

Abstract
Relatively little is known at the functional genomic level about the global host response to human immunodeficiency virus type 1 (HIV-1) infection. Microarray analyses by several laboratories, including our own, have revealed that HIV-1 infection causes significant changes in host mRNA abundance and regulation of several cellular biological pathways. However, it remains unclear what consequences these changes bring about at the protein level. Here we report the expression levels of ∼3,200 proteins in the CD4+ CEMx174 cell line after infection with the LAI strain of human immunodeficiency virus type 1 (HIV-1); the proteins were assessed using liquid chromatography-mass spectrometry coupled with stable isotope labeling and the accurate mass and time tag approach. Furthermore, we found that 687 (21%) proteins changed in abundance at the peak of virus production at 36 h postinfection. Pathway analysis revealed that the differential expression of proteins was concentrated in select biological pathways, exemplified by ubiquitin-conjugating enzymes in ubiquitination, carrier proteins in nucleocytoplasmic transport, cyclin-dependent kinase in cell cycle progression, and pyruvate dehydrogenase of the citrate cycle pathways. Moreover, we observed changes in the abundance of proteins with known interactions with HIV-1 viral proteins. Our proteomic analysis captured changes in the host protein milieu at the time of robust virus production, depicting changes in cellular processes that may contribute to virus replication. Continuing analyses are expected to focus on blocking virus replication by targeting these pathways and their effector proteins.
Human immunodeficiency virus type-1 (HIV-1) infection has dramatic effects on host cell physiology. Infection causes the host cell to produce large quantities of viral RNAs and proteins, alters the progression of the host cell cycle (49, 72), leads to interactions between viral and host cell proteins (25), and can result in profound alterations in host cell morphology, such as syncytium formation. One might expect that such profound functional and morphological changes in host cells would be associated with significant changes in the patterns of host cell gene expression. To this end, using genome-wide mRNA profiling, several studies have described changes in host gene expression due to HIV-1 infection (31, 89). Moreover, increased gene expression changes have also been associated with increased viral loads in viremic patients (17).
Virus replication is likely to unleash a battery of stresses with the abrupt introduction and accumulation of foreign proteins synthesized in infected cells. For example, replication of the hepatitis C virus genome induces endoplasmic reticulum (ER) stress, triggering the host unfolded-protein response (85). Similarly, the accumulation of viral proteins in the ER during herpes simplex virus infection also activates the kinase PERK, a hallmark of ER stress response (15, 55). Given the effects of viral infections on host protein synthesis and processing mechanisms over the course of viral infections, it is not surprising that protein abundance changes have been reported for viral infections. For instance, in hepatitis C virus infection models, abundance changes of proteins, particularly in relation to lipid metabolism, have been reported through the use of mass spectrometry (MS) (45) and in conjunction with stable isotope labeling (54). As described below, we have adopted both approaches for protein expression profiling in our HIV infection system to maximize the number of proteins surveyed.
Several lines of evidence suggest that the flux of protein synthesis and degradation may also be modulated during HIV-1 infection. Potential effects on host protein synthesis can be gleaned from the inhibition of PKR-induced eIF2 phosphorylation by HIV Tat (8) as well as from the down-regulation of PKR itself in HIV-1 infection (79). HIV-1 and other members of the retrovirus family, including HIV-2, simian immunodeficiency virus, mouse Moloney murine leukemia virus, and mouse mammary tumor virus, all encode viral proteases that have been reported to cleave the initiation factor eIF4G (4, 64), a component of the eIF4F translation initiation complex. We have also previously reported a decrease in total protein synthesis concomitant with degradation of cellular mRNA (1). More recently, protein expression profiling by gel electrophoresis coupled to MS has revealed the differential expression of cellular proteins associated with HIV-1 infection in monocyte-derived macrophages (18) as well as with the expression of HIV-1 Tat in human T cells (20) and astrocytes (71).
Here we report the first large-scale quantitative analysis of a CD4+ cell line in a model of acute HIV-1 infection, in order to capture cellular protein abundance changes and gain insights into cellular processes that are associated with robust virus production. High-sensitivity liquid chromatography (LC), coupled with MS and the accurate mass and time (AMT) tag approach, has allowed for the relative quantification of 3,255 host proteins. We report that while 79% of the proteins detected did not change in abundance, most of the protein abundance changes that did occur were concentrated in select pathways, suggesting that specific cellular functions may be implicated in robust virus production. Systematic analysis revealed changes in functional categories, such as nuclear transport, ubiquitination, cell cycle progression, and the citrate cycle. While some of the processes have been implicated in HIV-1 virion processing and packaging, the concerted quantitative changes of individual constituents can be assessed in only a large-scale profiling analysis. We will discuss the contributions of these changes in the context of host cells undergoing robust virus production.
MATERIALS AND METHODS
Cell line and virus.
Human CEMx174 cells (ATCC) were incubated in the LAI strain of HIV-1 (HIV-1LAI) (provided by Jean-Marie Bechet and Luc Montagnier through the AIDS Research and Reference Reagent Program, Division of AIDS, NIAID, NIH) (7, 91) at two 50% tissue culture infective doses (TCID50) per cell at 37°C for 2 h. Mock-infected cultures were incubated in virus-free media in like manners. Cells were then rinsed twice with fresh RPMI 1640 medium supplemented with 10% fetal bovine serum and subsequently incubated for 12 to 42 h as indicated. Infections were repeated to obtain biological replicates. A total of 108 cells were harvested for protein extraction by washing twice with phosphate-buffered saline and resuspending in 10 mM KH2PO4.
Assessment of intracellular p24 gag expression by flow cytometry.
CEMx174 cells were infected as described above and harvested at the times indicated following HIV and mock infections. Cell viability was measured based on trypan blue-excluding cells as a percentage of the total cell count. Harvested cells were fixed and stained using the Fix & Perm kit (Caltag) and anti-p24 KC57-fluorescein isothiocyanate (FITC) antibody (Beckman-Coulter) as described previously (31, 89). Fluorescence was detected on the FL1 channel on the logarithmic scale using a FC500 flow cytometer (Beckman-Coulter). A total of 10,000 events were gated using a forward- versus side-scatter plot for analysis using WinMDI, version 2.8.
Protein digestion.
Cells in 1 ml of phosphate buffer (pH 7.0) were lysed by 5 min of sonication in an ice-water bath. Equal volumes of trifluoroethanol (Sigma-Aldrich) were added to each sample, and the samples were incubated at 60°C for 1 h and then sonicated for 2 min as described previously (93), with the resulting protein extract quantified by bicinchoninic acid assay (Pierce). Protein disulfide bonds were reduced by 5 mM tributylphosphine (Sigma-Aldrich) with 30 min of incubation at 60°C. Samples were diluted fivefold with 50 mM NH4HCO3 (pH 7.8) to reduce the trifluoroethanol concentration to 10% prior to the addition of sequencing-grade modified trypsin (Promega) at a ratio of 1:50 (wt/wt, enzyme/protein). Samples were digested at 37°C overnight with gentle shaking. The digests were purified by solid-phase extraction C18 columns (Supelco).
Fractionation and LC-tandem MS (LC-MS/MS) analyses for generating peptide AMT tag databases.
To facilitate subsequent quantitative analyses, peptide AMT tag databases were initially created for the CEMx174 cell line. Global tryptic peptides were pooled from mock- and HIV-1-infected samples, and the pooled sample was subjected to cysteinyl (Cys) peptide enrichment, as described previously (51), to generate a Cys fraction and a noncysteinyl (non-Cys) fraction. Briefly, approximately 3 mg of the pooled peptide sample was used for cysteinyl peptide enrichment. The peptides were initially lyophilized and then redissolved in 120 μl of coupling buffer that consisted of 50 mM Tris-HCl, 1 mM EDTA, pH 7.5. After reducing with 5 mM dithiothreitol at 37°C for 1 h, the sample was diluted to 600 μl with coupling buffer divided into three equal aliquots and applied to three Handee Mini-spin columns (Pierce), each containing ∼100 μl of preequilibrated thiopropyl Sepharose 6B resins (Amersham Biosciences). The cysteinyl peptides were captured by the beads during 2 h of incubation at room temperature with gentle shaking. The unbound portion of the peptide sample was collected as the non-Cys fraction. Nonspecifically bound peptides were removed from the beads by stringent washing first with the coupling buffer, followed by 2 M NaCl, 80% acetonitrile-0.1% trifluoroacetic acid (TFA) solution, and 100% methanol, and then again with the coupling buffer. The captured cysteinyl peptides (Cys fraction) were released by incubating the beads with 120 μl of 20 mM freshly prepared dithiothreitol solution at room temperature for 30 min. The released peptides were further alkylated with 40 mM of iodoacetamide for 1 h at room temperature in the dark; alkylated peptide samples were then desalted using solid-phase extraction C18 columns. After lyophilization, both non-Cys and Cys fractions were resuspended in 600 μl of 10 mM ammonium formate in 25% acetonitrile, pH 3.0, and fractionated by strong cation exchange fractionation using a 200-mm by 2.1-mm Polysulfoethyl A column (PolyLC) as previously described (92). Thirty fractions were collected for each of the non-Cys and Cys samples.
Each strong cation exchange fraction was analyzed with an automated, custom-built, capillary reversed-phase LC system coupled online to a linear trapping quadrupole ion trap mass spectrometer using an electrospray ionization interface as previously described (92). The SEQUEST algorithm was used to search the MS/MS data against that for humans in the International Protein Index (IPI) database (version 2.29, April, 2004; available at http://www.ebi.ac.uk/IPI). For enriched cysteinyl peptide samples, a static mass modification for cysteinyl residues that corresponded to alkylation with iodoacetamide (57.0215 Da) was applied during the SEQUEST analysis. Criteria (established with probability-based evaluations using sequence-reversed database searching to provide confidence of >95% at the unique peptide level [74]) used to filter raw SEQUEST results were follows: (i) an Xcorr value of ≥1.6 for charge state plus one full tryptic peptide, (ii) an Xcorr value of ≥2.4 for charge state plus two full tryptic peptides and an Xcorr value of ≥4.3 for charge state plus two partial tryptic peptides, and (iii) an Xcorr value of ≥3.2 for charge state plus three full tryptic peptides and an Xcorr value of ≥4.7 for charge state plus three partial tryptic peptides. A delta correlation value (ΔCn) of >0.1 was used in all cases. Two AMT tag databases for the non-Cys peptides and Cys peptides were generated by including peptides that passed these criteria. The peptide retention times from each LC-MS/MS analysis were normalized to a range of 0 to 1 using a predictive peptide LC-normalized elution time (NET) model and linear regression as previously reported (69). Both the calculated accurate monoisotopic mass and NET of the identified peptides were included in the AMT tag database. To evaluate the proteome coverage of the AMT tag databases, we annotated the proteins based on gene ontology cellular components and biological function assignments by FatiGO (3).
16O/18O labeling and quantitative LC-FTICR analyses.
Trypsin-catalyzed 16O/18O labeling was carried out as previously described (75) by incubating the tryptic peptides in either 18O-enriched water (95%; ISOTEC) for HIV-infected samples or regular 16O water for mock-infected samples with immobilized trypsin (Applied Biosystems) as a catalyst for oxygen exchange at the tryptic C termini. The 16O/18O-labeled samples were mixed with equal amounts of mock- and HIV-1-infected samples, and the mixed sample was further fractionated into Cys and non-Cys peptides as described above. The Cys and non-Cys peptide samples were then analyzed using a custom-built capillary LC system coupled online, using an in-house-manufactured electrospray ionization interface, to an 11.5 T Fourier transform ion cyclotron resonance (FTICR) mass spectrometer. The analyses of quantitative LC-FTICR data sets were the same as previously described (75), which includes deisotoping, feature finding, 16O/18O peptide pair finding, and isotopic ratio calculation. Peptide identities were assigned by matching the normalized elution time and accurate mass measurements to the AMT tag database within a 5-ppm mass error and a 2% NET error. For the assessment of labeling efficiency by the 16O and 18O isotopes, identical aliquots of cell lysates were labeled with either isotope. Peptides were then quantified based on 16O-to-18O ratios, and deviations from the expected ratio of 1:1 were determined to exceed no more than 1.19-fold (Table 1). All quantified peptides were rolled up to nonredundant protein groups by using ProteinProphet (60), and the abundance ratio for each protein group was calculated by averaging the ratio of multiple unique peptides stemming from the same protein group.
TABLE 1.
Precision and accuracy of relative abundance measurements by 16O/18O labelinga
| Infected material type (replicate number) | Mean log (18O/16O) ratio | SD |
|---|---|---|
| HIV (1) | −0.06 | 0.04 |
| HIV (2) | −0.07 | 0.04 |
| Mock (1) | −0.05 | 0.03 |
| Mock (2) | −0.05 | 0.04 |
The precision (mean ratio) and accuracy (standard deviation) of calculating peptide ratios are shown. Cell lysates were divided into identical aliquots, one labeled with 16O and the other labeled with 18O. Duplicate analyses were performed with both HIV- and mock-infected cell lysates.
