350 mM fraction). Imidazole in the NTAIL-containing fraction was then removed by dialysis against Tris-HCl buffer (50 mM [pH 8]), 500 mM NaCl. The polyhistidine tag was cleaved off using a TEV protease/NTAIL mass ratio of 1:100 overnight at 20°C. The NTAIL constructs were further purified by a second Ni column equilibrated in the same buffer as the first one. The unbound fractions of this column containing the NTAIL constructs (without the His tag) were concentrated and loaded on a Superdex 75 (1.6 cm by 60 cm) column (GE Healthcare) equilibrated in potassium phosphate buffer (50 mM [pH 6]) and 500 mM NaCl. Eluted fractions of each column were analyzed by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) for the presence of the recombinant protein and verification of protein purity.
Protein concentrations were calculated using the following theoretical absorption coefficients at 280 nm (obtained by using ProtParam at the EXPASY server): 12,490 M−1 cm−1 for NTAIL and 5,500 M−1 cm−1 for the two shorter NTAIL constructs.
Mass spectrometry.
Matrix-assisted laser desorption ionization-time of flight (MALDI-TOF) experiments, used to verify the mass of the purified proteins, were performed on a Voyager Elite spectrometer (Perspective).
Biochemical binding assays.
For binding experiments, ∼5 μg of purified His-tagged SeV PX was immobilized on nickel beads (50 μl) (QIAGEN) and further incubated for 60 min at 20°C with 8 μg of purified SeV NTAIL in 50 mM Tris-HCl (pH 8) in the presence of 0, 100, 300, or 500 mM NaCl. Beads were washed with the same buffer, and immobilized material was eluted with 30 μl of 500 mM imidazole. Samples were heat denatured before SDS-PAGE analysis.
NMR spectroscopy. (i) NMR sample preparation.
All NMR samples (NTAIL constructs and PX) used were prepared in potassium phosphate buffer (50 mM [pH 6]) with 500 mM NaCl, 0.02% NaN3, protease inhibitor cocktail [Complete; Boehringer Mannheim]) and 10% D2O. Dithiothreitol was added to the PX sample because of the presence of a single cysteine residue. All samples had a protein concentration around 1 mM unless described differently.
(ii) NMR experiments.
NMR spectra were acquired at 25°C on a 600-MHz Varian Inova spectrometer equipped with a triple-resonance (1H, 13C, 15N) probe including shielded z-gradients. 2D 1H-15N correlation spectra of the three NTAIL constructs were typically recorded using spectral widths of 7620 × 1,300 Hz, 512 × 170 complex points, and offsets of 4.7 × 118 ppm. For {1H}-15N steady-state heteronuclear nuclear Overhauser effect (NOE) experiments (20) a 3-s 1.7-kHz WALTZ16 decoupling scheme centered at the amide proton frequencies was used to saturate the amide proton signals, which was replaced by a 3-s delay in the reference experiment. The recycle delay in both experiments was set to 2 s. Heteronuclear NOE values per amide group were obtained from the ratio between signal intensities in the saturated and the reference experiments, where the standard deviation in the noise was taken as a measure for the error in the signal intensity. 2D HET-SOFAST experiments were recorded as described by Schanda et al. (34), using a 0.2-s recycle delay and selective 15N pulses centered around 8.0 ppm. A 1H 140° PC9 excitation pulse with an average field strength of 0.36 kHz and a REBURP refocusing pulse with an average field strength of 0.87 kHz were used. To keep the acquisition time short, only 384 complex points were used in the 1H dimension. Information on the structural compactness (34) was obtained from the ratio between the signal intensity in an experiment where the aliphatic protons were selectively inverted and that of a reference experiment.
Resonance assignment of the short construct NTAIL(443-501) was performed using a series of triple-resonance NMR experiments: a CBCA(CO)NH experiment which correlates (Cβi-1 and Cαi-1 to HNi and Ni), and a CBCANH experiment which provides, in addition, the correlation of Cβi and Cαi to HNi and Ni. Carbon side-chain resonances were extracted from an (H)C(C) total correlated spectroscopy (TOCSY)-(CO)NH experiment. Biopack sequences provided by Varian were used with minor modification. Due to the unfolded character of NTAIL, the chemical shift dispersion for all nuclei is limited. Therefore, semiconstant time acquisition was performed along the 15N dimension. The spectral widths along the 13C, 15N, and 1H dimensions were, respectively, 10,000 Hz, 1,125 Hz, and 6,000 Hz. Data were processed using nmrPipe (12) and analyzed using nmrview (17). For the indirect dimensions, the time-domain signal was extended using linear prediction by 30 data points prior to apodization and fast Fourier transform (FFT). Arginine side-chain NɛHɛ resonances were assigned using a Arg-(H)C(C)TOCSY-NɛHɛ experiment (32). The chemical shift assignment was carried out in a semiautomated manner using a home-modified version of Smartnotebook (version 3.2) (38) and has been deposited in the Biological Magnetic Resonance Data Bank (BMRB) under accession no. 15123. HN-HN NOEs were obtained from a 3D (H)N-NOE spectroscopy (NOESY)-(H)N-heteronuclear single quantum correlation (HSQC) experiment (150-ms mixing time) recorded on a Varian Inova 800-MHz spectrometer equipped with a cryogenically cooled probe.
Secondary Cα chemical shift values (Δδsec Cα) were calculated using the sequence-specific random coil Cα chemical shift values provided by Wishart et al. (43).
