

Abstract
Phosphoinositide-dependent kinase l (PDK1) phosphorylates and activates multiple AGC serine kinases, including protein kinase B (PKB), p70Ribosomal S6 kinase (S6K) and p90Ribosomal S6 kinase (RSK). PDK1 is required for thymocyte differentiation and proliferation, and herein, we explore the molecular basis for these essential functions of PDK1 in T lymphocyte development. A key finding is that PDK1 is required for the expression of key nutrient receptors in T cell progenitors: CD71 the transferrin receptor and CD98 a subunit of L-amino acid transporters. PDK1 is also essential for Notch-mediated trophic and proliferative responses in thymocytes. A PDK1 mutant PDK1 L155E, which supports activation of PKB but no other AGC kinases, can restore CD71 and CD98 expression in pre-T cells and restore thymocyte differentiation. However, PDK1 L155E is insufficient for thymocyte proliferation. The role of PDK1 in thymus development thus extends beyond its ability to regulate PKB. In addition, PDK1 phosphorylation of AGC kinases such as S6K and RSK is also necessary for thymocyte development.
Keywords: Notch, PDK1, PKB, RSK, Thymus
Introduction
T cell development in the thymus produces peripheral T cells with a useful repertoire of T cell antigen receptors (TCRs) and is coordinated by extracellular signals from antigen receptors, cytokines and stromal cells. These stimuli link via tyrosine kinases to a diverse network of GTPases and serine kinases. The importance of serine kinases for thymocyte development is revealed by studies of phosphoinositide-dependent kinase l (PDK1), a kinase that phosphorylates a key ‘T' loop site within the catalytic domain of AGC serine kinases (Hinton et al, 2004). PDK1 phosphorylates and activates multiple AGC kinases, including phosphatidyl inositol-3 kinase (PI3K)-controlled serine kinases such as Akt or protein kinase B (PKB), the 70-kDa ribosomal S6 kinase-1 (S6K1) and the 90-kDa ribosomal S6 kinase (RSK). Deletion of PDK1 in T cell progenitors causes a block in T cell development (Hinton et al, 2004). PDK1-deficient thymocytes fail to differentiate past the pre-T cell stage and do not make peripheral T cells.
The TCR has variable α/β subunits that recognise peptide/MHC complexes and the selection of cells that have successfully rearranged their TCRβ gene locus is an essential step in T cell development (von Boehmer et al, 1999; Michie and Zuniga-Pflucker, 2002). This occurs in T cell precursors, which do not express the major histocompatibility complex (MHC) receptors CD4 and CD8 (double-negative (DN) thymocytes). T cell progenitors initiate rearrangements of the TCRβ locus and if successful produce a TCRβ chain that permits surface expression of the pre-TCR complex comprising TCRβ, pre-Tα and CD3 antigens. The pre-TCR, in combination with Notch, then induces proliferation and differentiation into CD4/CD8 double-positive (DP) thymocytes (Wolfer et al, 2002; Maillard et al, 2003; Ciofani et al, 2004; Schmitt et al, 2004a; Tanigaki et al, 2004; Ciofani and Zuniga-Pflucker, 2005; Laky et al, 2006). The process is referred to as β-selection, because only cells that have successfully rearranged their TCRβ locus undergo proliferative expansion and differentiate to DPs.
The pre-TCR activates PDK1-regulated kinases (Hinton et al, 2006; Mao et al, 2007) and these are clearly essential for thymocyte development as PDK1-null pre-T cells do not proliferate or differentiate yet express TCRβ subunits and have a functioning pre-TCR (Hinton et al, 2004). Loss of PDK1 causes a size reduction of β-selected pre-T cells indicative of a metabolic defect (Hinton et al, 2004). In this context, Notch signals support metabolism throughout β-selection (Ciofani and Zuniga-Pflucker, 2005) and the failed development of PDK1-null cells could thus reflect defective trophic responses to Notch. A number of facts are consistent with this hypothesis: deletion of floxed Notch1 alleles via Cre excision blocks thymocyte development at the same stage as PDK1 deletion (Wolfer et al, 2002); Notch-ligand interactions in pre-T cells activate the PDK1 substrate PKB (Ciofani and Zuniga-Pflucker, 2005); expression of a constitutively active PKB mutant can partially substitute for Notch and maintain thymocyte metabolism during β-selection (Ciofani and Zuniga-Pflucker, 2005); and PKB serine kinases are required for the transition of DN thymocytes to the DP stage, partly by enhancing the proliferation and survival of cells undergoing β-selection (Mao et al, 2007). A key question then is whether the impact of PDK1 loss on thymocyte development stems only from its key role in regulating PKB and/or reflects the unresponsiveness of cells to Notch-induced trophic signals.
To address these issues, the present study compares the development of wild–type (WT) and PDK1-null T cell progenitors in an in vitro model that uses OP9 stromal cells expressing the Notch ligand delta-like 1 (OP9-DL1 cells) to drive thymocyte differentiation (Schmitt et al, 2004b; Schmitt and Zuniga-Pflucker, 2006). To determine the contribution of the PDK1/PKB pathway to thymocyte development, we studied the differentiation of thymocytes whose WT PDK1 allele were substituted with a PDK1 L155E mutant, that permits phosphorylation of PKB, but not other substrates such as S6K1, PKC, SGK or RSK (Collins et al, 2003, 2005). The substitution of leucine (L) 155 in PDK1 with glutamate (E) disrupts the integrity of an important PDK1 domain termed the PIF-binding pocket. This domain is not required for PKB phosphorylation, but is necessary for PDK1 to interact with carboxy-terminal hydrophobic motifs in substrates such as S6K1 and RSK (Biondi et al, 2000, 2001; Frodin et al, 2000, 2002). The PDK1 L155E mutant can thus support normal activation of PKB, but not S6K1 and RSK activity (Collins et al, 2003). The value of PDK1 L155E in dissecting the contribution of different PDK1 substrates has been demonstrated in vivo (Collins et al, 2003; Bayascas et al, 2006). It can substitute for WT PDK1 in insulin responses in skeletal muscle demonstrating that PKB is the relevant target for PDK1 in these cells (Bayascas et al, 2006). However, PDK1 L155E does not support normal murine embryo development, indicating that PDK1 activation of PKB is not sufficient for all PDK1 functions (McManus et al, 2004).