Identification of protein signatures.
Four independent infections were performed to provide biological replicates. Peptide relative abundances in the form of 16O-to-18O (mock-to-HIV) ratios were generated along with standard errors (SE) with the use of the Rosetta elucidator system for differential analysis (Rosetta Biosoftware). We employed an error model developed for the stable isotope-labeling approach to estimate technical variations in the process as described previously (87). In brief, in a manner similar to that with the error model for microarray data (78), additive and multiplicative measurement errors were inferred from the relative peptide abundance measurements. An error-weighted averaging method (84, 87) was adopted to combine multiple peptide measurements, including those from Cys and non-Cys peptide fractions as well as from the biological replicates, to estimate the protein expression level and its error. The weighting factor is inversely proportional to the variance of the peptide measurement. A P value of less than 0.05 was chosen to identify signature proteins exhibiting differential expression.
Determination of functional pathway enrichment pathway analysis.
To identify functional categories represented by the signature genes, we used the Ingenuity Pathways knowledge base (IPKB) and the associated analytical tools (11). We also interrogated our data based on membership in canonical maps defined in the GeneGo MetaCore database (62). IPI identifiers were matched with Entrez gene identifications based on definitions in the cross-reference file for humans (available at ftp://ftp.ebi.ac.uk/pub/databases/IPI/current/). The hypergeometric distributions of signature proteins from a particular functional pathway or map were assigned P values by IPKB and MetaCore. IPKB pathways and MetaCore maps with P values of >1.1 × 10−3 and > 3.8 × 10−3, respectively, were excluded from our analyses, as their total expectation within the respective databases would be greater than 5% by chance alone, as described previously (16, 86).
Sodium dodecyl sulfate-polyacrylamide gel electrophoresis and Western blotting.
HIV-1- and mock-infected lysates were collected at the times indicated, with total proteins solubilized with M-PER detergent (Pierce). Fifty micrograms of total proteins from each condition was resolved by sodium dodecyl sulfate-polyacrylamide gel electrophoresis on 4 to 20% Tris-glycine gel (NuSep). Proteins were then electrotransferred to polyvinylidene difluoride membranes for Western blotting. Antibodies against Ran, caspase-3 (BD Pharmingen), dilipoamide dehydrogenase (DLD), translocated promoter region (TPR) (Abnova), Importin α3 and β (MBL), ubiquitin-activating enzyme E1 (UBE1), ubiquitin, and actin (Abcam) and peroxidase-conjugated secondary antibodies (Jackson ImmunoResearch) were used for probing, the ECL Plus chemiluminescence assay was used for signal visualization, and the Typhoon PhosphorImager and ImageQuant software (Amersham Biosciences) were used for quantification. Digital images were adjusted for brightness and contrast uniformly throughout each blot using Photoshop Elements 2.0 software (Adobe).
Propidium iodide DNA profiles by flow cytometry.
HIV- and mock-infected cells were harvested at the times indicated. As positive control, cells were treated with staurosporine (ST) (Calbiochem) at a concentration of 20 or 200 ng/ml for 12 h. Ethanol-fixed cells were stained with a propidium iodide (PI) buffer (5 μg PI and 70 kU RNase [Calbiochem] per milliliter phosphate-buffered saline, pH 7.4) as described previously (5, 36). PI fluorescence was detected on the FL3 channel on the linear scale on an FC500 flow cytometer (Beckman Coulter). Six thousand events were gated using a dual density plot of FL3-height and FL3-width to exclude cell debris from analysis. Based on PI staining, cells with hypodiploid DNA contents were deemed to represent apoptotic cells (32, 66).
RESULTS
HIV-1 infection model.
To study changes in cellular protein abundance associated with HIV-1 infection, we sought to minimize the number of uninfected CEMx174 cells by using two TCID50 per cell. We monitored virus production and cell viability between 6 and 44 h after HIV or mock infection to span across the completion of one virus replication cycle, which presumably included the period of peak virus production. We detected intracellular p24 expression after 16 h postinfection (p.i.), with an increase in both the percentage of positively stained cells as well as signal intensity approaching 42 h p.i. (Fig. 1B) (see viromics.washington.edu/publications/ericchan/HIV_Figures_1.pdf). This result is in line with those of previous studies of HIV life cycle kinetics (77). In parallel, a trypan blue exclusion assay was used to determine cell viability (Fig. 1B). We observed that peak virus production occurred at 36 h p.i., with 94.1% of the cells actively producing virus before cell viability decreased dramatically (Fig. 1A and B). Therefore, our subsequent analysis using two TCID50 at 36 h p.i. should reflect protein abundance changes at the peak of virus production, with close to all cultured cells actively producing virus.
FIG. 1.
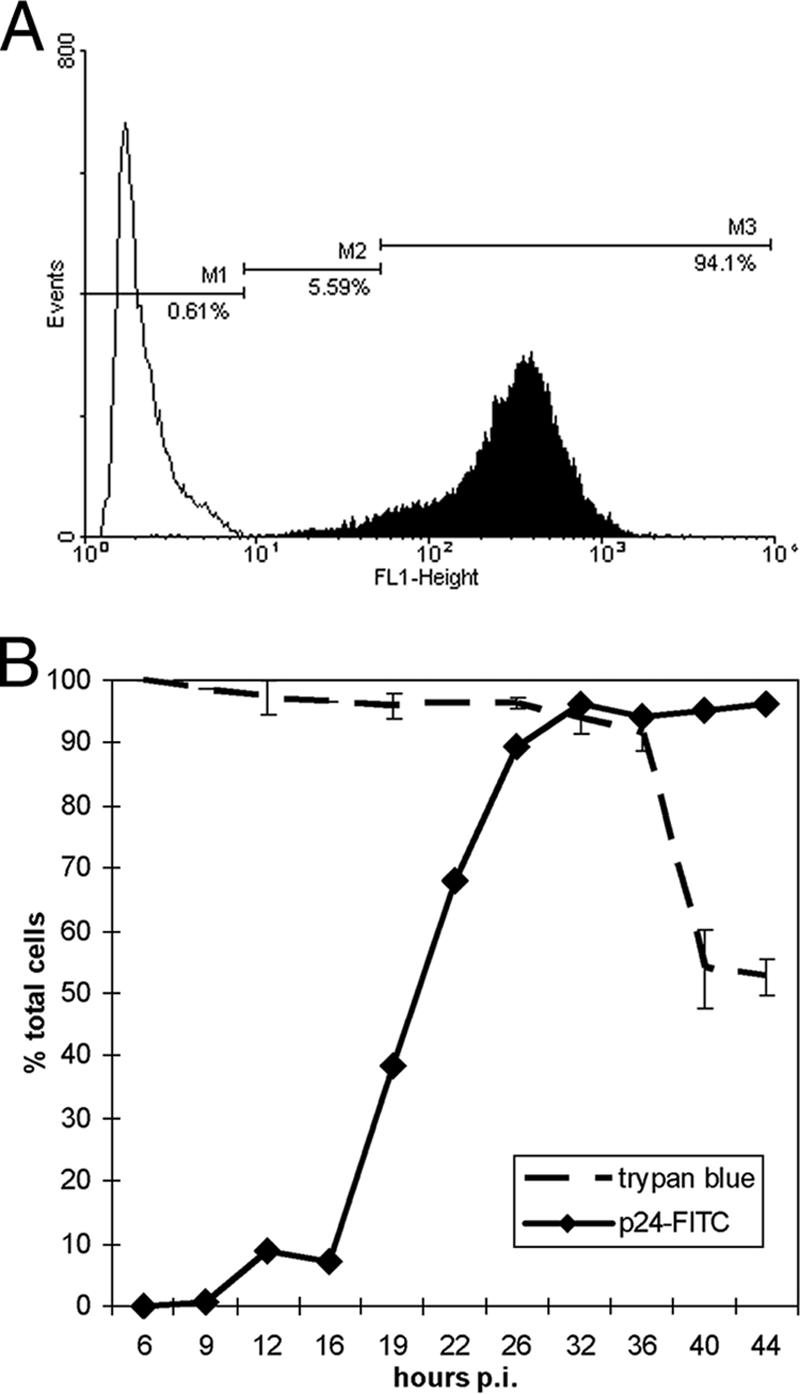
Intracellular gag p24 level and cell viability following HIV-1 infection of CEMx174. Cells were exposed to HIV-1LAI at a multiplicity of infection of 2TCID50/cell or to virus-free culture medium and harvested for intracellular p24-FITC and trypan blue staining. (A) Histograms of p24-FITC signal collected on the FL1 logarithmic scale on the flow cytometer. Open and filled areas represent mock and HIV infection, respectively, at 36 h p.i. The M1 marker denotes the background fluorescence level in the mock-infected sample, while the M2 and M3 markers denote low- and high-intensity p24-FITC signals, respectively. The M3 population represents active gag p24 production. (B) Summary of p24-FITC (left axis) and trypan blue (right axis) staining results. Individual p24-FITC staining results are shown at viromics.washington.edu/publications/ericchan/HIV _Figures_1.pdf. Most p24-FITC signals were within the M2 population up to 16 h p.i. in the HIV-infected samples, while the M3 population increased from 19 h p.i. onward. Error bars represent SE calculated from three independent samplings for trypan blue exclusion as visualized by light microscopy. A noticeable drop in cell viability occurred after 36 h p.i.
Abundance changes of proteins upon HIV-1 infection.
We chose to monitor protein abundance changes at 36 h p.i. because it was close to the time of peak virus production before cell viability plummeted (Fig. 1B). We further noted that protein recoveries from 108 viable cells produced 3.1 and 2.8 mg of proteins in the HIV- and mock-infected samples, respectively, as determined by the bicinchoninic acid assay. Given that cell viability and protein yields were comparable in virally infected and mock-infected cells, equal amounts of HIV- and mock-infected protein extracts were used in all subsequent quantitative LC-FTICR analyses by 18O labeling. In all, 11,028 unique peptides were quantified, representing 3,255 unique host proteins (see viromics.washington.edu/publications/ericchan/HIV_Tables _1.zip). A total of 687 host proteins (21%) exhibited significant changes in abundance upon infection; 344 and 343 of these signature proteins were up- and down-regulated upon infection, respectively (Tables 2 and 3 and Fig. 2).
TABLE 2.
Top 10% up-regulated signature proteinsa
| IPI number | Protein description | Log (ratio) |
|---|---|---|
| IPI00259478 | Hypothetical protein | 0.76 |
| IPI00027626 | T-complex protein 1, zeta subunit | 0.74 |
| IPI00019353 | Putative lipid kinase | 0.72 |
| IPI00306382 | Secretory carrier-associated membrane protein 3 | 0.72 |
| IPI00102059 | Kinase-like protein | 0.70 |
| IPI00328188 | Fatty acid synthase | 0.70 |
| IPI00026665 | Glutaminyl-tRNA synthetase | 0.69 |
| IPI00017855 | Aconitate hydratase, mitochondrial precursor | 0.69 |
| IPI00014439 | Dihydropteridine reductase | 0.69 |
| IPI00328347 | Pyruvate kinase, M2 isozyme | 0.68 |
| IPI00238209 | Hypothetical protein KIAA1068 | 0.67 |
| IPI00002493 | Pyrroline-5-carboxylate reductase | 0.66 |
| IPI00000792 | Quinone oxidoreductase | 0.66 |
| IPI00026941 | Serine protease 23 precursor | 0.65 |
| IPI00299254 | Translation initiation factor IF-2 | 0.65 |
| IPI00011118 | Ribonucleoside-diphosphate reductase M2 chain | 0.64 |
| IPI00166749 | Mitochondrial processing peptidase alpha subunit, mitochondrial precursor | 0.63 |
| IPI00257508 | Dihydropyrimidinase-related protein 2 | 0.63 |
| IPI00293102 | Protein phosphatase 2A, regulatory subunit B′ | 0.63 |
| IPI00013212 | Tyrosine protein kinase CSK | 0.62 |
| IPI00060440 | Zinc finger protein GLI4 | 0.62 |
| IPI00166711 | Rho guanine nucleotide exchange factor 1 | 0.61 |
| IPI00295625 | Succinyl-CoA ligase (GDP-forming) alpha-chain, mitochondrial precursor | 0.60 |
| IPI00302458 | Exportin 7 | 0.58 |
| IPI00333541 | Filamin A | 0.58 |
| IPI00024251 | Fibroblast growth factor 13 | 0.58 |
| IPI00032328 | Kininogen precursor | 0.58 |
| IPI00334907 | Phosphatidylinositol transfer protein, beta | 0.58 |
| IPI00334627 | Annexin A2 | 0.57 |
| IPI00174390 | Hypothetical protein FLJ23843 | 0.57 |
| IPI00294744 | Poly(A)-specific RNase (deadenylation nuclease) | 0.57 |
| IPI00001757 | RNA-binding protein 8A | 0.57 |
| IPI00005024 | Myb-binding protein 1A | 0.57 |
| IPI00294158 | Biliverdin reductase a precursor | 0.57 |
Shown are the 34 most up-regulated signature proteins (P < 0.05).