NMR titration and KD determination. (i) 15N-PX and unlabeled NTAIL.
Unlabeled NTAIL from two batches (115 μl at 3.1 mM and 100 μl at 1.7 mM) was gradually added to a 507-μl 0.47 mM 15N-labeled PX sample. After each addition, a 2D 1H-15N HSQC spectrum was recorded to follow the effect of the interaction of NTAIL on the PX signal positions and intensities. The final NTAIL concentration was 0.73 mM, corresponding to a 2.2 excess with respect to PX, taking into account the lower PX concentration (0.33 mM) due to dilution of the sample.
(ii) 15N-NTAIL and unlabeled PX.
The titration of 15N-NTAIL with unlabeled PX was performed in two stages. First, 490 μl of 1 mM PX was added in nine steps to a 535-μl 0.3 mM 15N-NTAIL sample. This sample was concentrated back to 500 μl, followed by addition of another 400 μl 1 mM PX in three steps, reaching a final ratio [NTAIL]/[PX] ratio of 1:4.9.
(iii) 15N NTAIL(443-501) and unlabeled PX.
A 15N-NTAIL(443-501) 490-μl 0.6 mM sample was used, and 250 μl of a 2 mM unlabeled PX sample was gradually added to this sample, following the chemical shift changes from 1H-15N HSQC spectra.
(iv) KD determination.
The dissociation constant KD can be estimated from the changes in chemical shifts of the 15N-labeled protein (A) caused by addition of the unlabeled binding partner (B), by fitting the chemical shift changes to the following equation for a two-state model in fast exchange:
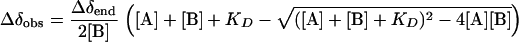 |
(1) |
where
 |
(2) |
is the combined value of observed proton and nitrogen chemical shift changes of one peak at a certain ratio of A to B, R is a scaling factor set to 6.5 determined from the ratio of variances in amide proton and nitrogen chemical shifts as observed in the BioMagResBank, and Δδend is the final chemical shift difference between free and complexed protein A.
RESULTS
SeV NTAIL belongs to the class of intrinsically disordered proteins.
The SeV NTAIL nucleotide sequence showed a high frequency of E. coli rare codons clustered in the middle of the sequence, which is likely to cause expression problems in E. coli. Indeed, no SeV NTAIL production was obtained when the natural gene was expressed in E. coli BL21(DE3) nor in E. coli strains supplemented with tRNA genes for rare codons [BL21(DE3)RIL and Rosetta(DE3)]. To improve SeV NTAIL expression, an optimized NTAIL synthetic gene was produced in which 70% of the codons were changed, including all of the E. coli rare codons. This synthetic gene was then cloned in an expression vector. NTAIL and the two shorter constructs, NTAIL(443-524) and NTAIL(443-501), were produced from the synthetic gene in E. coli BL21(DE3) as described in Materials and Methods. A high-level production of the three SeV NTAIL constructs was obtained in both LB medium and M9 minimal medium. For SeV NTAIL as well as NTAIL(443-524) and NTAIL(443-501), the protein was mainly recovered in the soluble fraction of bacterial lysates. The proteins were purified to homogeneity in three steps. The identity of the recombinant products was confirmed by mass spectrometry analysis and N-terminal sequencing. SeV NTAIL and the two shorter constructs displayed an abnormally slow migration in SDS-PAGE even after heat denaturation (Fig. 2). They migrated with apparent molecular masses (MMs) of ∼21, 16, and 12 kDa, respectively, for NTAIL, NTAIL(443-524), and NTAIL(443-501), whereas their MMs are 13.4, 8.9, and 6.7 kDa. SeV NTAIL, NTAIL(443-524), and NTAIL(443-501) were eluted from the gel filtration column as symmetrical peaks with elution volumes corresponding to globular proteins with masses of ∼31.4, 19.8, and 12.9 kDa, respectively (data not shown). This behavior has already been observed for other intrinsically disordered proteins and also for MV NTAIL (27). The same profile was observed regardless of the nature of the buffer (50 mM Tris-HCl [pH 8], 50 mM potassium phosphate [pH 6]) and the presence of different NaCl concentrations (0.2, 0.3, and 0.5 M). These first results confirm that SeV NTAIL does not adopt a well-folded structure (18).
FIG. 2.

Purified SeV NTAIL constructs. Shown are results of Coomassie blue staining of a 15% SDS-PAGE. Lane 1, size markers, indicated in kilodaltons; lane 2, NTAIL; lane 3, NTAIL(443-524); lane 4, NTAIL(443-501).
To further characterize SeV NTAIL and the two shorter constructs, several NMR experiments were recorded. The 2D 1H-15N HSQC spectrum of SeV NTAIL (Fig. 3A) represents a typical spectrum of an unfolded protein, since the signals are centered around 8.3 ppm in the 1H dimension. A total of 122 intense signals of the expected 126 backbone amides can be identified, as well as 9 less-intense signals, which probably result from a minor conformation (the mass spectrum of the sample only showed one component). Excluding these 9 lower-intensity signals, all peaks have narrow line widths, indicative of a flexible protein. In addition to the backbone signals, the expected side-chain signals of Trp (2 residues), Asn (3 residues), and Gln (4 residues) are present in the spectrum. Comparison of the 2D 1H-15N HSQC spectra of the three NTAIL constructs (Fig. 3) shows that the structural environments of the three proteins are identical, since the spectra superimpose perfectly. Only a very few signals (boxes in Fig. 3) are different, which presumably represent the amides of residues at the boundaries of the constructs. Interestingly, the set of smaller signals is present in both NTAIL and NTAIL(443-524) but absent in NTAIL(443-501).