The present results show that PDK1-null pre-T cells cannot respond to Notch-induced trophic signals, because Notch signals via PDK1 to induce and sustain expression of key nutrient receptors. In the absence of PDK1, pre-T cells are blocked at the DN stage of thymocyte differentiation. Expression of PDK1 L155E, which supports activation of PKB is able to replace WT PDK1 and restore nutrient receptor expression and pre-T cell differentiation, but does not restore normal thymus cellularity. These results identify an important role for the PDK1/PKB pathway during thymocyte differentiation, but show that the importance of PDK1 in the thymus cannot be ascribed solely to its role upstream of PKB. T cell development is thus equally dependent on PDK1 substrates that interact with PDK1 via its PIF domain.
Results
PDK1-deficient pre-T cells cannot respond to Notch signals and have defective expression of key nutrient receptors
To assess whether PDK1 is required for Notch-induced thymocyte growth, differentiation and proliferation, we compared the responses of WT versus PDK1-null pre-T cells in an in vitro system using OP9 stromal cells expressing the OP9-DL1. The OP9-DL1 system allows an assessment of Notch responsiveness in pre-T cells in vitro (Schmitt and Zuniga-Pflucker, 2002; Zuniga-Pflucker, 2004). PDK1-null pre-T cells were obtained from PDK1flΔneo/flΔneo Lck-Cre+ mice (hereafter referred to as T-PDK1−/− mice), which were generated by crossing mice with floxed PDK1 exons 3 and 4 on both alleles (PDK1flΔneo/flΔneo) (Lawlor et al, 2002) with mice expressing Cre recombinase under the control of the proximal p56lck proximal promoter (Lck-Cre+), which induces Cre expression in DN T cell progenitors in the thymus (Takahama et al, 1998; Hinton et al, 2004). DN thymocytes can be subdivided on the basis of differential surface expression of CD44 and CD25: the first T cell progenitors are CD44+/CD25− (DN1) followed sequentially by CD44+/CD25+ (DN2), CD44−/CD25+ (DN3) and CD44−/CD25− (DN4) populations. In T-PDK1−/−, thymocytes are blocked in development at the DN4 stage (Hinton et al, 2004).
WT DNs cultured on OP9-DL1 cells increase in cell size, whereas cells cultured on OP9 cells in the absence of DL-1 do not (Figure 1A). DNs cultured on OP9-DL1 cells also proliferate (70- to 80-fold in 6 days) and differentiate to DP thymocytes that co-express CD4 and CD8 (Figure 1B). In contrast, DN thymocytes cultured on OP9 cells in the absence of Notch ligand do not proliferate, although they differentiate to DPs (Figure 1B). T-PDK1−/− DN thymocytes fail to increase in cell size in response to OP9-DL1 (Figure 1C) and do not proliferate or differentiate to DPs (Figure 1D). The small size of T-PDK1−/− DN thymocytes cultured on OP9-DL1 cells (Figure 1A versus Figure 1C) parallels what is seen in ex vivo PDK1 deleted pre-T cells (Hinton et al, 2004), which are small compared to controls (Figure 1E).
Figure 1.

PDK1 is required for Notch-induced pre-T cell development. (A) Data show analysis of relative cell size (based on flow cytometric analysis of forward scatter) of G0/G1 thymocytes. WT DN3 thymocytes from were co-cultured for 2 days with OP9 or OP9-DL1 stromal cell monolayers. (B) Data show surface expression of CD4 and CD8 on thymocytes from WT mice before (input) and after culture on OP9 and OP9-DL1 for times indicated. DN thymocytes from WT mice were co-cultured for times indicated with OP9 or OP9-DL1. Numbers in brackets refer to fold proliferation. Numbers in quadrants indicate the percentage of each corresponding population. Data are representative of three independent experiments. (C) Data show analysis of relative cell size of T-PDK1−/− (PDK1flΔneo/− Lck-Cre+) DNs co-cultured for 2 days with OP9 or OP9-DL1 stromal cells. Data are representative of three independent experiments. (D) Data show CD4 and CD8 staining of T-PDK1−/− DNs following co-culture on OP9-DL1 for times indicated. Numbers in brackets refer to fold proliferation. Numbers in quadrants indicate the percentage of each corresponding population. Data are representative of three independent experiments. (E) The data shows analysis of relative cell size, based on flow cytometric analysis of forward scatter, of G0/G1 ex vivo DN4 thymocytes from control T-PDK1+/+ (PDK1flΔneo/flΔneo) and T-PDK1−/− (PDK1flΔneo/flΔneo Lck-Cre+) mice. Data are representative of three independent experiments. (F) Data show DL4–IgG fusion protein staining of ex vivo of T-PDK1−/− and normal littermate control DN4s. Geometric mean fluorescent intensities; T-PDK1−/−=68, T-PDK1+/+=110. Data are representative of two independent experiments. (G) The data show analysis of relative cell size, based on flow cytometric analysis of forward scatter, of region A (F) gated DN4 thymocytes taken immediately ex vivo from control T-PDK1+/+ (PDK1flΔneo/flΔneo) and T-PDK1−/− (PDK1flΔneo/flΔneo Lck-Cre+) mice.
PDK1-null pre-T cells thus do not grow, differentiate or proliferate in response to Notch. We examined whether this defect could be attributed to defective expression of Notch on T-PDK1−/− DNs. A DL4-IgG fusion protein was used to monitor Notch1 expression (Besseyrias et al, 2007). Figure 1F shows that WT and PDK1-null pre-T cells express Notch1. There is a two-fold reduction in Notch1 expression in PDK1-null cells, but does not explain failed development of T-PDK1−/− DNs as Notch1 haploinsufficient thymocytes develop normally (F Radtke, personal communication) and within WT DN4s, two-fold differences in Notch1 levels had no impact on cell size (Supplementary Figure 1). Moreover, PDK1−/− DN4s expressing identical levels of surface Notch1 as WT DN4s (Region A in Figure 1F) are still smaller (Figure 1G).