TABLE 3.
Top 10% down-regulated signature proteinsa
| IPI number | Protein description | Log (ratio) |
|---|---|---|
| IPI00103055 | Similar to RIKEN cDNA 1300003p13 gene | −0.86 |
| IPI00032590 | Katanin p60 subunit A like 1 | −0.59 |
| IPI00174025 | KIAA1010 protein | −0.55 |
| IPI00018262 | Acidic leucine-rich nuclear phosphoprotein 32 family member C | −0.55 |
| IPI00401260 | Hypothetical protein xp_376880 | −0.54 |
| IPI00030408 | Hypothetical protein | −0.51 |
| IPI00004524 | Grancalcin | −0.49 |
| IPI00218236 | Serine/threonine protein phosphatase pp1-beta catalytic subunit | −0.46 |
| IPI00000030 | Serine/threonine protein phosphatase 2A, 56-kDa regulatory subunit | −0.46 |
| IPI00175080 | Similar to hypothetical protein 4833421E05RIK | −0.46 |
| IPI00015955 | Exosome component RRP46 | −0.45 |
| IPI00012578 | Importin alpha-4 subunit | −0.44 |
| IPI00303962 | Hypothetical protein | −0.44 |
| IPI00218091 | Testis spermatocyte apoptosis-related gene 3 protein | −0.43 |
| IPI00004436 | U6 snRNA-associated Sm-like protein LSM1 | −0.41 |
| IPI00335140 | FLJ00355 protein | −0.41 |
| IPI00019385 | Translocon-associated protein, delta subunit precursor | −0.41 |
| IPI00007249 | Hypothetical protein KIAA0879 | −0.40 |
| IPI00072386 | Hypothetical protein FLJ10585 | −0.39 |
| IPI00000447 | Hypothetical protein | −0.38 |
| IPI00013183 | UBE1-like | −0.37 |
| IPI00017510 | Cytochrome c oxidase polypeptide II | −0.37 |
| IPI00299033 | Importin alpha-3 subunit | −0.37 |
| IPI00025717 | Metaxin 2 | −0.37 |
| IPI00019381 | Hypothetical protein FLJ10856 | −0.36 |
| IPI00020512 | Uncharacterized hematopoietic stem/progenitor cells protein MDS031 | −0.36 |
| IPI00164949 | Negative elongation factor C/D | −0.35 |
| IPI00384504 | Similar to Bcl-2-associated transcription factor | −0.35 |
| IPI00010201 | 26S proteasome non-ATPase regulatory subunit 8 | −0.35 |
| IPI00005517 | Ephrin-A5 precursor | −0.35 |
| IPI00025277 | Programmed cell death protein 6 | −0.35 |
| IPI00010240 | AD023 | −0.35 |
| IPI00009030 | Lysosome-associated membrane glycoprotein 2 precursor | −0.35 |
| IPI00016568 | Adenylate kinase isoenzyme 4, mitochondrial | −0.35 |
Shown are the 34 most down-regulated signature proteins (P < 0.05).
FIG. 2.

Distribution of the 687 host proteins up- or down-regulated at 36 h postinfection. Relative abundance is plotted as log10(HIV/mock) ratios as a function of log10(signal intensities) in arbitrary units. Green denotes the 344 down-regulated proteins, while red denotes the 343 up-regulated proteins. Blue denotes proteins without significant differential expression.
Characterization of protein abundance changes in relation to viral proteins.
To focus on protein activities that may be directly involved in virus production, we first looked for proteins with known interactions with viral proteins. There appeared to be an enrichment for cellular proteins with known interactions with HIV-1 integrase and Vpu (Table 4), and at a P value of less than 0.029, fewer than three false positives are expected of the 83 with known interactions with HIV-1 viral proteins, based on the NIAID HIV protein interaction database (available at http://www.ncbi.nlm.nih.gov/RefSeq/HIVInteractions/). Interestingly, about two-thirds of the protein interactors identified were down-regulated upon infection. We observed the uniform down-regulation of four members of the importin family (down 1.2- to 2.76-fold), presumably involved in the nuclear import of the integrase (30). On the other hand, integrase activities appeared to be supported by the up-regulation of the integrase interactor protein FLJ13963, which has been shown to ameliorate the cytopathic effects of the integration (67), as well as by the up-regulation of DNA-PK, possibly as a host response to the integration process and the ensuing DNA damage (22). The down-regulation of proteasome subunits would also reduce the proteolysis of integrase by the proteasome complex (55). Our results also showed mixed effects on Vpu activities. While Vpu has been shown to induce apoptosis via the inhibition of NF-κB activity, followed by the activation of caspase-3 and the reduction in TRAF-1 level (2), our observed increase in NF-κB and TRAF-1 and decrease in caspase-3 abundance levels seemed to depict a cellular environment not conducive to apoptosis, at least one that is not mediated by Vpu. This result also coincided with the observed down-regulation of CD95/Fas, which would otherwise initiate Vpu-mediated cell death (14). Taken together, the observed changes in protein abundance of known interactors of viral proteins have mixed effects on the functions of viral proteins and probably likewise on viral replication. Coincidentally, some of the effects contrary to those reported of the viral proteins may turn out to prolong host cell survival and therefore be beneficial to virus production, as exemplified by the repression of apoptosis and the induction of DNA repair pathways.
TABLE 4.
Signature proteins with known functional relations with HIV-1 viral proteinsa
| Description | Fold change | P value |
|---|---|---|
| Integrase partners | ||
| Importin alpha-4 subunit | −2.76 | 1.56 × 10−3 |
| 26S proteasome non-ATPase regulatory subunit 8 | −2.24 | 1.32 × 10−2 |
| 26S proteasome non-ATPase regulatory subunit 5 | −1.83 | 8.04 × 10−3 |
| Proteasome subunit beta type 2 | −1.48 | 4.00 × 10−5 |
| Importin alpha-7 subunit | −1.41 | 2.79 × 10−6 |
| Proteasome subunit alpha type 5 | −1.33 | 6.49 × 10−3 |
| 26S protease regulatory subunit 6a | −1.31 | 3.13 × 10−3 |
| 26S protease regulatory subunit S10B | −1.25 | 9.20 × 10−6 |
| Importin beta-1 subunit | −1.81 | 3.81 × 10−3 |
| Proteasome activator complex subunit 3 | −1.22 | 4.54 × 10−2 |
| Karyopherin (importin) beta 3 | −1.20 | 3.30 × 10−4 |
| Proteasome activator complex subunit 1 | −1.10 | 1.06 × 10−2 |
| Elongation factor 1-alpha 1 | 1.36 | 9.80 × 10−4 |
| 26S protease regulatory subunit 8 | 1.38 | 3.63 × 10−3 |
| Proteasome subunit alpha type 1 | 1.64 | 1.00 × 10−4 |
| Proteasome 26S ATPase subunit 2 | 2.11 | 4.97 × 10−2 |
| T-complex protein 1, delta subunit | 2.96 | 4.28 × 10−8 |
| DNA-dependent protein kinase catalytic subunit | 3.17 | 1.27 × 10−2 |
| Hypothetical protein FLJ13963 | 13.30 | 2.00 × 10−5 |
| Vpu partners | ||
| Tumor necrosis factor receptor superfamily member 6 precursor | −2.03 | 1.00 × 10−5 |
| Small glutamine-rich tetratricopeptide repeat-containing protein A | −1.23 | 2.20 × 10−4 |
| Apopain precursor | −1.64 | 1.67 × 10−3 |
| TNF receptor associated factor 1 | 1.31 | 1.10 × 10−2 |
| HLA class I histocompatibility antigen, CW-1 alpha chain precursor | −1.35 | 4.73 × 10−2 |
| NFKB1 protein | 1.30 | 4.98 × 10−2 |
Out of the total of 687 signature proteins, significant enrichment for interactions with HIV-1 integrase (P = 1.4×10−4) and Vpu (P = 0.029) was detected. Shown are the 25 signature protein interactors based on annotations in the NIAID HIV Interaction Project.
Functional characterization of cellular activities exhibiting protein abundance changes by pathway analysis.
Given that virus production reached an apex at 36 h p.i. without significant changes in cell viability, we wanted to examine other aspects of cell signaling and metabolism to determine whether cell functions are regulated to enable robust virus production. Previous microarray analyses have revealed the induction of cell division and transcription pathways (31, 55, 89). We wanted to understand the effects of changes in cellular protein abundance in the context of the cellular functions they partake in. If changes in protein abundance are reflections of global stress response to the introduction of viral proteins (19, 29), we would expect to see indiscriminate changes in cellular protein abundance, at least apart from pathways such as the unfolded protein response and translational control. To this end, we have resorted to the use of two commercially available databases of cellular metabolic and signaling pathways. Both GeneGo MetaCore and Ingenuity Pathways Analysis assign gene products to functional pathways based on manually curated literature findings. Pathway analysis by both programs reports the distribution of signature proteins in predefined pathways; if a pathway is populated with more signature proteins than expected, the pathway is deemed subject to regulation upon HIV-1 infection. For our analysis, we considered IPKB pathways and MetaCore maps with less than 5% expectation of having differential regulation occurring by chance alone, determined as described previously (16, 86).
Pathway analyses by MetaCore and Ingenuity revealed the significant enrichment of protein abundance in four maps and seven pathways, respectively (Table 5). The differences in the functional categories identified likely reflected the nature of manual curation in the two commercial databases. While both programs help point out functional categories represented by our signature proteins, the exclusion of a pathway does not preclude its association with virus production; rather, its association simply may not be evident from the existing body of literature as assessed by the two independent sources. Nevertheless, we focused on functional categories reported by the software tools for an unbiased assessment of cellular functions potentially associated with virus production. The most significantly regulated category based on MetaCore analysis was the RAN regulation pathway (Table 5 and Fig. 3). Central to the nucleocytoplasmic pathway, the GTP-binding protein Ran travels into and out of the nucleus when it is bound to GTP or GDP, respectively. Its increase in abundance was accompanied by the increase in abundance of the intranuclear filament protein TPR, which was detected in only the HIV-infected samples (Table 6). Furthermore, the concerted decrease in abundance of subunits of importins alpha and beta, which recognize nuclear localization signals on cargo proteins, and NTF2, which normally assembles at the nuclear pore complex following the docking of imported proteins (68), suggested that protein import to the nucleus may be impeded. In all, our data suggested that there was a bias favoring the export of proteins from the nucleus at the expense of import into the nucleus (Fig. 3).
TABLE 5.
Cellular pathways with greater-than-chance representation by signature proteinsa
| Analyzer | Pathway | Expectation |
|---|---|---|
| GeneGo MetaCore | RAN regulation pathway | 3.55 × 10−3 |
| Role of APC in cell cycle regulation | 4.96 × 10−3 | |
| Ligand-dependent transcription of retinoid-target genes | 7.68 × 10−3 | |
| Oxidative phosphorylation | 1.02 × 10−2 | |
| Role of 14-3-3 proteins in cell cycle regulation | 1.50 × 10−2 | |
| Role of Akt in hypoxia-induced HIF1 activation | 1.79 × 10−2 | |
| TCA | 2.64 × 10−2 | |
| Ingenuity | Protein ubiquitination pathway | 6.20 × 10−3 |
| Citrate cycle | 1.08 × 10−2 | |
| Valine, leucine, and isoleucine degradation | 2.52 × 10−2 | |
| ERK/MAPK signaling | 3.04 × 10−2 |
Shown are pathways with less than 5% expected chance of occurrence. Pathways were revealed by GeneGo MetaCore and Ingenuity Pathways Analysis. APC, anaphase-promoting complex.
FIG. 3.

RAN regulation pathway constituents exhibiting abundance changes, with a figure adapted from the GeneGo MetaCore database. In this pathway, importin subunits α4, α6, β1, and β3 as well as nuclear transport factor 2 and sentrin, were down-regulated at 36 h p.i., as indicated by the green arrows. At the same time, Ran GTPase and the intranuclear filament TPR were up-regulated, as indicated by the red arrows.
TABLE 6.