FIG. 3.

2D 1H-15N HSQC of (A) SeV NTAIL, (B) NTAIL(443-524), and (C) NTAIL(443-501) recorded at 25°C on a 600-MHz spectrometer. Positive contour levels are presented in black and negative contour levels in gray. The two negative signals around 108 ppm in the nitrogen dimension are folded signals that resonate around 129 ppm. Dashed lines connect the two signals from the Asn and Gln side-chain NH2 groups. The spectra are typical for a disordered protein, and the spectra of the different constructs superimpose perfectly. The few peaks that do not superimpose are indicated by gray boxes and presumably correspond to residues at the boundaries of the constructs.
In order to further characterize the structural properties of the NTAIL constructs, we measured both 15N heteronuclear NOE (hetNOE) (20) values that give information on fast local motions, as well as λNOE values from the HET-SOFAST (34) experiment, that report on “structural compactness” (data not shown). The majority of residues in the three constructs are very flexible, as evidenced by low hetNOE values (<0.35), and have an unstructured character, as evidenced by λNOE values between 0.6 and 1. However, in all three NTAIL constructs, the same 10 residues have hetNOE values between 0.35 and 0.6 and λNOE values ranging from 0.25 to 0.5, and thus represent a region that is less flexible and has a relatively high structural compactness.
SeV NTAIL binds to the surface created by the second (α2) and third (α3) helices of PX.
NMR experiments are necessarily performed at high protein concentrations, and we previously found that PX at such high concentrations is only stable in 0.5 M NaCl (3). In order to analyze the effect of ionic strength on the interaction between SeV PX and NTAIL, binding experiments were carried out using His-tagged PX immobilized on a metal affinity support, which was further incubated with NTAIL. Besides small amounts of NTAIL unspecifically recovered in the eluted fractions in the absence of PX (since no His tag is present in NTAIL), we observed roughly the same amount of NTAIL eluting with His-tagged PX whatever the ionic strength (between 0 and 500 mM NaCl) (Fig. 4). Therefore, all NMR experiments on mixtures of PX and NTAIL were performed at 0.5 M NaCl.
FIG. 4.

Binding of SeV NTAIL to PX is not influenced by ionic strength. Shown are the results of Coomassie blue staining by 15% SDS-PAGE. Lane M, size markers indicated in kilodaltons. The two following lanes contain purified PX and NTAIL proteins as specific markers. The next two lanes show two control experiments in which immobilized tagged PX on nickel beads was incubated without NTAIL and NTAIL was incubated without PX. Coelution experiments were performed with immobilized tagged PX incubated with NTAIL and washed with different NaCl concentrations (0, 100, 300, and 500 mM).
NMR chemical shifts are very sensitive to the local environment of the nuclear spin, and NMR is therefore a very useful technique to map protein interaction sites. While labeling only one of the two interacting proteins, the effect of the binding partner can be studied by recording 1H-15N HSQC spectra after addition of increasing amounts of the unlabeled binding partner. In the case of strong binding, the signals of the free protein will disappear and new signals of the complex will appear, which is referred to as “slow exchange.” When the two proteins only bind weakly, signals will move to a new position while adding the binding partner, reaching a plateau value when saturating the complex, which is referred to as “fast exchange.” In the case of intermediate exchange, signals will broaden during the course of the titration. In the case of PX, about 20 signals are affected by the addition of unlabeled NTAIL. Some peaks only change position, while others clearly show line broadening, which in some cases even causes the signal to become too broad to be detected above the noise level (Fig. 5A). The chemical shift changes along the protein sequence are shown in Fig. 5B, where negative bars indicate residues that could not be followed due to extreme line broadening. Clearly, only residues in the folded C-subdomain are affected, and more specifically the NTAIL binding site is located on the surface created by the second (α2) and third (α3) helices of PX, as shown in Fig. 5C.
FIG. 5.

Titration of 15N-PX with NTAIL. (A) Superimposition of seven 1H-15N HSQC spectra at relative ratios of NTAIL to PX that are indicated on the top right of the figure. This zoom illustrates PX resonances that shift (bold), broaden (boxed), or disappear (italic) upon NTAIL binding, which is typical for intermediate exchange. (B) Backbone amide chemical shift variations between the free and bound forms of PX. Chemical shift changes larger than the average chemical shift change plus 1 standard deviation (0.065 ppm) were considered significant and are represented by the orange (>0.065 ppm) and red (>0.13 ppm) bars. Some residues from the His tag (<474) are missing in the spectrum; all of the other gaps represent proline residues. Green bars correspond to residues showing extreme line broadening. (C) Ribbon representation of the lowest-energy structure of the PX C-subdomain (Protein Data Bank accession no. 1R4G), color coded according to the chemical shift changes shown in panel B. Balls represent the backbone amide groups that are affected by the presence of NTAIL. (D) Relative chemical shift changes for V559 versus the ratio between 15N-PX and NTAIL concentrations. The intersection at a ratio of 1:1 implies that one PX molecule binds to one NTAIL molecule.
The chemical shift changes as a function of PX and NTAIL concentrations were fitted to equation 1, using both the dissociation constant KD and the plateau value Δδend as fitting parameters. In this way, an average KD of 57 ± 18 μM was found, using 13 of the affected peaks. Using the Δδend values, a 1:1 stoichiometry of the complex could be determined, as shown for Val559 in Fig. 5D.