T lymphocyte cell size is dependent on the regulated expression of amino-acid transporters that include CD98 (42F) as a key component (Gottesdiener et al, 1988; Lindsten et al, 1988; Parmacek et al, 1989; Rathmell et al, 2000; Edinger and Thompson, 2002). The upregulation of CD71, the transferrin receptor, mediates iron uptake and is also critical for cell growth (Schneider et al, 1982; Trowbridge and Shackelford, 1986). Therefore, we considered whether defective growth response of PDK1-null pre-T cells might result from defective expression of nutrient transporters during thymus development. In initial experiments, we explored whether thymocyte differentiation is accompanied by changes in expression of CD71 and CD98. Figure 2A shows that DN3 thymocytes express low levels of CD98 and CD71, whereas surface expression of these nutrient transporters is high in DN4s. Progression through β-selection is thus accompanied by upregulation of CD71 and CD98. We then asked whether Notch controls CD71 and CD98 expression. Figure 2B shows that DN3 thymocytes cultured on OP9-DL1 cells upregulate expression of CD71 and CD98, but this does not happen when they are cultured on OP9 cells in the absence of Notch signalling. DN3s cultured on OP9-DL1 cells also increase in cell size (Figure 2B). DN4 thymocytes have high levels of CD71 and CD98 expression ex vivo (Figure 2A); they strikingly lose expression of these receptors if cultured on OP9 cells, but maintain CD71 and CD98 expression if cultured on OP9-DL1 cells (Figure 2C). Hence, sustained Notch signalling is required to maintain CD98 and CD71 surface expression on DN4 thymocytes. The cell size of DN4 thymocytes is also sustained by Notch: DN4s cultured on OP9 cells rapidly decrease in size, but maintain cell size when cultured on OP9-DL1 cells (Figure 2C).
Figure 2.
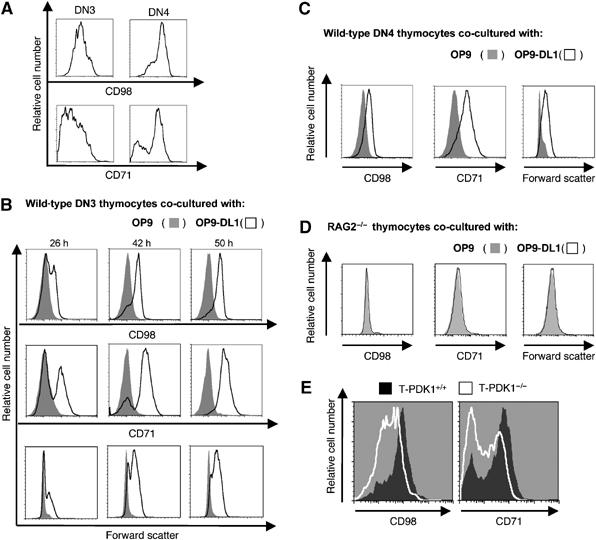
Nutrient receptor expression is regulated by Notch during thymocyte development and requires PDK1. (A) Data show CD98 and CD71 expression on WT DN3 and DN4 thymocytes. Data are representative of three independent experiments. (B) Data show CD98 and CD71 expression levels in sorted DN3 WT thymocytes co-cultured on OP9 and OP9-DL1 monolayers for times indicated. Analysis of relative cell size, based on flow cytometric analysis of forward scatter, is also shown. Data is representative of three independent experiments. (C) Data show expression of CD98 and CD71 and relative cell size based on flow cytometric analysis of forward scatter for sorted DN4 WT thymocytes co-cultured on OP9 and OP9-DL1 monolayers for 26 h. Data is representative of three independent experiments. (D) Data show expression of CD98 and CD71 and relative cell size based on flow cytometric analysis of forward scatter for RAG2−/− thymocytes co-cultured on OP9 and OP9-DL1 monolayers for 26 h. Data are representative of three independent experiments. (E) The data show flow cytometry for CD98 and CD71 surface expression on DN4 (CD25−CD44−) thymocytes taken immediately ex vivo from T-PDK1−/− (PDK1flΔneo/flΔneo Lck-Cre+) or WT mice. Data is representative of two independent experiments.
Notch1 thus regulates nutrient receptor expression on pre-T cells, but a key question is whether pre-TCR signalling is essential for T cells to become competent to respond to Notch1 in the context of CD98 and CD71 expression. To address this issue, we cultured DN3 cells from Recombinase gene 2-null mice (Rag2−/−) that lack a pre-TCR, on OP9-DL1 cells. Figure 2D shows that Rag2−/− DN3s fail to upregulate CD98 and CD71 when cultured on OP9-DL1 cells. Hence, Notch is not sufficient to regulate CD71 and CD98 expression in the absence of the pre-TCR, rather Notch is required to sustain expression of CD98 and CD71 on β-selected pre-T cells.
Next, we examined CD71 and CD98 expression in thymocytes from T-PDK1−/− mice. In T-PDK1−/− mice, there is partial deletion of PDK1 in DN3s, complete PDK1 loss in the DN4s and a failure of these cells to develop beyond the DN4 stage (Hinton et al, 2004). Figure 2E shows that CD98 and CD71 expression levels are defective in PDK1−/− DN4 thymocytes. DN4 thymocytes from T-PDK1−/− mice were uniformly low for CD98 expression and had a high frequency of CD71 low cells compared to WT controls. The loss of CD71 and CD98 expression is not reflective of a global signalling defect in T-PDK1−/− DN4s, because the upregulation of a number of other surface receptors occurs normally as PDK1-null DN3s transit to DN4s. T-PDK1−/− DN4s can thus upregulate CD2 and CD5 expression normally; a marker of a functioning pre-TCR (Hinton et al, 2004). The reduced expression of CD98 and CD71 on PDK1-null DN4s reveals that the expression of these key nutrient receptors is mediated by PDK1.
Is reduced expression of nutrient receptors sufficient to explain the defective growth responses of pre-T cells? Experiments to limit nutrient availability revealed that the ability of pre-T cells to increase cell size and develop in response to Notch was dependent on the availability of exogenous amino acids (data not shown). We also examined whether differences in levels of expression of nutrient receptors on the surface of pre-T cells had an impact on trophic responses in pre-T cells. The data (Supplementary Figure 2) show that WT DN4s expressing low levels of CD71 or CD98 are small cells compared to cells expressing high levels of CD71. Hence, expression of CD98 and CD71 is rate limiting for pre-T cell growth.
A PDK1 L155E mutant supports nutrient receptor expression and pre-T cell differentiation
There are multiple PDK1 substrates in T cells, including PKB, S6K1 and RSK. Expression of constitutively active PKB mutants can substitute for cytokines and induce expression of CD71 and CD98 in haematopoietic cells (Edinger and Thompson, 2002). PDK1 is necessary for PKB activation and hence the impact of PDK1 loss on thymocyte development could reflect loss of PKB activity in these cells. In this context, simultaneous loss of three PKB isoforms blocks thymocyte development at the DN/DP stage, similar to the effects of deleting PDK1 (Hinton et al, 2004; Mao et al, 2007). However, it is not known if PKB is the only effector of PDK1 in the thymus and it is important to address the involvement of additional PDK1 substrates in T cell development.