Distribution of signature proteins in the three most significantly regulated pathwaysa
| Pathway | Signature proteins | Fold change |
|---|---|---|
| Ubiquitination | Proteasome (prosome, macropain) 26S subunit, non-ATPase, 8 | −2.24 |
| Proteasome (prosome, macropain) 26S subunit, non-ATPase, 5 | −1.83 | |
| S-phase kinase-associated protein 1A (p19A) | −1.73 | |
| Proteasome (prosome, macropain) 26S subunit, non-ATPase, 14 | −1.62 | |
| Cell division cycle 34 | −1.59 | |
| Ubiquitin-conjugating enzyme E2G 2 (UBC7 homolog, yeast) | −1.55 | |
| Ubiquitin carboxyl-terminal hydrolase L5 | −1.53 | |
| Ubiquitin-conjugating enzyme E2B (RAD6 homolog) | −1.52 | |
| Proteasome (prosome, macropain) subunit, beta type, 2 | −1.48 | |
| Proteasome (prosome, macropain) subunit, alpha type, 5 | −1.33 | |
| Proteasome (prosome, macropain) 26S subunit, ATPase, 3 | −1.31 | |
| Ubiquitin-conjugating enzyme E2M (UBC12 homolog, yeast) | −1.30 | |
| Proteasome (prosome, macropain) 26S subunit, ATPase, 6 | −1.25 | |
| Thimet oligopeptidase 1 | 1.32 | |
| SGT1, suppressor of G2 allele of SKP1 (S. cerevisiae) | 1.34 | |
| Proteasome (prosome, macropain) 26S subunit, ATPase, 5 | 1.38 | |
| Ubiquitin specific peptidase 10 | 1.51 | |
| Proteasome (prosome, macropain) subunit, alpha type, 1 | 1.64 | |
| Cas-Br-M (murine) ecotropic retroviral transforming sequence | 1.97 | |
| Proteasome (prosome, macropain) 26S subunit, ATPase, 2 | 2.11 | |
| Ubiquitin specific peptidase 5 (isopeptidase T) | 2.17 | |
| TCA | ATP citrate lyase | 2.15 |
| Aconitase 2, mitochondrial | 1.45 | |
| Dihydrolipoamide dehydrogenase | 6.37 | |
| Isocitrate dehydrogenase 2 (NADP+), mitochondrial | 5.78 | |
| Isocitrate dehydrogenase 3 (NAD+) alpha | −1.50 | |
| Malate dehydrogenase 1, NAD (soluble) | 2.61 | |
| Cuccinate-CoA ligase, GDP-forming, alpha subunit | 1.67 | |
| Ran regulation | Karyopherin subunit A4 | −2.75 |
| Karyopherin subunit A6 | −1.41 | |
| Karyopherin subunit B1 | −1.81 | |
| Karyopherin subunit B3 | −1.20 | |
| Nuclear transport factor 2 | −1.32 | |
| SUMO1 | −1.26 | |
| RAN | 3.51 | |
| TPR | b |
The three most significantly regulated pathways are the Ran-regulated nucleocytoplasmic pathway, the ubiquitination pathway, and the TCA cycle. Numerical values represent fold changes of relative protein abundance.
HIV only.
Ingenuity Pathways Analysis reported ubiquitination as the most significantly regulated pathway. We observed the down-regulation of the E1 ubiquitin-activating enzyme as well as a few of the 30 known E2 ubiquitin-conjugating enzymes upon infection. As cdc34 mediates the degradation of cell cycle G1 checkpoint proteins and initiates entry into S phase (33, 70), its down-regulation renders further indication of a block in G1/S transition in our infection model. Additional evidence for cell cycle dysregulation came from the down-regulation of E2b, a human homolog of the yeast (Saccharomyces cerevisiae) RAD6 protein responsible for postreplicative DNA repair in late S/G2 phase (52), and of Ubc12, responsible for the neddylation and stabilization of human cullins, including cdc53/cullin (35, 44, 65). Apart from the ubiquitination of cell cycle regulators, our data also pointed to a potential increase in activities, based on the increased abundance of ubiquitin-specific peptidases. We also observed the up- and down-regulation of various subunits of the 20S proteasome core and the 19S regulator (Table 6). In fact, some of these changes appeared to be counterproductive, such as the up-regulation of 20S core subunit α1 and the down-regulation of subunit α5. While this result could point to an overall dysregulation and repression of proteolytic activities of the proteasome complex, it may also stem from the nature of the proteasome as a dynamic complex, with its 20S and 19S subunits performing nondegradative functions. For instance, we observed the simultaneous up- and down-regulation of the 19S ATPase subunits 2 and 3, respectively, the former being a positive (58, 82) and the latter a negative modulator (59) of viral Tat transactivation. Taken together, our observations in the ubiquitination pathway suggested that the regulated expression of components of the ubiquitination pathway likely reflects G1/S arrest in the cell cycle, accompanied by presumed increases in Tat transactivation and Gag monoubiquitination that coincide with peak virus production at 36 h p.i. (Fig. 1A).
Components of the citrate (trichloracetic acid [TCA]) cycle exhibited changes in abundance, as reported by both MetaCore and Ingenuity analyses (Table 5). The up-regulation of the E3 component of the pyruvate dehydrogenase complex DLD at 36 h p.i. suggested an overall induction in the generation of acetyl-CoA from pyruvate (43). The supply of acetyl-CoA, key to biosynthetic pathways, including those for lipogenesis and cholesterogenesis, may be further augmented by the elevated abundance of the ATP citrate lyase. The generation of acetyl-CoA for fatty acid synthesis coincided with the dysregulation in oxidative phosphorylation, evidenced by the decreased abundance of cytochrome c oxidase and NADH dehydrogenase subunits (see viromics.washington.edu/publications /ericchan/HIV_Tables_1.zip), presumably related to the inactivation of the energy regulatory switch AMP kinase in the presence of excess ATP (13). In all, our observations on components of the TCA cycle and oxidative phosphorylation suggested that an induction of ATP-consuming biosynthetic pathways occurred at the time of peak virus production.
Functional characterization of cellular activities exhibiting protein abundance changes by network analysis.
We have also sought to characterize our signature proteins based on protein-protein interactions. While pathway analyses revealed functional pathways with greater-than-chance occurrences of differential protein abundance, none of the pathways were fully populated with differentially expressed proteins, nor should any of them be, as changes in abundance do not capture other changes in protein expression, such as modification and relocalization. Nevertheless, de novo network construction by the Ingenuity software produced a network comprised entirely of 14 differentially expressed proteins de novo, connected based on physical or inferred interactions (Fig. 4). This represents a cluster of proteins and their underlying biological functions, subject to quantitative changes at the protein level. From the plasma membrane, signaling through the CD30/TNFRSF8 (57) and CD95/Fas appear to be impaired, as evidenced by the down-regulation of these positive regulators of apoptosis. Further indications of antiapoptosis can be found in the up-regulation of TRAF1, which has been shown to be down-regulated in Vpu-mediated apoptosis (2). The increase in TRAF1 and tumor necrosis factor alpha (TNF-α)-inducible primary response gene 3 (TNFAIP3) expressions corroborates with their induction by NF-κB (9, 40), which itself was up-regulated based on our observations; the induction also seemed to be specific to NF-kB activities, as the CD86 ligand, which otherwise has been shown to elevate TNF response (24), was in fact down-regulated. NF-κB activation is also supported by the elevated abundance of PRDX4 (37) and LCP2 (95). Also in the nucleus, the up-regulation of four SWI/SNF proteins suggests that their chromatin-remodeling properties may be a reflection of active transcription; moreover, SMARCB1/hSNF5 has been shown to be incorporated into virions, presumably to aid in DNA-joining activities of the integrase (46, 96). Taken together, our data (by network analysis and in agreement with our pathway analyses) indicated that peak virus production coincided with a repression in apoptosis induction. Transcriptional activation also seemed to be active, as evidenced by the elevated abundance of NF-κB and by signs of active remodeling of chromatins.
FIG. 4.
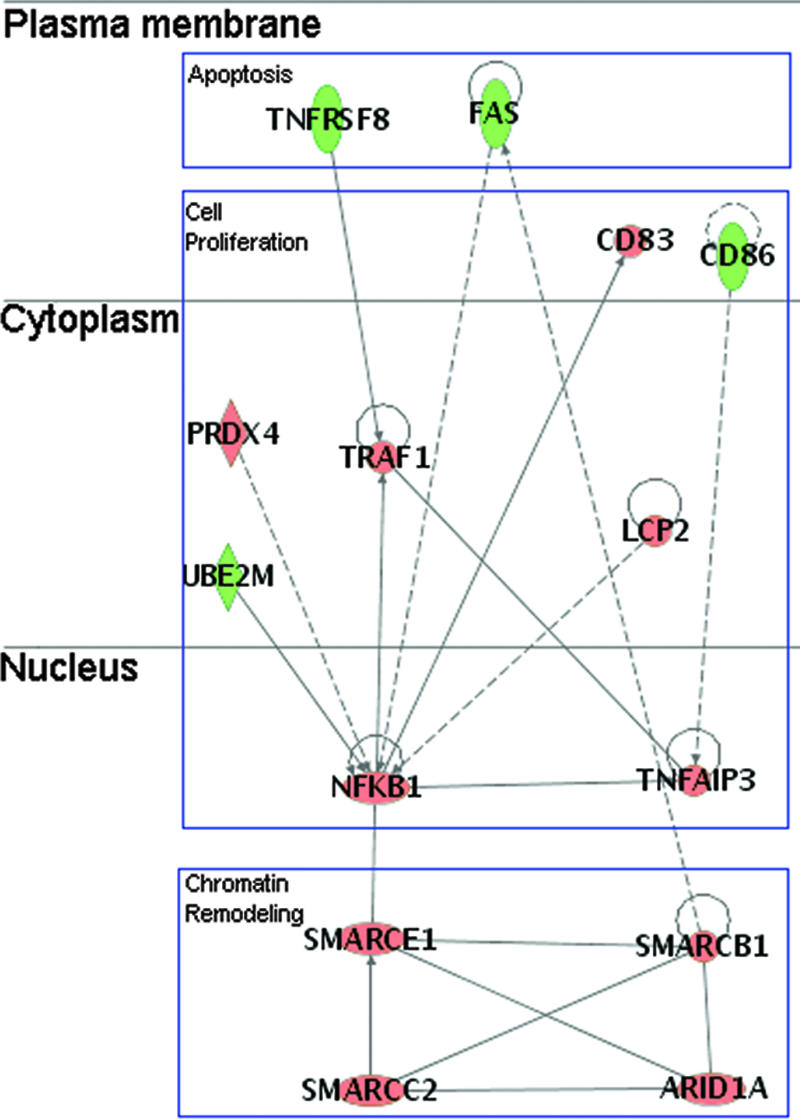
Network of proteins with known interactions recorded in IPKB, with P values of <10−36; proteins are laid out based on their subcellular localization. Three major categories of proteins were represented by the network, namely, apoptosis, cell proliferation, and chromatin remodeling. A line indicates interactions, with the arrowhead indicating directionality. The absence of arrowheads refers to a binding interaction. A dotted line indicates an inferred or indirect interaction. Red and green refer to up-regulation and down-regulation, respectively, as reported by MS analysis.
Orthogonal validation and analysis of temporal protein expression by Western blotting.
As our mass spectrometry approach uses tryptic peptide quantification as a proxy for protein abundance measurements, we have also sought to verify our proteomic results at the level of intact proteins by Western blotting. In particular, we have focused on constituents of the pathways discussed above, namely the nuclear transport, ubiquitination, and TCA pathways. In addition, given some of the observations that are contrary to the expected effects of infection, such as the apparent resistance to the nuclear import of the integrase preintegration complex, we wanted to examine the protein expression patterns prior to 36 h p.i., the recorded peak of virus production. In general, our Western blotting results were in agreement with the directionality of relative protein abundance changes reported by mass spectrometry (Fig. 5 and Table 7). In the nuclear transport pathway, both RAN and TPR exhibited increases in abundance from 12 to 36 h p.i.; the weak intensities of TPR suggest the protein could be of low abundance, in turn accounting for the absence of mass spectrometry signal in the mock-infected samples. On the other hand, while both importin α3 (KPNA3) and importin β1 (KNPB1) exhibited decreases in abundance at 36 h p.i., as reported by mass spectrometry and Western blotting, the decreases were not evident until between 18 and 24 h p.i. This suggests that the nuclear import machinery was available for the integration of the provirus. In the TCA pathway, DLD showed a gradual increase in abundance as we approached 36 h p.i., suggesting that the changes in energy demands could be accumulative over the course of infection. In the ubiquitination pathway, the UBE1 showed a slight decrease in abundance over the course of our analysis, in agreement with our MS results. Interestingly, free ubiquitin (Ubi-1), though not measured by MS, exhibited elevated abundance starting at around 18 h p.i. This is in agreement with the notion that the ubiquitination pathway was active in spite of any apparent increase in proteasomal activities inferred from the down-regulation of select proteasome subunits. Caspase-3 exhibited a decrease in abundance at only 12 and 36 h p.i., in agreement with our observations that apoptosis was not heightened over the course of our analysis.
FIG. 5.