The binding site of SeV NTAIL is only located in one region having a helical propensity.
In order to precisely define residues of NTAIL involved in PX binding, a similar NMR experiment was done involving 15N-NTAIL and unlabeled PX. About 20 NTAIL signals are affected by the presence of PX (Fig. 6). Some signals only change position, while others clearly show line broadening as was observed for PX resonances in presence of NTAIL. All of the affected NTAIL signals are not only found in the 2D 1H-15N HSQC of NTAIL(443-524) but also in the spectrum of the shortest construct, NTAIL(443-501), allowing us to conclude that the NTAIL binding region on PX is only located between residues 443 and 501 and thus that the extreme C terminus of SeV NTAIL does not interact with PX. Interaction between 15N-NTAIL(443-501) and unlabeled PX confirmed that the same resonances are affected by the presence of PX in both NTAIL and NTAIL(443-501). As we assigned the resonances of this shorter construct (Fig. 7), the PX binding site can now be mapped on the NTAIL(443-501) sequence, and comprises residues 472 to 493. In panels A to D of Fig. 8, an overview of the structural properties of NTAIL(443-501) is presented, where the PX interaction site, as identified from the titration with PX (Fig. 8E), is indicated by the gray area. Chemical shift deviations from random coil of the Cα atoms in the region 477 to 491 are higher than 0.9 ppm and indicate that this region, which falls within the PX interaction site, shows a helical tendency in uncomplexed NTAIL. This is confirmed by the higher hetNOE values that indicate reduced flexibility, low λNOE values that evidence the presence of a structured region, and strong HN-HN(i,i + 1) NOEs, as well as the presence of HN-HN(i,i + 2) NOEs, which are both typical for an α-helical structure. The region is slightly smaller than the α helix (residues 477 to 493) predicted by the consensus method used for protein secondary structure prediction (Jpred; http://www.compbio.dundee.ac.uk/∼www-jpred/). In Fig. 8F and G, the hetNOE and λNOE values of NTAIL(443-501) in complex with PX are shown. Several data points in the interaction site are lacking, since many resonances strongly broaden or even disappear upon interaction. Moreover, error bars are larger for all other residues in the interaction site as a result of the signal broadening. Nevertheless, the average hetNOE for residues in the PX interaction site (residues 472 to 493 [gray area in Fig. 8]) has changed from 0.29 ± 0.17 (21 residues) to 0.47 ± 0.20 (12 residues), clearly indicating the rigidification of this region in NTAIL upon interaction with PX. Regions of NTAIL that do not interact with PX remain very flexible. The observation of strong HN-HN(i,i + 1) NOEs for residues at the edges of the interaction site (Fig. 8H) (which thus experience less signal broadening) confirms that, in complex with PX, NTAIL(477-491) folds as an α-helix. The PX interaction site of NTAIL contains five arginine residues. Their side-chain NɛHɛ resonances are affected by binding to PX (white bars in Fig. 8E). Moreover, four out of the five arginine side chains become less flexible when bound to PX (white bars in Fig. 8B and F), indicating that these positively charged residues are directly involved in binding to PX.
FIG. 6.

Interaction of 15N NTAIL(401-524) with PX. An enlargement of the seven 1H-15N HSQC spectra with different relative ratios of NTAIL and PX is shown. Dashed arrows indicate peaks that change position upon addition of PX, whereas disappearing peaks are indicated by a dashed circle.
FIG. 7.

Assigned HSQC of NTAIL(443-501) recorded at 25°C on an 800-MHz spectrometer. Positive contour levels are presented in black and negative contour levels in gray. Backbone amide resonances are indicated in boldface and side-chain NɛHɛ (R, W, and Q) and NδHδ (N) are in italic. Arginine side chains are folded twice (−42 ppm), and tryptophan side chains are folded once (+21 ppm).
FIG. 8.

Characterization of NTAIL(443-501) free and in complex with PX. (A) Secondary Cα chemical shifts, where high positive values indicate helical propensity, (B) hetNOE, (C) λNOE, and (D) HN-HN NOEs of NTAIL(443-501) free. For the i,i + 1 NOEs in panel D, the thickness of the line indicates strong, medium, or weak. (E) Backbone and arginine side-chain combined NH chemical shift changes upon interaction with PX. Chemical shift changes superior to 0.045 ppm (dashed line) were considered significant. Open bars in panels B, E, and F are from arginine side-chain NɛHɛ resonances. (F and G) hetNOE (F) and HET-SOFAST (G) of NTAIL(443-501) in complex with PX. (H) HN-HN NOEs in complex with PX. Note that several data points are missing, as several resonances disappeared upon interaction with PX. The gray area highlights the region affected by PX binding.
DISCUSSION
Interaction between PX and NTAIL is governed by electrostatics.
During SeV RNA synthesis, the interaction between the polymerase complex (L+P) and the nucleocapsid (N/RNA) occurs via the binding of the C-terminal domains of P and N proteins. We have now characterized the C-terminal domain of SeV N protein (NTAIL), which, like MV NTAIL (4), belongs to the class of intrinsically disordered proteins, and precisely defined the binding regions within this latter domain and the C-terminal domain of P (PX). SeV NTAIL was shown to bind to the surface created by helices α2 and α3 of PX. A similar binding site was observed for MV NTAIL on XD involving the α2/α3 face of this latter domain (22). Only one region of SeV NTAIL, involving residues 472 to 493, was shown to bind to PX. Within this region, a stretch of residues (477 to 491) has a helical propensity in free NTAIL. Upon binding to PX, this stretch of residues rigidifies and gains in structural compactness, forming a regular α-helix. The extreme C terminus of SeV NTAIL does not interact with PX. This is in contrast with the observation of two interacting sites within MV NTAIL involved in XD binding (Fig. 1) (5, 29).