One way to explore the contribution of PKB to PDK1 function is to use ectopically expressed constitutively active PKB mutants, a strategy used to determine much of what we know about PKB in T cells (Jones et al, 2000; Parsons et al, 2001; Edinger and Thompson, 2002; Na et al, 2003; Rathmell et al, 2003; Patra et al, 2004). However, PKB mutants may not necessarily mimic the actions of endogenous PKB. One alternative and powerful strategy to explore the relative contributions of different PDK1 substrates to a biological response in vivo is to analyse mice with ‘knock-in' mutations of PDK1 alleles (Collins et al, 2003, 2005; McManus et al, 2004; Bayascas et al, 2006). One such PDK1 mutant, containing a L155E mutation, cannot support activation of S6K and RSK but allows activation of PKB (Collins et al, 2003; Figure 3A). Mice that are homozygous for the PDK1 L155E mutation do not survive beyond developmental stage E12. However, it is possible to generate thymocytes that selectively express PDK1 L155E (T-PDK1L155E/−) by backcrossing mice that express a single PDK1 L155E allele and a single PDK1 floxed allele (PDK1L155E/flΔneo) with mice expressing Cre recombinase under the control of the p56lck proximal promoter (Supplementary Figure 3). The presence of the single PDK1 floxed allele in all tissues allows normal mouse development. However, as progenitors enter the thymus and express Cre recombinase, the WT allele is deleted thereby generating pre-T cells that express a single PDK1 L155E allele (PDK1L155E/flΔneo Lck-Cre+, referred to as T-PDKL155E/−). A comparison of the ability of a single PDK L155E allele versus a single WT allele to support T cell development can then be assessed relative to the impact of complete PDK1 deletion. The crucial control for the analysis of pre-T cells that express the single L155E mutant allele are pre-T cells that express a single WT PDK1 allele (PDK1+/flΔneo Lck-Cre+, referred to as T-PDK1+/−). The impact of PDK1 haplo-insufficiency on thymocyte development has been described and of relevance to the present work is the fact that a single WT PDK1 allele restores DN4 cell size and cell cycle progression and allows thymocyte differentiation from DNs to DPs (Kelly et al, 2006).
Figure 3.
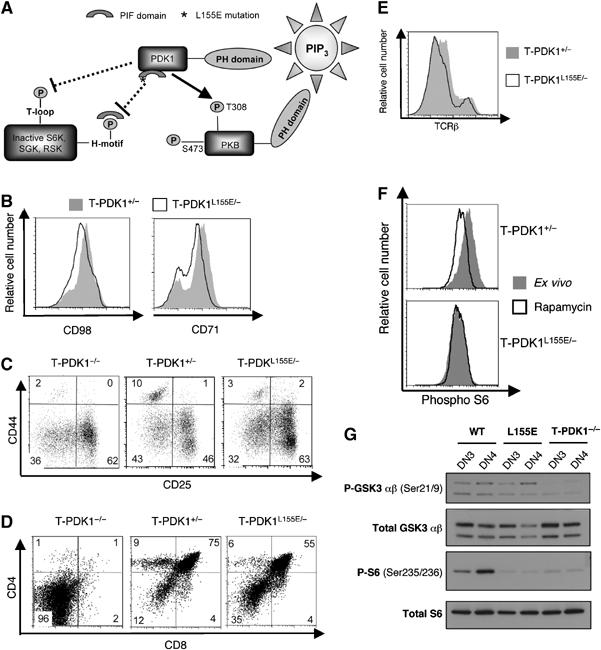
PDK1 L155E restores nutrient receptor expression and thymocyte differentiation. (A) A schematic representation of PDK1 L155E. A leucine (L) to glutamate (E) mutation at residue 155 in the PIF domain of PDK1 (denoted by *) prevents docking of PDK1 at the H-motif of substrates S6K, SGK and RSK. This prevents their T loop phosphorylation by PDK1 and subsequent activation. Dotted lines represent these inactivated pathways. PDK1 L155E mutation is permissive for PKB activation (solid line)(Collins et al, 2003). (B) Data show CD98 (left panel) and CD71 (right panel) expression on control T-PDK1+/− (PDK1+/flΔneoLck-Cre+) and T-PDK1L155E/− (PDK1L155E/flΔneoLck-Cre+) DN4 thymocytes. (C) Data show CD44 and CD25 expression on T-PDK1−/− (PDK1flΔneo/flΔneoLck-Cre+), T-PDK1+/− and T-PDK1L155E/− DNs. Data are representative of four independent experiments. (D) Flow cytometric analysis of CD4 and CD8 expression on T-PDK1−/−, T-PDK1+/− and T-PDK1L155E/− Thy1+ thymocytes is shown. Data are representative of four independent experiments. (E) Data show TCRβ staining of Thy1-gated T-PDK1+/− and T-PDK1L155E/− thymocytes (F) Data shows intracellular staining for phosphorylated S6 ribosomal protein in T-PDK1+/− and T-PDK1L155E/− DN4s. Data show phospho S6 staining in thymocytes immediately ex vivo (filled histogram) or treated with 20 nM rapamycin (open histogram) as an internal negative control to reduce S6 protein phosphorylation to basal levels. Data are representative of three independent experiments. (G) Data show western blot analysis of phosphoGSK3 α/β Ser21/9 and S6 ribosomal protein Ser235/6 in wild type (WT), T-PDK1L155E/− (L155E) and T-PDK1−/− DN3 and DN4 thymocytes.
Experiments with PDK1 L155E explore whether kinases that interact with PDK1 via its PIF domain are required for thymocyte development. Initially, we compared CD71 and CD98 expression in DN4 thymocytes from T-PDK1+/− and T-PDK1L155E/− mice. Figure 3B shows that expression of PDK1 L155E restores CD71 and CD98 expression to a level comparable to that seen in DN4s expressing WT PDK1. PDK1 L155E could also restore thymocyte differentiation in PDK1 null pre-T cells. The distribution of the different DN subpopulations in T-PDK1+/− and T-PDK1L155E/− mice was comparable (Figure 3C). Figure 3D shows that unlike T-PDK1−/− thymi, which have virtually no DPs and single positives (SPs), T-PDK1L155E/− thymi contain DP and SP subsets at relatively normal ratios. Mature WT SP thymocytes express high surface levels of the TCRβ and there was a normal frequency of these cells in T-PDKL155E/− thymi (Figure 3E). PDK1 L155E can thus substitute for PDK1 loss and is sufficient for normal CD71 and CD98 expression and for pre-T cell differentiation into DPs and SPs.