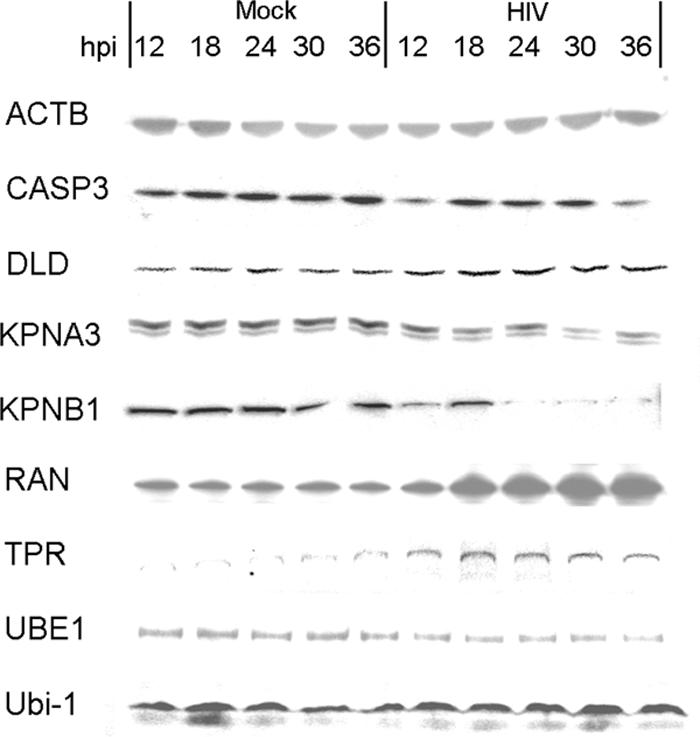
Protein abundance assayed by Western blotting. Chemiluminescent signals were quantified by densitometry (Table 7). Five micrograms of HIV- and mock-infected cell protein extracts was loaded per lane, with each lane representing one of five time points between 12 and 36 h p.i. All proteins, except ubiquitin (Ubi-1), had also been quantified by MS. Beta-actin (ACTB) appeared as a band at ∼42 kDa, caspase-3 (CASP3) appeared at ∼32 kDa, DLD appeared at ∼75 kDa, importin α3 (KPNA3) appeared as a doublet at ∼59 kDa, importin β (KPNB1) appeared at ∼97 kDa, Ran appeared at ∼30 kDa, translocated promoter region (TPR) appeared at >150 kDa, ubiquitin-activating enzyme E1 (UBE1) appeared at ∼110 kDa, and ubiquitin (Ubi-1) appeared at <10 kDa.
TABLE 7.
Comparison of protein abundances sampled by Western blotting with the MS resultsa
| Protein | Change (fold) for:
|
|||||
|---|---|---|---|---|---|---|
| Western blotting (densitometry) at time (h) p.i.
|
18O labeling at 36 h p.i.b | |||||
| 12 | 18 | 24 | 30 | 36 | ||
| ACTB | 1.18 | 1.12 | −1.18 | 1.31 | 1.12 | −1.20‡ |
| CASP3 | −1.70 | −1.11 | −1.40 | −1.70 | −2.10 | −1.64 |
| DLD | 1.41 | 1.34 | 2.10 | 1.95 | 2.27 | 6.31 |
| KPNA3 | −1.20 | −1.31 | −1.37 | −2.47 | −2.14 | −2.33 |
| KPNB1 | −1.93 | −1.53 | 1.21 | −2.70 | −2.75 | −1.81 |
| RAN | −1.21 | 1.47 | 1.82 | 2.12 | 2.89 | 3.51 |
| TPR | 2.78 | 3.10 | 2.51 | 1.94 | 1.92 | |
| UBE1 | −1.32 | −1.41 | −1.01 | 1.27 | −1.52 | −2.35 |
| Ubi-1 | 1.21 | −1.07 | 1.49 | 1.89 | 2.10 | N/D |
All values are derived from the protein abundance from the HIV-infected sample compared to that from the time-matched, mock-infected sample. Positive and negative fold changes indicate increases and decreases, respectively, in abundance upon infection. N/D, not detected; ‡, without significant differential expression (P ≥ 0.05).
For TPR, a signal was detected for the HIV-infected samples but not for the mock-infected samples.
Analysis of apoptosis and cell cycle distribution by DNA content.
The differential expression of requisite components of cell cycle regulation, including cyclin-dependent kinase (cdc34), ubiquitin E2 enzymes, and proteasome complex subunits, suggested that cell cycle progression may be affected amid robust virus production. On the other hand, the decrease in abundance of apoptosis effectors, such as caspase-3 and CD95/Fas, indicated that changes in cell death do not accompany such changes. To determine whether our differential expression analysis corroborates with functional consequences in regards to cell cycle progression and apoptosis, we performed PI staining to measure DNA content as an indicator of cell cycle stages (5, 32, 36, 66). As DNA fragmentation precedes cell death by the apoptotic pathway, hypodiploid DNA content would also be indicative of cell damage or death (61). We used staurosporine, a broad inhibitor of protein kinases and inducer of apoptosis in lymphocytes (10), to provide a qualitative benchmark of cell death and damage. Notably, the hypodiploid population characteristic of apoptotic or damaged cells was noticeable at 42 h p.i. and upon ST treatment, but not at earlier time points or upon mock infection (Fig. 6B) (viromics.washington.edu/publications/ericchan/HIV_Figures_1.pdf). Moreover, our flow cytometry results from three separate measurements revealed a moderate increase in the G1/G0 population between 24 h and 36 h p.i., but before 42 h p.i. (Fig. 6B; see the URL mentioned above). The drop in the G1/G0 population at 42 h p.i. (Fig. 6B) was accompanied by an increase in the sub-G1 population (Fig. 6A), in agreement with the increase in cells positively stained by trypan blue (Fig. 1B). In line with our MS results, the flow cytometric data suggested there might be a block to G1 exit while virus production accelerated between 24 and 36 h p.i., though the block was not accompanied by any surge in cell death.
FIG. 6.
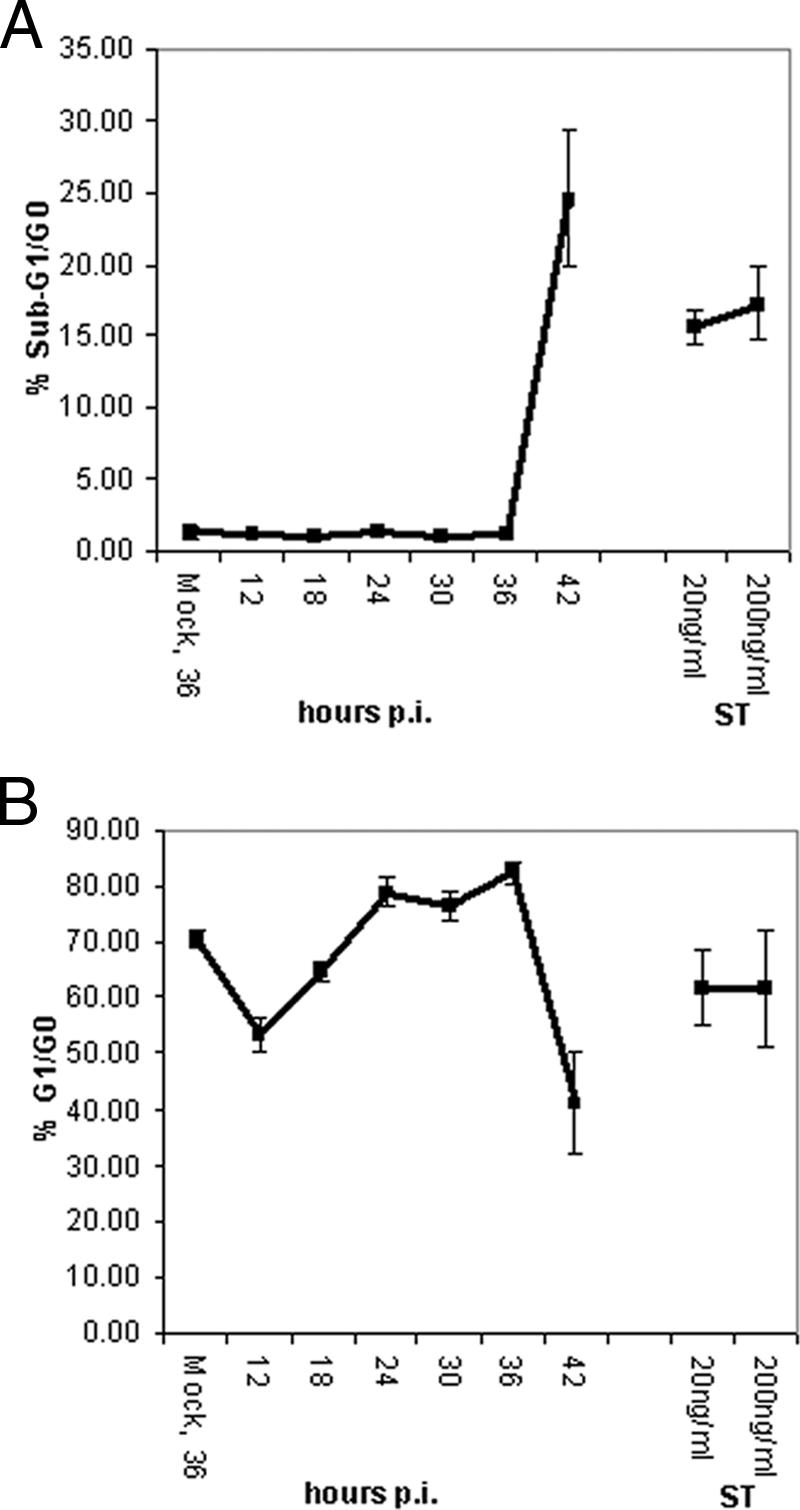
Cell death and cell cycle distribution assessed by PI staining. Diploid G1/G0 and hypodiploid cells were identified based on DNA contents. Hypodiploid cells were presumed to arise from the DNA fragmentation characteristic of apoptotic cells (32, 66). HIV-infected cells were collected at the times indicated; as positive controls for apoptosis, cells were treated with ST for 12 h at a concentration of 20 or 200 ng/ml. The PI signal was collected on the FL3 channel; sub-G1/apoptotic and G1/G0 cell populations were estimated by the markers M1 and M2, respectively (see viromics.washington.edu/publications/ericchan /HIV_Figures_1.pdf). Results shown from the 36-h mock infection are representative of the time-matched mock infection samples analyzed. Average percentages of (A) apoptotic, sub-G1/G0 and (B) G1/G0 cells were compared among the various conditions. Apoptotic cells were readily detected only at 42 h p.i. and in the positive controls. In addition, there was a moderate drift towards G1/G0 accumulation, relative to mock infection beginning at 24 h p.i. and prior to 42 h p.i. Error bars represent SE calculated from three separate measurements.
DISCUSSION
HIV-1 is known to co-opt host machinery as part of its life cycle, starting with the docking of envelope gp120 to the cell surface receptor CD4. In this study, the response of the human CD4+ cell line CEMx174 to HIV-1 infection was quantitatively measured at the proteome level. To our knowledge, this is the first quantitative analysis of host proteome changes in CD4+ cells actively producing HIV-1 virus. We have quantified 3,255 cellular proteins, of which 344 increased and 343 decreased in abundance at 36 h p.i., the peak of virus production in our infection system. While we cannot preclude the effects stemming simply from virus binding, we have confirmed that almost all of the cells were actively producing virus at 36 h p.i. (Fig. 1A); therefore, our comparison to mock-infected cells should be sufficient for revealing protein expression changes associated with robust virus replication.
We found these virus-producing cells to exhibit signs of cell cycle arrest, though not accompanied by programmed cell death. The extent of protein abundance changes coincided with active transcriptional activities, as exemplified by the induction of chromatin remodeling proteins. Traffic across the nuclear envelope also appeared to favor the export of proteins, which may include the viral Rev and matrix proteins, along with viral mRNA, as part of the process of the egress of progeny virions. As with the reported decrease in isocitrate dehydrogenase expression in the proteomic analysis of HIV-1 Tat-expressing astrocytes (71), we observed a similar decrease in the abundance of the cytosolic, NAD-dependent isocitrate dehydrogenase. However, we also observed the elevated abundance of the mitochondrial, NADH-dependent isocitrate dehydrogenase, along with other components of the TCA cycle (Table 6) (viromics.washington.edu/publications/ericchan/HIV _Tables_1.zip). Such differences could be attributed to the expression of Tat in the presence or absence of the full complement of HIV viral proteins expressed in the proper order. On the other hand, the disconcerted changes in expression of the TCA cycle constituents may reflect a partially dysregulated TCA cycle, perhaps confined to the oxidation of citrate in the cytoplasm. Interestingly, as earlier microarray analyses of CEM cells infected under similar conditions showed prominent changes in the sterol biosynthesis machinery (89), the decrease in oxidation of citrate in the cytoplasm would supply the citrate needed for the synthesis of cholesterol used for the budding of virions. At the same time, the overall induction of the TCA cycle activities would be in line with the increase in supply of cytosolic acetyl-CoA required for fatty acid and cholesterol synthesis via the transcriptional activation of sterol regulatory elements and, subsequently, Nef's induction of ATP citrate lyase (80, 90), also up-regulated based on our analysis (Table 6).