By displaying the NTAIL binding site on the 3D structure of the PX C-subdomain (Fig. 5C), we can now better explain the phenotypes observed for several mutants created by Tuckis et al. (42) (Fig. 9C). In these PX mutants, clustered charged or hydrophobic residues were changed to alanine residues and tested for various protein-protein interactions and for viral RNA synthesis. Four of the mutants fall into the C-subdomain of PX. Of these mutants, the first one, with mutations (L524A, V525A, and I526A) affecting residues within helix α1 that is not involved in NTAIL binding, was completely inactive. Two of the mutations (L524 and V525) are located in the hydrophobic core of PX and may destabilize the PX structure leading to a complete loss of function. Two other mutants with mutations affecting residues within helix α3 behave differently. One, located at the N terminus (D549A, E551A, and K553A), retains essentially wild-type activity, while the other, located at the C terminus of helix 3 (E560A, E561A, and D562A) is completely inactive in vitro. That the first mutant has an unaltered activity can be explained by the fact that the N-terminal part of helix α3 is almost not affected by NTAIL binding. Only the amides of L552 and K553, both facing to the interior of the protein, display chemical shift changes upon interaction with NTAIL. Most probably, these chemical shift changes are caused by an interaction with a side chain of NTAIL that is directed towards the hydrophobic core. In addition, both side chains of the mutated residues D549 and K553 are not located on but point away from the binding site. In contrast, mutations in the C-terminal part of helix α3 concern three negatively charged residues located in a region that is highly affected by the presence of NTAIL. These residues create a negative surface potential (Fig. 9) necessary for the interaction with NTAIL, which contains in its PX helical binding region several positively charged arginine residues (477-SDIERRIAMRLAERR-491). The interaction between SeV NTAIL and PX is thus mainly driven by electrostatic interactions.
FIG. 9.

Comparison of the NTAIL binding site on the SeV PX C-subdomain (top) and on MV XD (bottom). (A and D) Surface representation displaying only the heavy atoms color coded according to the surface potential from red (negative) to blue (positive) and (B and E) surface representation where hydrophobic patches are colored yellow. (C) Ribbon representation of SeV PX C-subdomain showing the residues that were simultaneously mutated by Tuckis et al. (42). (F) Ribbon representation of MV XD in complex with MV NTAIL (22). All images were created using MOLMOL (25).
The last mutant, located within helix α2 (R533A, E535A, and K536A) is still active, although the in vitro transcription activity was found to be 50% lower. The mutations are located in the region that is involved in NTAIL binding. However, the two positively charged arginine and lysine residues are both located at the opposite site of the α2/α3 face, and mutating them to an alanine residue should not influence binding. Only the side chain of E535 points towards the binding site and enlarges the negatively charged patch created by the glutamic acid residues on helix α3. This does probably influence the interaction between PX and NTAIL, which might be the cause of the lower activity of this mutant. Another mutation that appeared to affect both transcription and replication in vitro was the mutation to alanine residues of N506 and R509 in the unfolded N-subdomain of PX. We have unambiguously shown that this domain does not directly interact with NTAIL and the fact that this mutation influences the function of the polymerase must thus result from an indirect effect. The unstructured character (2) and the intrinsic dynamics (unpublished data) of this subdomain are supposed to be important for placing the polymerase onto the nucleocapsid, and it is possible that this mutant has an altered flexibility which is not optimal for the action of the P tetramer.
As mentioned above, the NTAIL helix is placed by its positively charged arginine residues in the binding site on the mainly negatively charged PX α2/α3 face. More specifically we showed that four NTAIL arginine side chains (R482, R486, R490, and R491) become less flexible upon interaction with PX, while the flexibility of R481 is virtually unaltered. R482, R486, and R490 are located in the region that has helical propensity in free NTAIL, and they are thus all positioned on the same side of the helix as they are spaced by four residues which makes about one helical turn. R491 is located just outside of the helical region and could well line up with the other arginines to create one positively charged surface area. This surface can thus interact with the negatively charged area on PX mainly created by E535 on helix α2 and both E561 and D562 on helix α3, by binding of the NTAIL helix in a parallel fashion with respect to helix α3 of PX. Such a parallel arrangement was also found for the MV NTAIL/XD complex (22).
Because of solubility problems with PX, all measurements had to be performed at a salt concentration of 0.5 M. It is possible that the affinity between PX and NTAIL is affected at this salt concentration and that the measured KD of 57 μM is an overestimation compared to when this value is measured under low-salt conditions. However, evidence has accumulated over the years indicating that some surface salt bridges can be relatively insensitive to NaCl (see reference 30 and references therein).
SeV N and P C-terminal domains form a low-affinity complex.