A genomic PCR analysis verified that Cre-mediated deletion of the flΔneo allele had occurred in T-PDKL155E/− DPs (Supplementary Figure 4). There was also functional loss of PDK1 as judged by failed phosphorylation of the ribosomal S6 subunit in T-PDK1L155E/−DN4 thymocytes. S6 phosphorylation is mediated by S6K1, which requires phosphorylation at its ‘T loop' site by PDK1. A crucial step that enables this phosphorylation event is the docking of PDK1 via its PIF domain at a hydrophobic-motif in S6K. The L155E mutant of PDK1 is thus defective in activating S6K1 (Collins et al, 2003). The activity of S6K1 can be monitored by quantification of phosphorylation of its substrate, the ribosomal S6 subunit, by intracellular staining with specific phosphorylated S6 antisera. Basal levels of S6 phosphorylation are high in WT DN4s, but absent following PDK1 deletion (Hinton et al, 2004). Figure 3F shows that expression of a single WT PDK1 allele can support activation of S6K1 in pre-T cells, but the PDK1 L155E allele cannot. These data were confirmed by western blot analysis (Figure 3G). Western blot data also confirm that PDK1 L155E can support PKB activity as determined by the normal phosphorylation of GSK3α on its PKB substrate sequence Serine 21 in both WT and PDK1 L155E pre-T cells (Figure 3G). In contrast, GSK3α Serine 21 phosphorylation is absent in PDK1-null pre-T cells.
PDK1 L155E is not sufficient for cell growth or proliferation of pre-T cells
PDK1 L155E substitutes for WT PDK1 in the context of CD98 and CD71 expression and DN to DP differentiation. However, DN to DP transition is normally accompanied by a large proliferative expansion of β-selected pre-T cells and there was no such proliferative expansion in T-PDK1L155E/− mice (Figure 4A). T-PDK1L155E/− mice thus have very low numbers of DPs and SPs and peripheral α/β T cells compared to control mice (Figure 4B). PDK1 L155E thus supports DN to DP differentiation, but does not restore thymus cellularity. In accordance with the failed proliferative expansion of T-PDK1L155E/− thymocytes, there was a decrease in the frequency of cells in S/G2 phases of the cell cycle in T-PDK1L155E/− DN4s (Figure 4C). Moreover, the normal increase in cell size that accompanies the DN3 to DN4 transition is lost in T-PDK1L155E/− mice (Figure 4D). Hence, expression of a single WT PDK1 allele can maintain normal growth and proliferation of pre-T cells, whereas PDK1 L155E cannot. Thus, PDK1 activation of PKB is sufficient for pre-T cell differentiation, but PDK1 substrates that interact with PDK1 via its PIF domain are required for optimal pre-T cell growth and proliferative expansion.
Figure 4.
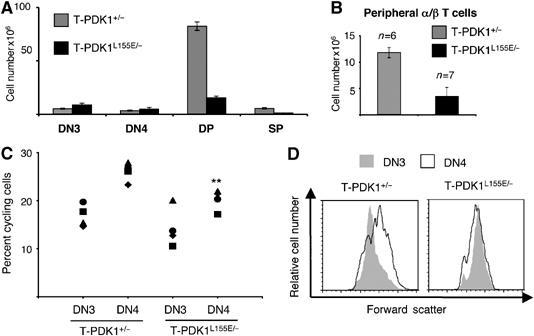
PDK1 L155E does not restore thymus cellularity. (A) Data show T-PDK1+/− (PDK1+/flΔneoLck-Cre+) and T-PDK1L155E/− (PDK1L155E/flΔneoLck-Cre+) thymocyte subpopulation numbers. For DN thymocytes; T-PDK1+/− (n=4) and T-PDK1L155E/− (n=3) mice, for DP thymocytes; T-PDK1+/− (n=4) and T-PDK1L155E/− (n=8) mice, for SP thymocytes; T-PDK1+/− (n=4) and T-PDK1L155E/− (n=6) mice. (B) Data show numbers of α/β T cells from T-PDK1+/− and T-PDK1L155E/− mice. (C) Data show the percentage of cells in the proliferative (S and G2) stages of the cell cycle in T-PDK1+/− (n=4) and T-PDK1L155E/− (n=4) DN3 and DN4 cells. (**P-value of significance 0.0074). (D) Data show analysis of relative cell size based on flow cytometric analysis of forward scatter for T-PDK1+/− and T-PDK1L155E/− DN3 and DN4 thymocytes. Data are representative of four independent experiments.
Notch-induced proliferation but not differentiation of T cell progenitors is mTOR and RSK dependent
The PDK1 PIF domain is required for the phosphorylation of multiple kinases, such as RSK and S6K1 (Collins et al, 2003). We examined the role of RSK in pre-T cell development using a recently described specific RSK inhibitor BI-D1870 (Sapkota et al, 2007). This inhibitor inhibits all four RSK isoforms, but not other related AGC kinases (Sapkota et al, 2007). Figure 5A shows that RSK inhibition does not prevent the DN to DP differentiation of WT pre-T cells, but does inhibit Notch-induced proliferative expansion of these pre-T cell subpopulations (Figure 5B). Inhibition of RSK with BI-D1870 thus allows cells to differentiate, but does not allow them to proliferate normally. We also considered the possible contribution of S6K1 to thymus development. S6K1 must be phosphorylated by PDK1 at its T loop site to become activated, but also has an additional requirement for PDK1 function as its activation is dependent on PKB, which regulates S6K1 via modulation of TSC-1/2 function and mTOR (mammalian target of rapamycin) (Inoki et al, 2002; Jaeschke et al, 2002; Manning et al, 2002). In this respect, in mature T cells, S6K1 couples PKB and mTOR to the control of T cell cycle progression (Brennan et al, 1999).
Figure 5.