We also observed differential expression in the ubiquitination pathway, accompanied by changes in the abundance of proteasome components. As a dynamic complex, individual proteasomal subunits may exhibit differences in expression relative to each other, as we observed with the up-regulation of three proteasome 19S and 20S subunits, amidst a larger number of down-regulated proteasome subunits. This is not without precedence, as is exemplified by the maturation of select 20S beta subunits during immunoproteasome formation upon gamma interferon treatment (39). In addition, as the proteasome appears to pose a block to replication reportedly countered by the viral Nef protein (73), the observed down-regulation of select proteasome subunits (Table 4) (see viromics.washington.edu/publications/ericchan/HIV_Tables_1.zip) may also point to an alleviation in such blocks. Furthermore, some of the ubiquitination activities may also be integrated with cell cycle progression. Evidence for G1/S arrest stemmed from the down-regulation of cdc34, the ubiquitin E2-conjugating enzyme which normally degrades and lifts the block by checkpoint proteins on S-phase entry (33, 70). Cell cycle arrest was also evident from the down-regulation of another E2 enzyme, Ubc12, responsible for the neddylation of cdc53/cullin and its association in an SCF ubiquitin ligase complex (27, 50); the complex in turn regulates the degradation of G1 cyclins and transition into S phase (94). A prolonged G1 stage may in fact be beneficial to virus production, as HIV-1 long-terminal repeat activity has been shown to peak during an elongated G1-phase via the disruption of cdk2-cyclin E interaction by Tat expression (33, 49, 70). HIV-1 Vpr also induces G1 (63) and G2 (76) arrest. The increase in TPR abundance (Table 6) could also be indicative of G1 arrest, as this nuclear structural protein begins to accumulate in the nuclear envelope early in the G1 phase (48). While Vpr has been attributed to both the induction (63) or suppression (21) of apoptosis, our data at 36 h p.i. points to the latter, as evidenced by the reduced abundance of caspase-3 (Fig. 5), CD95/Fas, and CD30/TNFRSF8 (Fig. 4). Results from our flow cytometric analysis also pointed to a moderate increase in the G1/G0 population, without any increase in apoptosis (Fig. 6A and B). The up-regulation of MAP2K2 in the ERK/MAPK pathway (Table 5) (42) is known to block Fas-mediated apoptosis, while the up-regulation of EGLN1 in the HIF1 response pathway (Table 5) suppresses HIF1-alpha transcription, which may augment p53 level and potentiate cells for apoptosis (47, 88). Furthermore, apoptosis may be repressed by the down-regulation of β-tubulin (see viromics.washington.edu/publications/ericchan/HIV_Tables_1.zip), which was also reported in earlier proteomic analyses of Tat-expressing, Jurkat cells and astrocytes (20, 71), as the binding of Tat to tubulin is believed to trigger apoptosis (12, 23). While our results appeared to contradict the known apoptosis-inducing effects of Vpr and Vpu, it bears noting that CD4 cell apoptosis is reported to be predominant in uninfected, bystander lymphocytes (28, 56), which are not included in our model, as close to all of the cells were producing virus at 36 h p.i. It should be noted that the reported resistance to Vpr-induced apoptosis by G1 arrest (5) is in line with our observation of a moderate accumulation of G1/G0 cells, without appreciable increases in cell death.
The import of integrase for provirus integration also appeared to be impaired, based on the down-regulation of importin α and β subunits (38, 53) (Fig. 3). Likewise, the import of Rev and Vpr via the use of importins may be impaired. However, alternative transporters may be available, as has been reported for Rev (6) and the simian immunodeficiency virus Vpx protein (83), which is partially homologous to Vpr. In view of the fact that the up-regulation of TPR and Ran, accompanied by the down-regulation of importin subunits, appeared to support traffic out of the nucleus, we speculate that viral egress was probably the dominant process at the point of peak virus production. The apparently dramatic increase in TPR abundance may promote the export of RNA polymerase II transcripts, including both host and HIV viral mRNA (81). Changes in the abundance of ubiquitin-specific peptidases, such as Usp10 (Table 5) (see viromics.washington.edu/publications /ericchan/HIV_Tables_1.zip), may facilitate the breakdown of polyubiquitin chains and favor the monoubiquitination of cellular proteins by the E2-conjugating enzyme Tsg101, as in the case of the androgen receptor (26) and the HIV gag protein (34). Together with a presumable increase in cholesterol biosynthesis, these changes set in place a cellular environment suitable for the budding of virions. To reconcile the impediment to the import of integrase with the robust virus production we observed, we may point to the fact that integrase is an inherently unstable protein subject to degradation via the N-end rule (55), and that other HIV viral proteins, such as Vpr and Vpu, are subject to temporal expression predicated on Tat transactivation of the viral long-terminal repeat; therefore, it is that unlikely we could capture all the purported effects of HIV viral proteins on host protein expression and function at a single time point. To this end, we expanded our analysis to a select set of proteins involved in the pathways discussed (Fig. 5); indeed, the differential expression of proteins as reflected by their measured abundances involved in nuclear transport appeared to change midcourse, further indicating that the possible impediment to viral integration did not occur early on amidst the normal availability of importins α and β up to around 24 h p.i. On the other hand, differential expression of DLD in the TCA cycle progressed consistently as virus production was reaching the peak at 36 h p.i., perhaps indicative of a persistent need for an increased energy supply to meet the needs by parasitic virus production. Nevertheless, by beginning our profiling efforts at the time of peak virus production at 36 h p.i., we were able to witness these active factories of progeny virions maximizing cholesterol and presumably viral RNA syntheses, while resisting apoptosis.
In all, our data indicated that the CEMx174 cells in our infection model exhibited signs of antiproliferation, as exemplified by arrest at the G1/G0 stage and decrease in the NF-kB-associated cell proliferation program. On the other hand, while cell cycle dysregulation is a common cause of apoptosis, these virus-producing cells exhibited no significant signs of apoptosis, as confirmed by flow cytometric analysis. To reconcile the known apoptosis-inducing effects of viral proteins, such as Env and Vpr, we speculate that cell survival signals may have overcome the apoptotic signals long enough to sustain virus replication. Changes in the nuclear transport pathway also seemed to be skewed towards the export of progeny virions. Our analysis at 36 h p.i. has probably captured the final moments of the cells as virus producers, as these cells eventually succumbed to extensive cell death at 40 to 42 h p.i.
Protein expression profiling by MS provides a catalog of proteins at the disposal of the host cell for sustaining rapid virus production. While mRNA profiling by microarrays reveals the transcriptional programs affected by infection, proteomic profiling has the ability to capture posttranscriptional changes not reflected at the mRNA level, such as changes in protein turnover rates. Although the approach does not distinguish protein expression patterns that are causal to robust virus production from those that are simply associated with the overall change in cell signaling and metabolism, it provides leads for follow-up assays to establish causality. As proteins do not function in isolation, pathway analysis helps unveil the concerted changes of protein networks that are most likely to be revealing of host mechanisms contributory to viral production. Extending the approach to in vivo models of HIV-1 infection is going to yield more biologically relevant insights into pathogenesis at the systems level; however, animal variations call for comparisons with well-controlled in vitro systems in order to separate causal markers from noise. Moreover, cell culture studies are more effective at elucidating the primary effects of infection in virus-producing cells; therapeutic interventions that alter the microenvironment optimal for virus production may help lower the viral set point and thereby prolong the state of nonprogression to AIDS (41). Even without establishing causality, proteomic profiling also promises to reveal diagnostic markers for assessing the efficacy of and subject responsiveness to treatments.
Acknowledgments
We are grateful to Mike McCune (UCSF), Jim Mullins (University of Washington), Silvia Vega (Rosetta Biosoftware), and Angélique van ′t Wout (Sanquin Research) for their helpful comments.
We thank the National Institute on Drug Abuse (grant 1P30DA01562501 to M.G.K.), the NIH National Center for Research Resources (grants RR018522 to R.D.S. and RR00166 to M.G.K.), and the Environmental Molecular Sciences Laboratory at PNNL for use of the instrumentation applied in this research. The Environmental Molecular Sciences Laboratory is a national scientific user facility sponsored by the Department of Energy's Office of Biological and Environmental Research. Pacific Northwest National Laboratory is operated by Battelle Memorial Institute for the U.S. Department of Energy under contract no. DE-AC06-76RLO 1830. E.Y.C. was supported by an NIH Viral Oncology training grant (NIH T32CA09229) and a fellowship from the Merck research laboratories.
Footnotes
Published ahead of print on 9 May 2007.
REFERENCES
- 1.Agy, M. B., M. Wambach, K. Foy, and M. G. Katze. 1990. Expression of cellular genes in CD4 positive lymphoid cells infected by the human immunodeficiency virus, HIV-1: evidence for a host protein synthesis shut-off induced by cellular mRNA degradation. Virology 177:251-258. [DOI] [PubMed] [Google Scholar]
- 2.Akari, H., S. Bour, S. Kao, A. Adachi, and K. Strebel. 2001. The human immunodeficiency virus type 1 accessory protein Vpu induces apoptosis by suppressing the nuclear factor κB-dependent expression of antiapoptotic factors. J. Exp. Med. 194:1299-1312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Al-Shahrour, F., R. Diaz-Uriarte, and J. Dopazo. 2004. FatiGO: a web tool for finding significant associations of gene ontology terms with groups of genes. Bioinformatics 20:578-580. [DOI] [PubMed] [Google Scholar]
- 4.Alvarez, E., L. Menendez-Arias, and L. Carrasco. 2003. The eukaryotic translation initiation factor 4GI is cleaved by different retroviral proteases. J. Virol. 77:12392-12400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Andersen, J. L., J. L. DeHart, E. S. Zimmerman, O. Ardon, B. Kim, G. Jacquot, S. Benichou, and V. Planelles. 2006. HIV-1 Vpr-induced apoptosis is cell cycle dependent and requires Bax but not ANT. PLoS Pathogens 2:e127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Arnold, M., A. Nath, J. Hauber, and R. H. Kehlenbach. 2006. Multiple importins function as nuclear transport receptors for the Rev protein of human immunodeficiency virus type 1. J. Biol. Chem. 281:20883-20890. [DOI] [PubMed] [Google Scholar]
- 7.Barre-Sinoussi, F., J. C. Chermann, F. Rey, M. T. Nugeyre, S. Chamaret, J. Gruest, C. Dauguet, C. Axler-Blin, F. Vezinet-Brun, C. Rouzioux, W. Rozenbaum, and L. Montagnier. 1983. Isolation of a T-lymphotropic retrovirus from a patient at risk for acquired immune deficiency syndrome (AIDS). Science 220:868-871. [DOI] [PubMed] [Google Scholar]