The interaction between the polymerase complex (L+P) and the nucleocapsid (N/RNA) occurs via the binding of the C-terminal domains of P and N proteins. During RNA synthesis, the P tetramer is proposed to “cartwheel” along the N/RNAs, by on-and-off interactions via its PX domains with successive NTAIL domains, while the polymerase L, remaining fixed, transcribes or replicates the template RNA (23). Therefore, a weak binding affinity between SeV PX and NTAIL is necessary for movement of the L protein along the nucleocapsid. Whereas displacement of P on the nucleocapsid occurs in steps of N subunits (i.e., per 6 nucleotides), L moves one nucleotide at a time. In this context, the complex between PX and NTAIL should be a temporary complex that should be able to exist long enough for L to synthesize new RNA, but short enough for moving on to the next N-protomer. In the case of MV, a KD in the 0.1 μM range was found for the complex between the entire MV NTAIL and XD (5). However, when using a smaller MV NTAIL construct comprising residues 477 to 505 that represents the region that folds upon binding, a KD of 13 μM was found (21, 22). This difference in affinity was explained by the existence of an extra 9-residue binding site in MV NTAIL located at the extreme C terminus, which keeps its unfolded character during the interaction. As was discussed by Bourhis et al. (5), the high affinity between MV XD and NTAIL probably does not allow the polymerase to move along the nucleocapsid template as would be needed for RNA synthesis. Therefore, they propose the intervention of other cellular/viral cofactors that mediate the interaction. In this study, we have shown that SeV NTAIL only contains one binding region comprising residues 472 to 493 and that the C-terminal end is not involved in PX binding. The low affinity between SeV PX and NTAIL ensures that the individual PX/NTAIL complexes are easily disrupted, which thus allows the polymerase to progress along the N/RNA template. During RNA synthesis, the four possible PX/NTAIL complexes are probably not all formed at the same moment. P probably uses one or more free PX domains to reach for the subsequent NTAIL domain(s). The rather low PX/NTAIL affinity in SeV would thus allow the polymerase to properly perform its task without the eventual need for additional viral or host factors. The affinity of the whole tetrameric polymerase for the N/RNA template, when all four PX domains interact with four NTAIL domains, is obviously higher and ensures that the polymerase does stay tightly attached to the inactive nucleocapsid when it is packaged into a virus particle.
N and P interactions in paramyxoviruses are different on a molecular level.
On a global level, the interactions between the C-terminal domains of N and P proteins from SeV and MV are very similar, giving rise to a four-helix bundle. MV and SeV NTAIL are both intrinsically unstructured containing a specific region which shows restricted motion before undergoing induced folding upon binding. The MV XD and SeV PX C-subdomains not only adopt a similar three-helix bundle fold (3, 16, 22) but present the same binding site for their respective NTAIL partner located on the α2/α3 face (22). On a molecular level, however, the binding sites on both complexes are quite different (Fig. 9). While the binding interface of the MV NTAIL-XD complex is dominated by hydrophobic amino acids as shown by the hydrophobic cleft that is built up by XD α2/α3 helices (Fig. 9E) and which provides a complementary surface for the hydrophobic side of the induced MV NTAIL α helix (486-QDSRRSADALLRLQAMAGI-504) (22), we determined that interaction between SeV NTAIL and PX is mainly driven by electrostatic interactions, as shown by the negative surface created by PX α2/α3 helices (Fig. 9A) and the positively charged NTAIL binding sequence.
This result shows once more the great facility of adaptation and evolution of viruses. It gives a good illustration of how selection pressure allowed the C-terminal domains of N and P to evolve concomitantly within the paramyxovirus family in order to lead to protein complexes having the same 3D fold and thus the same function, but with very limited sequence identity. This study also points out that the design of antiviral agents blocking the interaction between the C-terminal domains of P and N proteins and thus the synthesis of RNA within the paramyxovirus family would require a specific strategy for each genus.
Acknowledgments
This work was supported by the CNRS, the UJF, and the CEA. K.H. was funded by a fellowship from The Netherlands Organization for Scientific Research (NWO) as well as by a postdoctoral fellowship from the French Ministère délégué à la Recherche.
We thank Joe Curran for providing us with a clone of SeV natural NTAIL gene. We also thank Anne Chouquet for technical help in cloning the NTAIL construct and Isabel Ayala for helping us with the purifications. We thank Marjolaine Noirclerc-Savoye and Benoit Gallet (RoBioMol at the Institut de Biologie Structurale, Grenoble, France) for the subcloning of the two shorter NTAIL constructs, David Lemaire and Bernard Dublet for MALDI-TOF experiments, Jean-Pierre Andrieu for N-terminal sequencing, and Paul Schanda for help with SOFAST experiments.
Footnotes
Published ahead of print on 25 April 2007.