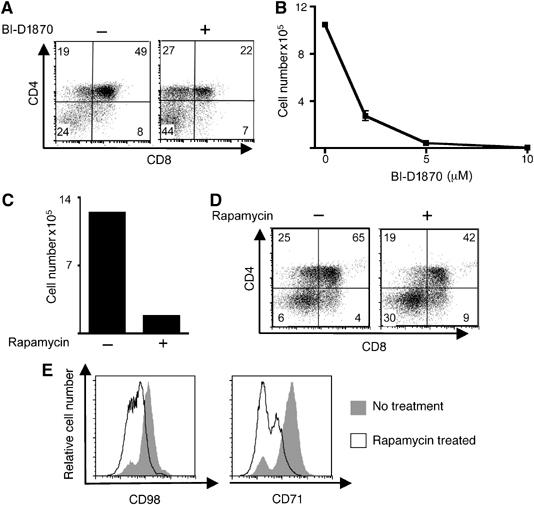
Inhibition of either mTOR or RSK permits thymocyte differentiation but not proliferation. (A) The data show surface expression of CD4 and CD8 on DN WT thymocytes following 3 days co-culture with 5 ng/ml IL7 on OP9-DL1 monolayers in the presence or absence of 10 μM BI-D1870. The data are representative of two independent experiments. (B) Data show numbers of cells following 6 days co-culture of DN thymocytes on OP9-DL1 in the presence of BI-D1870 at concentrations indicated. (C) Data show the number of cells following 6 days co-culture of DN thymocytes co-cultured on OP9-DL1 monolayers with 5 ng/ml IL7 in the presence or absence of 20 nM rapamycin. Data are representative of three independent experiments. (D) Data show CD4 and CD8 expression on DN3 WT thymocytes following 6 days co-culture with 5 ng/ml IL7 on OP9-DL1 in the presence or absence of 20 nM rapamycin. Data are representative of three independent experiments. (E) Data show CD98 (left panel) and CD71 (right panel) surface expression on DN3 thymocytes, following 2 days co-culture with 20 nM rapamycin on OP9-DL1. Data are representative of three independent experiments.
Previous studies have shown that inhibition of mTOR with rapamycin reduces thymus cellularity in vivo (Chiang and Abraham, 2005; Holz et al, 2005). Figure 5C and D show that rapamycin blocks Notch-dependent proliferative expansion of pre-T cells in vitro, but not DN to DP differentiation. As rapamycin immediately inactivates S6K1, these results are consistent with a role for S6K1 in thymus development. However, important caveats are that rapamycin can disrupt sustained activation of PKB (Sarbassov et al, 2006). Moreover, mTOR is needed for PKB activation of S6K1, but can also regulate S6K1-independent pathways (Dann and Thomas, 2006). Accordingly, the ability of rapamycin to inhibit T cell proliferation may be explained by a requirement for mTOR signalling to mediate PKB function, rather than reflecting loss of S6K1 activity. In this context, in a pro-B cell line, PKB could induce expression of CD98 and CD71 via a mTOR-dependent pathway (Edinger and Thompson, 2002). To explore the mTOR requirement for nutrient receptor expression in pre-T cells, we examined the impact of rapamycin on CD98 and CD71 expression in the OP9 DL-1 system. The data (Figure 5E) show that the ability of Notch signals to sustain expression of CD98 and CD71 is partially sensitive to rapamycin inhibition. Hence, the expression of nutrient receptors in pre-T cells is regulated by PDK1- and mTOR-sensitive pathways.
Discussion
The differentiation and proliferation of pre-T cells in the thymus is dependent on sustained Notch receptor/ligand interactions (Schmitt and Zuniga-Pflucker, 2005; Guidos, 2006; Visan et al, 2006). The present work shows that Notch-mediated trophic and proliferative responses are lost following deletion of PDK1. As T cell progenitors progress through β-selection, there is induced cell surface expression of CD98, an essential subunit of mammalian L-type amino-acid transporters and CD71, the transferrin receptor. The expression of these nutrient receptors is induced by the pre-TCR in a Notch- and PDK1-dependent manner. Moreover, sustained Notch signalling is required to maintain CD71 and CD98 expression as pre-T cells progress through β-selection. Strikingly, PDK1 deletion prevents expression of these key nutrient receptors and prevents Notch-induced growth responses. The observations that PDK1 regulates the expression of key amino-acid and iron transporters during thymocyte development and is required for Notch trophic responses offer an explanation as to why the loss of PDK1 has such a major impact on T cell development. The loss of nutrient receptor regulation in PDK1-null T cells will thus render the cells metabolically insufficient to support the demands made by the massive proliferative expansion occurring as thymocytes make the DN to DP transition.
To investigate PDK1 substrates that control expression of CD98 and CD71, we looked at the expression of these nutrient receptors in pre-T cells expressing a PDK1 L155E allele that can activate PKB, but not AGC kinases that bind to PDK1 via its PIF domain. PDK1 L155E restores expression of CD71 and CD98 and also supports both the differentiation of pre-T cells to DP and mature SP cells. PKB thus has key functions regulating the expression of amino-acid transporters and transferrin receptors during thymocyte development. However, PDK1 L155E did not restore normal thymus cellularity. Hence, PKB activation is sufficient for thymocyte differentiation, but not sufficient for optimal proliferation. Accordingly, kinases that interact with PDK1 via its PIF domain are necessary for cell growth and proliferation of pre-T cells in the thymus. The role of PDK1 during thymocyte development is thus multifaceted and not simply a reflection of its role as an upstream activator of PKB.
Serine/threonine kinases that interact with PDK1 via its PIF domain include RSK and S6K1. The present results identify RSK as an important signalling molecule for pre-T cell development, as inhibition of this kinase inhibited proliferative expansion of pre-T cells, but did not prevent their differentiation. The importance of the PDK1 PIF domain could thus be explained by its role in RSK regulation. However, there is still the question of the involvement of S6K1. It is difficult to assess a thymus autonomous role for S6Ks in mice doubly deficient for S6Ks, as these show perinatal lethality (Pende et al, 2004). Nevertheless, we tested the role of another S6K regulator mTOR in thymocyte growth and proliferation by examining the impact of rapamycin on Notch-induced thymocyte development. These experiments revealed that the inhibition of mTOR with rapamycin prevents optimal nutrient receptor expression and growth and proliferation of DN thymocytes, but permits their differentiation. Accordingly, there is a dual requirement for PDK1 and mTOR for cell growth and proliferation during thymocyte development.
In summary, the present study shows that PDK1 is important during thymocyte development, because it regulates expression of key nutrient receptors on the surface of pre-T cells and mediates Notch-induced cell growth and proliferative responses. PDK1 was first identified as a key activator of PKB, but was also found to mediate activation of other AGC kinases such as RSK and S6K1 (Biondi et al, 2000, 2001; Frodin et al, 2000, 2002). A PDK1 L155E mutant, with a disrupted PIF pocket can support normal activation of PKB and restore nutrient receptor expression and DP and SP thymocyte differentiation in PDK1-null cells. However, PDK1 L155E cannot support normal thymocyte proliferation. The importance of PDK1 for β-selection thus reflects the role of multiple AGC serine kinases in this process rather than just reflecting PKB regulation.