- 8.Brand, S. R., R. Kobayashi, and M. B. Mathews. 1997. The Tat protein of human immunodeficiency virus type 1 is a substrate and inhibitor of the interferon-induced, virally activated protein kinase, PKR. J. Biol. Chem. 272:8388-8395. [DOI] [PubMed] [Google Scholar]
- 9.Brouard, S., P. O. Berberat, E. Tobiasch, M. P. Seldon, F. H. Bach, and M. P. Soares. 2002. Heme oxygenase-1-derived carbon monoxide requires the activation of transcription factor NF-κB to protect endothelial cells from tumor necrosis factor-α-mediated apoptosis. J. Biol. Chem. 277:17950-17961. [DOI] [PubMed] [Google Scholar]
- 10.Bruno, S., B. Ardelt, J. S. Skierski, F. Traganos, and Z. Darzynkiewicz. 1992. Different effects of staurosporine, an inhibitor of protein kinases, on the cell cycle and chromatin structure of normal and leukemic lymphocytes. Cancer Res. 52:470-473. [PubMed] [Google Scholar]
- 11.Calvano, S. E., W. Xiao, D. R. Richards, R. M. Felciano, H. V. Baker, R. J. Cho, R. O. Chen, B. H. Brownstein, J. P. Cobb, S. K. Tschoeke, C. Miller-Graziano, L. L. Moldawer, M. N. Mindrinos, R. W. Davis, R. G. Tompkins, S. F. Lowry, and the Inflammation and Host Response to Injury Large Scale Collaborative Research Program. 2005. A network-based analysis of systemic inflammation in humans. Nature 437:1032-1037. [DOI] [PubMed] [Google Scholar]
- 12.Campbell, G. R., E. Pasquier, J. Watkins, V. Bourgarel-Rey, V. Peyrot, D. Esquieu, P. Barbier, J. de Mareuil, D. Braguer, P. Kaleebu, D. L. Yirrell, and E. P. Loret. 2004. The glutamine-rich region of the HIV-1 Tat protein is involved in T-cell apoptosis. J. Biol. Chem. 279:48197-48204. [DOI] [PubMed] [Google Scholar]
- 13.Carling, D. 2005. AMP-activated protein kinase: balancing the scales. Biochimie 87:87-91. [DOI] [PubMed] [Google Scholar]
- 14.Casella, C. R., E. L. Rapaport, and T. H. Finkel. 1999. Vpu increases susceptibility of human immunodeficiency virus type 1-infected cells to Fas killing. J. Virol. 73:92-100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cheng, G., Z. Feng, and B. He. 2005. Herpes simplex virus 1 infection activates the endoplasmic reticulum resident kinase PERK and mediates eIF-2α dephosphorylation by the γ134.5 protein. J. Virol. 79:1379-1388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Choi, S., X. Liu, P. Li, T. Akimoto, S. Y. Lee, M. Zhang, and Z. Yan. 2005. Transcriptional profiling in mouse skeletal muscle following a single bout of voluntary running: evidence of increased cell proliferation. J. Appl. Physiol. 99:2406-2415. [DOI] [PubMed] [Google Scholar]
- 17.Chun, T. W., J. S. Justement, R. A. Lempicki, J. Yang, G. Dennis, Jr., C. W. Hallahan, C. Sanford, P. Pandya, S. Liu, M. McLaughlin, L. A. Ehler, S. Moir, and A. S. Fauci. 2003. Gene expression and viral prodution in latently infected, resting CD4+ T cells in viremic versus aviremic HIV-infected individuals. Proc. Natl. Acad. Sci. USA 100:1908-1913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ciborowski, P., Y. Enose, A. Mack, M. Fladseth, and H. E. Gendelman. 2004. Diminished matrix metalloproteinase 9 secretion in human immunodeficiency virus-infected mononuclear phagocytes: modulation of innate immunity and implications for neurological disease. J. Neuroimmunol. 157:11-16. [DOI] [PubMed] [Google Scholar]
- 19.Clemens, M. J. 2005. Translational control in virus-infected cells: models for cellular stress responses. Sem. Cell Dev. Biol. 16:13-20. [DOI] [PubMed] [Google Scholar]
- 20.Coiras, M., E. Camafeita, T. Urena, J. A. Lopez, F. Caballero, B. Fernandez, M. R. Lopez-Huertas, M. Perez-Olmeda, and J. Alcami. 2006. Modifications in the human T cell proteome induced by intracellular HIV-1 Tat protein expression. Proteomics 6(Suppl. 1):S63-S73. [DOI] [PubMed] [Google Scholar]
- 21.Conti, L., G. Rainaldi, P. Matarrese, B. Varano, R. Rivabene, S. Columba, A. Sato, F. Belardelli, W. Malorni, and S. Gessani. 1998. The HIV-1 Vpr protein acts as a negative regulator of apoptosis in a human lymphoblastoid T cell line: possible implications for the pathogenesis of AIDS. J. Exp. Med. 187:403-413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Daniel, R., R. A. Katz, and A. M. Skalka. 1999. A role for DNA-PK in retroviral DNA integration. Science 284:644-647. [DOI] [PubMed] [Google Scholar]
- 23.de Mareuil, J., M. Carre, P. Barbier, G. R. Campbell, S. Lancelot, S. Opi, D. Esquieu, J. D. Watkins, C. Prevot, D. Braguer, V. Peyrot, and E. P. Loret. 2005. HIV-1 Tat protein enhances microtubule polymerization. Retrovirology 2:5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Diehn, M., A. A. Alizadeh, O. J. Rando, C. L. Liu, K. Stankunas, D. Botstein, G. R. Crabtree, and P. O. Brown. 2002. Genomic expression programs and the integration of the CD28 costimulatory signal in T cell activation. Proc. Natl. Acad. Sci. USA 99:11796-11801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Emerman, M., and M. H. Malim. 1998. HIV-1 regulatory/accessory genes: keys to unraveling viral and host cell biology. Science 280:1880-1884. [DOI] [PubMed] [Google Scholar]
- 26.Faus, H., H. A. Meyer, M. Huber, I. Bahr, and B. Haendler. 2005. The ubiquitin-specific protease USP10 modulates androgen receptor function. Mol. Cell. Endocrinol. 245:138-146. [DOI] [PubMed] [Google Scholar]
- 27.Feldman, R. M. R., C. C. Correll, K. B. Kaplan, and R. J. Deshaies. 1997. A complex of Cdc4p, Skp1p, and Cdc53p/cullin catalyzes ubiquitination of the phosphorylated CDK inhibitor Sic1p. Cell 91:221-230. [DOI] [PubMed] [Google Scholar]
- 28.Finkel, T. H., G. Tudor-Williams, N. K. Banda, M. F. Cotton, T. Curiel, C. Monks, T. W. Baba, R. M. Ruprecht, and A. Kupfer. 1995. Apoptosis occurs predominantly in bystander cells and not in productively infected cells of HIV- and SIV-infected lymph nodes. Nat. Med. 1:129-134. [DOI] [PubMed] [Google Scholar]
- 29.Gale, M., Jr., S. L. Tan, and M. G. Katze. 2000. Translational control of viral gene expression in eukaryotes. Microbiol. Mol. Biol. Rev. 64:239-280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gallay, P., T. Hope, D. Chin, and D. Trono. 1997. HIV-1 infection of nondividing cells through the recognition of integrase by the importin/karyopherin pathway. Proc. Natl. Acad. Sci. USA 94:9825-9830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Geiss, G. K., R. E. Bumgarner, M. C. An, M. B. Agy, A. B. van 't Wout, E. Hammersmark, V. S. Carter, D. Upchurch, J. I. Mullins, and M. G. Katze. 2000. Large-scale monitoring of host cell gene expression during HIV-1 infection using cDNA microarrays. Virology 266:8-16. [DOI] [PubMed] [Google Scholar]
- 32.Geley, S., B. L. Hartmann, R. Hattmannstorfer, M. Loffler, M. J. Ausserlechner, D. Bernhard, R. Sgonc, E. M. Strasser-Wozak, M. Ebner, B. Auer, and R. Kofler. 1997. p53-induced apoptosis in the human T-ALL cell line CCRF-CEM. Oncogene 15:2429-2437. [DOI] [PubMed] [Google Scholar]
- 33.Goebl, M. G., J. Yochem, S. Jentsch, J. P. McGrath, A. Varshavsky, and B. Byers. 1988. The yeast cell cycle gene CDC34 encodes a ubiquitin-conjugating enzyme. Science 241:1331-1335. [DOI] [PubMed] [Google Scholar]
- 34.Goff, A., L. S. Ehrlich, S. N. Cohen, and C. A. Carter. 2003. Tsg101 control of human immunodeficiency virus type 1 Gag trafficking and release. J. Virol. 77:9173-9182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gong, L., and E. T. Yeh. 1999. Identification of the activating and conjugating enzymes of the NEDD8 conjugation pathway. J. Biol. Chem. 274:12036-12042. [DOI] [PubMed] [Google Scholar]
- 36.Gottlieb, R. A., J. Nordberg, E. Skowronski, and B. M. Babior. 1996. Apoptosis induced in Jurkat cells by several agents is preceded by intracellular acidification. Proc. Natl. Acad. Sci. USA 93:654-658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Haridas, V., J. Ni, A. Meager, J. Su, G. L. Yu, Y. Zhai, H. Kyaw, K. T. Akama, J. Hu, L. J. Van Eldik, and B. B. Aggarwal. 1998. TRANK, a novel cytokine that activates NF-κB and c-Jun N-terminal kinase. J. Immunol. 161:1-6. [PubMed] [Google Scholar]
- 38.Hearps, A. C., and D. A. Jans. 2006. HIV-1 integrase is capable of targeting DNA to the nucleus via an importin alpha/beta-dependent mechanism. Biochem. J. 398:475-484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Heink, S., D. Ludwig, P. M. Kloetzel, and E. Kruger. 2005. IFN-γ-induced immune adaptation of the proteasome system is an accelerated and transient response. Proc. Natl. Acad. Sci. USA 102:9241-9246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hinata, K., A. M. Gervin, Z. Y. Jennifer, and P. A. Khavari. 2003. Divergent gene regulation and growth effects by NF-κB in epithelial and mesenchymal cells of human skin. Oncogene 22:1955-1964. [DOI] [PubMed] [Google Scholar]
- 41.Ho, D. D. 1996. Viral counts count in HIV infection. Science 272:1124-1125. [DOI] [PubMed] [Google Scholar]
- 42.Holmstrom, T. H., I. Schmitz, T. S. Soderstrom, M. Poukkula, V. L. Johnson, S. C. Chow, P. H. Krammer, and J. E. Eriksson. 2000. MAPK/ERK signaling in activated T cells inhibits CD95/Fas-mediated apoptosis downstream of DISC assembly. EMBO J. 19:5418-5428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Holness, M. J., and M. C. Sugden. 2003. Regulation of pyruvate dehydrogenase complex activity by reversible phosphorylation. Biochem. Soc. Trans. 31:1143-1151. [DOI] [PubMed] [Google Scholar]
- 44.Hori, T., F. Osaka, T. Chiba, C. Miyamoto, K. Okabayashi, N. Shimbara, S. Kato, and K. Tanaka. 1999. Covalent modification of all members of human cullin family proteins by NEDD8. Oncogene 18:6829-6834. [DOI] [PubMed] [Google Scholar]
- 45.Jacobs, J. M., D. L. Diamond, E. Y. Chan, M. A. Gritsenko, W. Qian, M. Stastna, T. Baas, D. G. Camp, R. L. Carithers, Jr., R. D. Smith, and M. G. Katze. 2005. Proteome analysis of liver cells expressing a full-length hepatitis C virus (HCV) replicon and biopsy specimens of posttransplantation liver from HCV-infected patients. J. Virol. 79:7558-7569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kalpana, G. V., S. Marmon, W. Wang, G. R. Crabtree, and S. P. Goff. 1994. Binding and stimulation of HIV-1 integrase by a human homolog of yeast transcription factor SNF5. Science 266:2002-2006. [DOI] [PubMed] [Google Scholar]
- 47.Kim, J. Y., H. J. Ahn, J. H. Ryu, K. Suk, and J. H. Park. 2004. BH3-only protein Noxa is a mediator of hypoxic cell death induced by hypoxia-inducible factor 1α. J. Exp. Med. 199:113-124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Krull, S., J. Thyberg, B. Bjorkroth, H. R. Rackwitz, and V. C. Cordes. 2004. Nucleoporins as components of the nuclear pore complex core structure and Tpr as the architectural element of the nuclear basket. Mol. Biol. Cell 15:4261-4277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kundu, M., S. Sharma, A. De Luca, A. Giordano, J. Rappaport, K. Khalili, and S. Amini. 1998. HIV-1 Tat elongates the G1 phase and indirectly promotes HIV-1 gene expression in cells of glial origin. J. Biol. Chem. 273:8130-8136. [DOI] [PubMed] [Google Scholar]
- 50.Lammer, D., N. Mathias, J. M. Laplaza, W. Jiang, Y. Liu, J. Callis, M. Goebl, and M. Estelle. 1998. Modification of yeast Cdc53p by the ubiquitin-related protein Rub1p affects function of the SCFCdc4 complex. Genes Dev. 12:914-926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Liu, T., W. J. Qian, E. F. Strittmatter, D. G. Camp, G. A. Anderson, B. D. Thrall, and R. D. Smith. 2004. High-throughput comparative proteome analysis using a quantitative cysteinyl-peptide enrichment technology. Anal. Chem. 76:5345-5353. [DOI] [PubMed] [Google Scholar]
- 52.Lyakhovich, A., and M. P. Shekhar. 2004. RAD6B overexpression confers chemoresistance: RAD6 expression during cell cycle and its redistribution to chromatin during DNA damage-induced response. Oncogene 23:3097-3106. [DOI] [PubMed] [Google Scholar]