REFERENCES
- 1.Abraham, G., D. P. Rhodes, and A. K. Banerjee. 1975. The 5′ terminal structure of the methylated mRNA synthesized in vitro by vesicular stomatitis virus. Cell 5:51-58. [DOI] [PubMed] [Google Scholar]
- 2.Bernado, P., L. Blanchard, P. Timmins, D. Marion, R. W. Ruigrok, and M. Blackledge. 2005. A structural model for unfolded proteins from residual dipolar couplings and small-angle X-ray scattering. Proc. Natl. Acad. Sci. USA 102:17002-17007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Blanchard, L., N. Tarbouriech, M. Blackledge, P. Timmins, W. P. Burmeister, R. W. Ruigrok, and D. Marion. 2004. Structure and dynamics of the nucleocapsid-binding domain of the Sendai virus phosphoprotein in solution. Virology 319:201-211. [DOI] [PubMed] [Google Scholar]
- 4.Bourhis, J. M., K. Johansson, V. Receveur-Brechot, C. J. Oldfield, K. A. Dunker, B. Canard, and S. Longhi. 2004. The C-terminal domain of measles virus nucleoprotein belongs to the class of intrinsically disordered proteins that fold upon binding to their physiological partner. Virus Res. 99:157-167. [DOI] [PubMed] [Google Scholar]
- 5.Bourhis, J. M., V. Receveur-Brechot, M. Oglesbee, X. Zhang, M. Buccellato, H. Darbon, B. Canard, S. Finet, and S. Longhi. 2005. The intrinsically disordered C-terminal domain of the measles virus nucleoprotein interacts with the C-terminal domain of the phosphoprotein via two distinct sites and remains predominantly unfolded. Protein Sci. 14:1975-1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Calain, P., and L. Roux. 1993. The rule of six, a basic feature for efficient replication of Sendai virus defective interfering RNA. J. Virol. 67:4822-4830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Çevik, B., J. Kaesberg, S. Smallwood, J. A. Feller, and S. A. Moyer. 2004. Mapping the phosphoprotein binding site on Sendai virus NP protein assembled into nucleocapsids. Virology 325:216-224. [DOI] [PubMed] [Google Scholar]
- 8.Curran, J., R. Boeck, N. Lin-Marq, A. Lupas, and D. Kolakofsky. 1995. Paramyxovirus phosphoproteins form homotrimers as determined by an epitope dilution assay, via predicted coiled coils. Virology 214:139-149. [DOI] [PubMed] [Google Scholar]
- 9.Curran, J., H. Homann, C. Buchholz, S. Rochat, W. Neubert, and D. Kolakofsky. 1993. The hypervariable C-terminal tail of the Sendai paramyxovirus nucleocapsid protein is required for template function but not for RNA encapsidation. J. Virol. 67:4358-4364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Curran, J., and D. Kolakofsky. 1988. Scanning independent ribosomal initiation of the Sendai virus X protein. EMBO J. 7:2869-2874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Curran, J., J.-B. Marq, and D. Kolakofsky. 1995. An N-terminal domain of the Sendai paramyxovirus P protein acts as a chaperone for the NP protein during the nascent chain assembly step of genome replication. J. Virol. 69:849-855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Delaglio, F., S. Grzesiek, G. W. Vuister, G. Zhu, J. Pfeifer, and A. Bax. 1995. NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR 6:277-293. [DOI] [PubMed] [Google Scholar]
- 13.Egelman, E. H., S.-S. Wu, M. Amrein, A. Portner, and G. Murti. 1989. The Sendai virus nucleocapsid exists in at least four different helical states. J. Virol. 63:2233-2243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Horikami, S. M., and S. A. Moyer. 1995. Alternative amino acids at a single site in the Sendai virus L protein produce multiple defects in RNA synthesis in vitro. Virology 211:577-582. [DOI] [PubMed] [Google Scholar]
- 15.Hunt, D. M., E. F. Smith, and D. W. Buckley. 1984. Aberrant polyadenylation by a vesicular stomatitis virus mutant is due to an altered L protein. J. Virol. 52:515-521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Johansson, K., J. M. Bourhis, V. Campanacci, C. Cambillau, B. Canard, and S. Longhi. 2003. Crystal structure of the measles virus phosphoprotein domain responsible for the induced folding of the C-terminal domain of the nucleoprotein. J. Biol. Chem. 278:44567-44573. [DOI] [PubMed] [Google Scholar]
- 17.Johnson, B. A., and R. A. Blevins. 1994. NMRView: a computer program for the visualization and analysis of NMR data. J. Biomol. NMR 4:603-614. [DOI] [PubMed] [Google Scholar]
- 18.Karlin, D., F. Ferron, B. Canard, and S. Longhi. 2003. Structural disorder and modular organization in Paramyxovirinae N and P. J. Gen. Virol. 84:3239-3252. [DOI] [PubMed] [Google Scholar]
- 19.Karlin, D., S. Longhi, V. Receveur, and B. Canard. 2002. The N-terminal domain of the phosphoprotein of Morbilliviruses belongs to the natively unfolded class of proteins. Virology 296:251-262. [DOI] [PubMed] [Google Scholar]
- 20.Kay, L. E., D. A. Torchia, and A. Bax. 1989. Backbone dynamics of proteins as studied by 15N inverse detected heteronuclear NMR spectroscopy: application to staphylococcal nuclease. Biochemistry 28:8972-8979. [DOI] [PubMed] [Google Scholar]