Materials and methods
Mice
Mice (5- to 7-week-old) (backcrossed for at least seven generations to C57BL6) were maintained in SPF conditions under Home Office project license PPL60/3116. T-PDK1−/− mice (PDK1flΔneo/flΔneo Lck-Cre+ or PDK1flΔneo/− Lck-Cre+ were generated as described previously (Takahama et al, 1998; Lawlor et al, 2002; Hinton et al, 2004). T-PDK1−/− mice have partial deletion of PDK1 in DN3 thymocytes and complete ablation of PDK1 in DN4 pre-T cells (Hinton et al, 2004). Control mice used for analyses of T-PDK1−/− mice were age-matched WT littermates or PDK1flΔneo/flΔneo where indicated.
Mice containing a knock-in mutation of PDK1, wherein leucine (L) at residue 155 was changed to glutamate (E) (PDK1L155E/+) (Collins et al, 2003) were crossed with PDK1flΔneo/flΔneo mice to generate mice expressing a single PDK1 L155E allele and a single PDK1 floxed allele (PDK1L155E/flΔneo). These were then backcrossed with mice expressing Cre recombinase under the control of the p56lck proximal promoter in T cell precursors to generate PDK1L155E/flΔneo Lck-Cre+ mice (referred to as T-PDK1L155E/−). Control mice PDK1+/flΔneo Lck-Cre+ (referred to as T-PDK+/−) were generated by crossing mice expressing a single PDK1 floxed allele (PDK1+/flΔneo) with mice expressing Cre recombinase under the control of the p56lck proximal promoter.
PCR analysis
Cre-mediated deletion of the floxed PDK1 allele from DP sorted thymocytes in T-PDK1L155E/− mice was confirmed by PCR using primers p99 (5′-ATC CCA AGT TAC TGA GTT GTG TTG GAA G) and p100 (5′-TGT GGA CAA ACA GCA ATG AAC ATA CAC GC). A PCR product of 200 bp was generated for WT and PIF alleles and a 250 bp band was generated when the flΔneo allele was present. A 200 bp product generated by PCR analysis using primers p80 (5′-CTA TGC TGT GTT ACT TCT TGG AGC ACA G) and p100 was indicative of deletion of exons 3 and 4 of PDK1.
Flow cytometric analysis
Antibodies conjugated to fluorescein isothiocyanate, phycoerythrin, allophycocyanin and biotin were obtained from either Pharmingen (San Diego, CA, USA) or eBioscience (San Diego, CA, USA). TriColour-conjugated antibodies were obtained from Caltag (Burlingame, CA, USA). Cells were stained for surface expression of the following markers using the antibodies in parentheses: CD4 (RM4-5), CD8 (53-6.7), CD25 (7D4), CD44 (IM7), CD71 (C2), CD98 (RL388), Thy1.2 (53-2.1), TCR β (H57-597), B220 (RA3-6B2) and TCR γ/δ (GL3). Cells were stained with saturating concentrations of antibody in accordance with the manufacturer's instructions. Data were acquired on either a FACS Calibur (Becton Dickinson, Franklin Lakes, NJ, USA) or an LSR1 flow cytometer (Becton Dickinson) using CellQuest software and were analysed using either CellQuest (Becton Dickinson) or FlowJo (Treestar, San Carlos, CA, USA) software. Viable cells were gated according to their forward scatter and side scatter profiles. CD4 and CD8 DN subsets were gated by lineage exclusion (lineage−) of all CD4, CD8 DP and SP cells and TCR γ/δ. DN3s and DN4s were further defined as CD25+CD44− and CD25−CD44− thymocytes, respectively. Mature SP thymocytes were defined as Thy-1+, TCRβhi and positive for CD4 or CD8 expression. A DL4 IgG fusion protein was used to monitor Notch1 expression as described previously (Besseyrias et al, 2007). Cellular DNA content was measured by DAPI staining of saponin permeabilised cells. Phospho-S6 levels in ex vivo thymocytes was assessed as described previously (Hinton et al, 2004).
OP9 cultures and assay
OP9 bone marrow stromal cells expressing OP9-DL1 (Schmitt et al, 2004b) and control OP9 cells were a gift from Juan Carlos Zúñiga-Pflücker (Toronto, Canada). OP9 cells were maintained in αMEM supplemented with 50 μM 2-mercaptoethanol, 100 U/ml penicillin, 1 mg/ml streptomycin and 20% heat-inactivated FBS. Sorted thymus DN3 (CD25+CD44−) and DN4 (CD25−CD44−) subsets were co-cultured on OP9 and OP9-DL1 monolayers for times indicated in Figure legends. DN3 and DN4 thymocytes were purified by first depleting thymic populations of CD4+ and CD8+ cells using an AutoMACs magnetic cell sorter (Miltenyi Biotech, Auburn, CA, USA) before sorting to a purity greater than 95%, using a FACS VantageSE cell sorter (Becton Dickinson). On day of harvest thymocytes were filtered through 50 μm filters to remove OP9 cells before developmental progression of T lineage cells was assessed.
Western blot analysis
Sorted DN3 and DN4 thymocytes were lysed on ice in NP-40 lysis buffer (50 mM Hepes (pH 7.4), 75 mM NaCl, 1% Nonidet P-40, 10 mM sodium fluoride, 10 mM iodoacetimide, 1 mM EDTA, 40 mM β-glycerophosphate, protease inhibitors, 1 mM phenylmethylsulfonyl fluoride, 100 μM sodium orthovanadate). Lysates were centrifuged at 1600 g for 15 min at 4°C. Protein samples were separated by sodium dodecyl sulphate 4–12% polyacrylamide gel electrophoreisis, transferred to nitrocellulose membrane and detected by western blot analysis using standard techniques. Blots were probed with antibodies that recognise phosphorylated GSK3 α/β on Ser21/9 (Cell Signaling Technologies, Danvers, MA, USA), total GSK3 α/β (Upstate, Hampshire, UK), phosphorylated S6 ribosomal protein on Ser235/236 (Cell Signaling Technologies) and total S6 ribosomal protein (Cell Signaling Technologies).
Supplementary Material
Supplementary Figure 1
Supplementary Figure 2
Supplementary Figure 3
Supplementary Figure 4
Supplementary Figure Legends
Acknowledgments
This work was supported by Wellcome Trust Programme Grant 60/3116 and Wellcome Trust Principal Research Fellowship (DAC). EM was supported by a Marie Heim-Vögtlin Fellowship PMP DB-115611/1. We thank Juan Carlos Zúñiga-Pflücker for the generous gift of OP9 cell lines and for the seminal work that established this model; members of Biological Services Unit for mouse care; and our co-workers for critical reading of the manuscript.