- 53.Maertens, G., P. Cherepanov, Z. Debyser, Y. Engelborghs, and A. Engelman. 2004. Identification and characterization of a functional nuclear localization signal in the HIV-1 integrase interactor LEDGF/p75. J. Biol. Chem. 279:33421-33429. [DOI] [PubMed] [Google Scholar]
- 54.Mannova, P., R. Fang, H. Wang, B. Deng, M. W. McIntosh, S. M. Hanash, and L. Beretta. 2006. Modification of host lipid raft proteome upon hepatitis C virus replication. Mol. Cell. Proteomics 5:2319-2325. [DOI] [PubMed] [Google Scholar]
- 55.Mulder, L. C. F., and M. A. Muesing. 2000. Degradation of HIV-1 integrase by the N-end rule pathway. J. Biol. Chem. 275:29749-29753. [DOI] [PubMed] [Google Scholar]
- 56.Muro-Cacho, C. A., G. Pantaleo, and A. S. Fauci. 1995. Analysis of apoptosis in lymph nodes of HIV-infected persons. Intensity of apoptosis correlates with the general state of activation of the lymphoid tissue and not with stage of disease or viral burden. J. Immunol. 154:5555-5566. [PubMed] [Google Scholar]
- 57.Muta, H., L. H. Boise, L. Fang, and E. R. Podack. 2000. CD30 signals integrate expression of cytotoxic effector molecules, lymphocyte trafficking signals, and signals for proliferation and apoptosis. J. Immunol. 165:5105-5111. [DOI] [PubMed] [Google Scholar]
- 58.Nacken, W., A. J. Kingsman, S. M. Kingsman, F. Sablitzky, and C. Sorg. 1995. A homologue of the human MSS1 gene, a positive modulator of HIV-1 gene expression, is massively expressed in Xenopus oocytes. Biochim. Biophys. Acta 1261:293-295. [DOI] [PubMed] [Google Scholar]
- 59.Nelbock, P., P. J. Dillon, A. Perkins, and C. A. Rosen. 1990. A cDNA for a protein that interacts with the human immunodeficiency virus Tat transactivator. Science 248:1650-1653. [DOI] [PubMed] [Google Scholar]
- 60.Nesvizhskii, A. I., A. Keller, E. Kolker, and R. Aebersold. 2003. A statistical model for identifying proteins by tandem mass spectrometry. Anal. Chem. 75:4646-4658. [DOI] [PubMed] [Google Scholar]
- 61.Nicoletti, I., G. Migliorati, M. C. Pagliacci, F. Grignani, and C. Riccardi. 1991. A rapid and simple method for measuring thymocyte apoptosis by propidium iodide staining and flow cytometry. J. Immunol. Methods 139:271-279. [DOI] [PubMed] [Google Scholar]
- 62.Nikolsky, Y., S. Ekins, T. Nikolskaya, and A. Bugrim. 2005. A novel method for generation of signature networks as biomarkers from complex high throughput data. Toxicol. Lett. 158:20-29. [DOI] [PubMed] [Google Scholar]
- 63.Nishizawa, M., M. Kamata, R. Katsumata, and Y. Aida. 2000. A carboxy-terminally truncated form of the human immunodeficiency virus type 1 Vpr protein induces apoptosis via G1 cell cycle arrest. J. Virol. 74:6058-6067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ohlmann, T., D. Prevot, D. Decimo, F. Roux, J. Garin, S. J. Morley, and J. L. Darlix. 2002. In vitro cleavage of eIF4GI but not eIF4GII by HIV-1 protease and its effects on translation in the rabbit reticulocyte lysate system. J. Mol. Biol. 318:9-20. [DOI] [PubMed] [Google Scholar]
- 65.Osaka, F., H. Kawasaki, N. Aida, M. Saeki, T. Chiba, S. Kawashima, K. Tanaka, and S. Kato. 1998. A new NEDD8-ligating system for cullin-4A. Genes Dev. 12:2263-2268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Oyaizu, N., T. W. McCloskey, M. Coronesi, N. Chirmule, V. S. Kalyanaraman, and S. Pahwa. 1993. Accelerated apoptosis in peripheral blood mononuclear cells (PBMCs) from human immunodeficiency virus type-1 infected patients and in CD4 cross-linked PBMCs from normal individuals. Blood 82:3392-3400. [PubMed] [Google Scholar]
- 67.Parissi, V., A. Caumont, V. Richard de Soultrait, C. H. Dupont, S. Pichuantes, and S. Litvak. 2000. Inactivation of the SNF5 transcription factor gene abolishes the lethal phenotype induced by the expression of HIV-1 integrase in yeast. Gene 247:129-136. [DOI] [PubMed] [Google Scholar]
- 68.Paschal, B. M., and L. Gerace. 1995. Identification of NTF2, a cytosolic factor for nuclear import that interacts with nuclear pore complex protein p62. J. Cell Biol. 129:925-937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Petritis, K., L. J. Kangas, P. L. Ferguson, G. A. Anderson, L. Pasa-Tolic, M. S. Lipton, K. J. Auberry, E. F. Strittmatter, Y. Shen, R. Zhao, and R. D. Smith. 2003. Use of artificial neural networks for the accurate prediction of peptide liquid chromatography elution times in proteome analyses. Anal. Chem. 75:1039-1048. [DOI] [PubMed] [Google Scholar]
- 70.Plon, S. E., K. A. Leppig, H. N. Do, and M. Groudine. 1993. Cloning of the human homolog of the CDC34 cell cycle gene by complementation in yeast. Proc. Natl. Acad. Sci. USA 90:10484-10488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Pocernich, C. B., D. Boyd-Kimball, H. F. Poon, V. Thongboonkerd, B. C. Lynn, J. B. Klein, V. Calebrese, A. Nath, and D. A. Butterfield. 2005. Proteomics analysis of human astrocytes expressing the HIV protein Tat. Mol. Brain Res. 133:307-316. [DOI] [PubMed] [Google Scholar]
- 72.Poon, B., K. Grovit-Ferbas, S. A. Stewart, and I. S. Chen. 1998. Cell cycle arrest by Vpr in HIV-1 virions and insensitivity to antiretroviral agents. Science 281:266-269. [DOI] [PubMed] [Google Scholar]
- 73.Qi, M., and C. Aiken. 2007. Selective restriction of Nef-defective human immunodeficiency virus type 1 by a proteasome-dependent mechanism. J. Virol. 81:1534-1536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Qian, W. J., T. Liu, M. E. Monroe, E. F. Strittmatter, J. M. Jacobs, L. J. Kangas, K. Petritis, D. G. Camp II, and R. D. Smith. 2005. Probability-based evaluation of peptide and protein identifications from tandem mass spectrometry and SEQUEST analysis: the human proteome. J. Proteome Res. 4:53-62. [DOI] [PubMed] [Google Scholar]
- 75.Qian, W. J., M. E. Monroe, T. Liu, J. M. Jacobs, G. A. Anderson, Y. Shen, R. J. Moore, D. J. Anderson, R. Zhang, S. E. Calvano, S. F. Lowry, W. Xiao, L. L. Moldawer, R. W. Davis, R. G. Tompkins, D. G. Camp II, R. D. Smith, and the Inflammation and the Host Response to Injury Large Scale Collaborative Research Program. 2005. Quantitative proteome analysis of human plasma following in vivo lipopolysaccharide administration using 16O/18O labeling and the accurate mass and time tag approach. Mol. Cell. Proteomics 4:700-709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Re, F., D. Braaten, E. K. Franke, and J. Luban. 1995. Human immunodeficiency virus type 1 Vpr arrests the cell cycle in G2 by inhibiting the activation of p34cdc2-cyclin B. J. Virol. 69:6859-6864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Reddy, B., and J. Yin. 1999. Quantitative intracellular kinetics of HIV type 1. AIDS Res. Hum. Retrovir. 15:273-283. [DOI] [PubMed] [Google Scholar]
- 78.Roberts, C. J., B. Nelson, M. J. Marton, R. Stoughton, M. R. Meyer, H. A. Bennett, Y. D. He, H. Dai, W. L. Walker, T. R. Hughes, M. Tyers, C. Boone, and S. H. Friend. 2000. Signaling and circuitry of multiple MAPK pathways revealed by a matrix of global gene expression profiles. Science 287:873-880. [DOI] [PubMed] [Google Scholar]
- 79.Roy, S., M. G. Katze, N. T. Parkin, I. Edery, A. G. Hovanessian, and N. Sonenberg. 1990. Control of the interferon-induced 68-kilodalton protein kinase by the HIV-1 tat gene product. Science 247:1216-1219. [DOI] [PubMed] [Google Scholar]
- 80.Sato, R., A. Okamoto, J. Inoue, W. Miyamoto, Y. Sakai, N. Emoto, H. Shimano, and M. Maeda. 2000. Transcriptional regulation of the ATP citrate-lyase gene by sterol regulatory element-binding proteins. J. Biol. Chem. 275:12497-12502. [DOI] [PubMed] [Google Scholar]
- 81.Shibata, S., Y. Matsuoka, and Y. Yoneda. 2002. Nucleocytoplasmic transport of proteins and poly(A)+ RNA in reconstituted Tpr-less nuclei in living mammalian cells. Genes Cells 7:421-434. [DOI] [PubMed] [Google Scholar]
- 82.Shibuya, H., K. Irie, J. Ninomiya-Tsuji, M. Goebl, T. Taniguchi, and K. Matsumoto. 1992. New human gene encoding a positive modulator of HIV Tat-mediated transactivation. Nature 357:700-702. [DOI] [PubMed] [Google Scholar]
- 83.Singhal, P. K., P. R. Kumar, M. R. K. S. Rao, M. Kyasani, and S. Mahalingam. 2006. Simian immunodeficiency virus Vpx is imported into the nucleus via importin alpha-dependent and -independent pathways. J. Virol. 80:526-536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Stoughton, R. S., and H. Dai. May 2002. Statistical combining of cell expression profiles. U.S. patent 6,351,712.
- 85.Tardif, K. D., K. Mori, and A. Siddiqui. 2002. Hepatitis C virus subgenomic replicons induce endoplasmic reticulum stress activating an intracellular signaling pathway. J. Virol. 76:7453-7459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Tavazoie, S., J. D. Hughes, M. J. Campbell, R. J. Cho, and G. M. Church. 1999. Systematic determination of genetic network architecture. Nat. Genet. 22:281-285. [DOI] [PubMed] [Google Scholar]
- 87.Tian, Q., S. B. Stepaniants, M. Mao, L. Weng, M. C. Feetham, M. J. Doyle, E. C. Yi, H. Dai, V. Thorsson, J. Eng, D. Goodlett, J. P. Berger, B. Gunter, P. S. Linseley, R. B. Stoughton, R. Aebersold, S. J. Collins, W. A. Hanlon, and L. E. Hood. 2004. Integrated genomic and proteomic analyses of gene expression in mammalian cells. Mol. Cell. Proteomics 3:960-969. [DOI] [PubMed] [Google Scholar]
- 88.To, K. K. W., and L. E. Huang. 2005. Suppression of hypoxia-inducible factor 1α (HIF-1α) transcriptional activity by the HIF prolyl hydroxylase EGLN1. J. Biol. Chem. 280:38102-38107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.van 't Wout, A. B., G. K. Lehrman, S. A. Mikheeva, G. C. O'Keeffe, M. G. Katze, R. E. Bumgarner, G. K. Geiss, and J. I. Mullins. 2003. Cellular gene expression upon human immunodeficiency virus type 1 infection of CD4+-T-cell lines. J. Virol. 77:1392-1402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.van 't Wout, A. B., J. V. Swain, M. Schindler, U. Rao, M. S. Pathmajeyan, J. I. Mullins, and F. Kirchhoff. 2005. Nef induces multiple genes involved in cholesterol synthesis and uptake in human immunodeficiency virus type 1-infected T cells. J. Virol. 79:10053-10058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Wain-Hobson, S., J. P. Vartanian, M. Henry, N. Chenciner, R. Cheynier, S. Delassus, L. P. Martins, M. Sala, M. T. Nugeyre, D. Guetard, D. Klatzmann, J.-C. Gluckmann, W. Rosenbaum, F. Barre′-Sinoussi, and L. Montagnier. 1991. LAV revisited: origins of the early HIV-1 isolates from Institut Pasteur. Science 252:961-965. [DOI] [PubMed] [Google Scholar]
- 92.Wang, H., W. J. Qian, M. H. Chin, V. A. Petyuk, R. C. Barry, T. Liu, M. A. Gritsenko, H. M. Mottaz, R. J. Moore, D. G. Camp, A. H. Khan, D. J. Smith, and R. D. Smith. 2006. Characterization of the mouse brain proteome using global proteomic analysis complemented with cysteinyl-peptide enrichment. J. Proteome Res. 5:361-369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Wang, H., W. J. Qian, H. M. Mottaz, T. R. W. Clauss, D. J. Anderson, R. J. Moore, D. G. Camp, A. H. Khan, D. M. Sforza, M. Pallavicini, D. J. Smith, and R. D. Smith. 2005. Development and evaluation of a micro- and nanoscale proteomic sample preparation method. J. Proteome Res. 4:2397-2403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Willems, A. R., S. Lanker, E. E. Patton, K. L. Craig, T. F. Nason, N. Mathias, R. Kobayashi, C. Wittenberg, and M. Tyers. 1996. Cdc53 targets phosphorylated G1 cyclins for degradation by the ubiquitin proteolytic pathway Cell 86:453-463. [DOI] [PubMed] [Google Scholar]
- 95.Yamamoto, T., T. Matsuda, A. Junicho, H. Kishi, A. Yoshimura, and A. Muraguchi. 2001. Hematopoietic cell-specific adapter proteins, SLP-76 and BLNK, effectively activate NF-AT as well as NF-κB by Syk and Tec PTKs in non-lymphoid cell lines. FEBS Lett. 491:272-278. [DOI] [PubMed] [Google Scholar]
- 96.Yung, E., M. Sorin, A. Pal, E. Craig, A. Morozov, O. Delattre, J. Kappes, D. Ott, and G. V. Kalpana. 2001. Inhibition of HIV-1 virion production by a transdominant mutant of integrase interactor 1. Nat. Med. 7:920-926. [DOI] [PubMed] [Google Scholar]