- 21.Kingston, R. L., W. A. Baase, and L. S. Gay. 2004. Characterization of nucleocapsid binding by the measles virus and mumps virus phosphoproteins. J. Virol. 78:8630-8640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kingston, R. L., D. J. Hamel, L. S. Gay, F. W. Dahlquist, and B. W. Matthews. 2004. Structural basis for the attachment of a paramyxoviral polymerase to its template. Proc. Natl. Acad. Sci. USA 101:8301-8306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kolakofsky, D., P. Le Mercier, F. Iseni, and D. Garcin. 2004. Viral DNA polymerase scanning and the gymnastics of Sendai virus RNA synthesis. Virology 318:463-473. [DOI] [PubMed] [Google Scholar]
- 24.Kolakofsky, D., T. Pelet, D. Garcin, S. Hausmann, J. Curran, and L. Roux. 1998. Paramyxovirus RNA synthesis and the requirement for hexamer genome length: the rule of six revisited. J. Virol. 72:891-899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Koradi, R., M. Billeter, and K. Wüthrich. 1996. MOLMOL: a program for display and analysis of macromolecular structures. J. Mol. Graph. 14:29-32, 51-55. [DOI] [PubMed] [Google Scholar]
- 26.Kuroya, N., and M. Ishida. 1953. Newborn virus pneumonitis (type Sendai). II. Isolation of new virus possessing hemagglutinin activity. Yokohama Med. Bull. 4:217-233. [PubMed] [Google Scholar]
- 27.Longhi, S., V. Receveur-Brechot, D. Karlin, K. Johansson, H. Darbon, D. Bhella, R. Yeo, S. Finet, and B. Canard. 2003. The C-terminal domain of the measles virus nucleoprotein is intrinsically disordered and folds upon binding to the C-terminal moiety of the phosphoprotein. J. Biol. Chem. 278:18638-18648. [DOI] [PubMed] [Google Scholar]
- 28.Marion, D., N. Tarbouriech, R. W. Ruigrok, W. P. Burmeister, and L. Blanchard. 2001. Assignment of the 1H, 15N and 13C resonances of the nucleocapsid-binding domain of the Sendai virus phosphoprotein. J. Biomol. NMR 21:75-76. [DOI] [PubMed] [Google Scholar]
- 29.Morin, B., J. M. Bourhis, V. Belle, M. Woudstra, F. Carriere, B. Guigliarelli, A. Fournel, and S. Longhi. 2006. Assessing induced folding of an intrinsically disordered protein by site-directed spin-labeling electron paramagnetic resonance spectroscopy. J. Phys. Chem. B 110:20596-20608. [DOI] [PubMed] [Google Scholar]
- 30.Perez-Jimenez, R., R. Godoy-Ruiz, B. Ibarra-Molero, and J. M. Sanchez-Ruiz. 2004. The efficiency of different salts to screen charge interactions in proteins: a Hofmeister effect? Biophys. J. 86:2414-2429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Poch, O., I. Sauvaget, M. Delarue, and N. Tordo. 1989. Identification of four conserved motifs among the RNA-dependent polymerase encoding elements. EMBO J. 8:3867-3874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rao, N. S., P. Legault, D. R. Muhandiram, J. Greenblatt, J. L. Battiste, J. R. Williamson, and L. E. Kay. 1996. NMR pulse schemes for the sequential assignment of arginine side-chain Hɛ protons. J. Magn. Reson. B 113:272-276. [DOI] [PubMed] [Google Scholar]
- 33.Ryan, K. W., and D. W. Kingsbury. 1988. Carboxyl-terminal region of Sendai virus P protein is required for binding to viral nucleocapsids. Virology 167:106-112. [DOI] [PubMed] [Google Scholar]
- 34.Schanda, P., V. Forge, and B. Brutscher. 2006. HET-SOFAST NMR for fast detection of structural compactness and heterogeneity along polypeptide chains. Magn. Reson. Chem. 44:177-184. [DOI] [PubMed] [Google Scholar]
- 35.Sidhu, M. S., J. P. Menonna, S. D. Cook, P. C. Dowling, and S. A. Udem. 1993. Canine distemper virus L gene: sequence and comparison with related viruses. Virology 193:50-65. [DOI] [PubMed] [Google Scholar]
- 36.Six, C., F. Franke, K. Mantey, C. Zandotti, F. Freymuth, F. Wild, I. Parent du Chatelet, and P. Malfait. 2005. Measles outbreak in the Provence-Alpes-Cote d'Azur region, France, January-July 2003. Eur. Surveill. 10:46-48. [PubMed] [Google Scholar]
- 37.Skiadopoulos, M. H., S. R. Surman, J. M. Riggs, W. R. Elkins, M. St. Claire, M. Nishio, D. Garcin, D. Kolakofsky, P. L. Collins, and B. R. Murphy. 2002. Sendai virus, a murine parainfluenza virus type 1, replicates to a level similar to human PIV1 in the upper and lower respiratory tract of African green monkeys and chimpanzees. Virology 297:153-160. [DOI] [PubMed] [Google Scholar]
- 38.Slupsky, C. M., R. F. Boyko, V. K. Booth, and B. D. Sykes. 2003. Smartnotebook: a semi-automated approach to protein sequential NMR resonance assignments. J. Biomol. NMR 27:313-321. [DOI] [PubMed] [Google Scholar]
- 39.Tarbouriech, N., J. Curran, C. Ebel, R. W. Ruigrok, and W. P. Burmeister. 2000. On the domain structure and the polymerization state of the Sendai virus P protein. Virology 266:99-109. [DOI] [PubMed] [Google Scholar]
- 40.Tarbouriech, N., J. Curran, R. W. Ruigrok, and W. P. Burmeister. 2000. Tetrameric coiled coil domain of Sendai virus phosphoprotein. Nat. Struct. Biol. 7:777-781. [DOI] [PubMed] [Google Scholar]
- 41.Testa, D., and A. K. Banerjee. 1977. Two methyltransferase activities in the purified virions of vesicular stomatitis virus. J. Virol. 24:786-793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tuckis, J., S. Smallwood, J. A. Feller, and S. A. Moyer. 2002. The C-terminal 88 amino acids of the Sendai virus P protein have multiple functions separable by mutation. J. Virol. 76:68-77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wishart, D. S., C. G. Bigam, A. Holm, R. S. Hodges, and B. D. Sykes. 1995. 1H, 13C and 15N random coil NMR chemical shifts of the common amino acids. I. Investigations of nearest-neighbor effects. J. Biomol. NMR 5:67-81. [DOI] [PubMed] [Google Scholar]