References
- Bayascas JR, Sakamoto K, Armit L, Arthur JS, Alessi DR (2006) Evaluation of approaches to generate PDK1 tissue specific knock-in mice. J Biol Chem 281: 28772–28781 [DOI] [PubMed] [Google Scholar]
- Besseyrias V, Fiorini E, Strobl LJ, Zimber-Strobl U, Dumortier A, Koch U, Arcangeli ML, Ezine S, Macdonald HR, Radtke F (2007) Hierarchy of Notch-Delta interactions promoting T cell lineage commitment and maturation. J Exp Med 204: 331–343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biondi RM, Cheung PC, Casamayor A, Deak M, Currie RA, Alessi DR (2000) Identification of a pocket in the PDK1 kinase domain that interacts with PIF and the C-terminal residues of PKA. EMBO J 19: 979–988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biondi RM, Kieloch A, Currie RA, Deak M, Alessi DR (2001) The PIF-binding pocket in PDK1 is essential for activation of S6 K and SGK, but not PKB. EMBO J 20: 4380–4390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brennan P, Babbage JW, Thomas G, Cantrell D (1999) p70(s6k) integrates phosphatidylinositol 3-kinase and rapamycin-regulated signals for E2F regulation in T lymphocytes. Mol Cell Biol 19: 4729–4738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiang GG, Abraham RT (2005) Phosphorylation of mammalian target of rapamycin (mTOR) at Ser-2448 is mediated by p70S6 kinase. J Biol Chem 280: 25485–25490 [DOI] [PubMed] [Google Scholar]
- Ciofani M, Zuniga-Pflucker JC (2005) Notch promotes survival of pre-T cells at the beta-selection checkpoint by regulating cellular metabolism. Nat Immunol 6: 881–888 [DOI] [PubMed] [Google Scholar]
- Ciofani M, Schmitt TM, Ciofani A, Michie AM, Cuburu N, Aublin A, Maryanski JL, Zuniga-Pflucker JC (2004) Obligatory role for cooperative signaling by pre-TCR and Notch during thymocyte differentiation. J Immunol 172: 5230–5239 [DOI] [PubMed] [Google Scholar]
- Collins BJ, Deak M, Arthur JS, Armit LJ, Alessi DR (2003) In vivo role of the PIF-binding docking site of PDK1 defined by knock-in mutation. EMBO J 22: 4202–4211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins BJ, Deak M, Murray-Tait V, Storey KG, Alessi DR (2005) In vivo role of the phosphate groove of PDK1 defined by knock-in mutation. J Cell Sci 118: 5023–5034 [DOI] [PubMed] [Google Scholar]
- Dann SG, Thomas G (2006) The amino acid sensitive TOR pathway from yeast to mammals. FEBS Lett 580: 2821–2829 [DOI] [PubMed] [Google Scholar]
- Edinger AL, Thompson CB (2002) Akt maintains cell size and survival by increasing mTOR-dependent nutrient uptake. Mol Biol Cell 13: 2276–2288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frodin M, Antal TL, Dummler BA, Jensen CJ, Deak M, Gammeltoft S, Biondi RM (2002) A phosphoserine/threonine-binding pocket in AGC kinases and PDK1 mediates activation by hydrophobic motif phosphorylation. EMBO J 21: 5396–5407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frodin M, Jensen CJ, Merienne K, Gammeltoft S (2000) A phosphoserine-regulated docking site in the protein kinase RSK2 that recruits and activates PDK1. EMBO J 19: 2924–2934 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gottesdiener KM, Karpinski BA, Lindsten T, Strominger JL, Jones NH, Thompson CB, Leiden JM (1988) Isolation and structural characterization of the human 4F2 heavy-chain gene, an inducible gene involved in T-lymphocyte activation. Mol Cell Biol 8: 3809–3819 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guidos CJ (2006) Synergy between the pre-T cell receptor and Notch: cementing the alphabeta lineage choice. J Exp Med 203: 2233–2237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinton HJ, Alessi DR, Cantrell DA (2004) The serine kinase phosphoinositide-dependent kinase 1 (PDK1) regulates T cell development. Nat Immunol 5: 539–545 [DOI] [PubMed] [Google Scholar]
- Hinton HJ, Clarke RG, Cantrell DA (2006) Antigen receptor regulation of phosphoinositide-dependent kinase 1 pathways during thymocyte development. FEBS Lett 580: 5845–5850 [DOI] [PubMed] [Google Scholar]
- Holz MK, Ballif BA, Gygi SP, Blenis J (2005) mTOR and S6K1 mediate assembly of the translation preinitiation complex through dynamic protein interchange and ordered phosphorylation events. Cell 123: 569–580 [DOI] [PubMed] [Google Scholar]
- Inoki K, Li Y, Zhu T, Wu J, Guan KL (2002) TSC2 is phosphorylated and inhibited by Akt and suppresses mTOR signalling. Nat Cell Biol 4: 648–657 [DOI] [PubMed] [Google Scholar]
- Jaeschke A, Hartkamp J, Saitoh M, Roworth W, Nobukuni T, Hodges A, Sampson J, Thomas G, Lamb R (2002) Tuberous sclerosis complex tumor suppressor-mediated S6 kinase inhibition by phosphatidylinositide-3-OH kinase is mTOR independent. J Cell Biol 159: 217–224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones RG, Parsons M, Bonnard M, Chan VS, Yeh WC, Woodgett JR, Ohashi PS (2000) Protein kinase B regulates T lymphocyte survival, nuclear factor kappaB activation, and Bcl-X(L) levels in vivo. J Exp Med 191: 1721–1734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly AP, Hinton HJ, Clarke RG, Cantrell DA (2006) Phosphoinositide-dependent kinase l (PDK1) haplo-insufficiency inhibits production of alpha/beta (alpha/beta) but not gamma delta (gamma/delta) T lymphocytes. FEBS Lett 580: 2135–2140 [DOI] [PubMed] [Google Scholar]
- Laky K, Fleischacker C, Fowlkes BJ (2006) TCR and Notch signaling in CD4 and CD8 T-cell development. Immunol Rev 209: 274–283 [DOI] [PubMed] [Google Scholar]
- Lawlor MA, Mora A, Ashby PR, Williams MR, Murray-Tait V, Malone L, Prescott AR, Lucocq JM, Alessi DR (2002) Essential role of PDK1 in regulating cell size and development in mice. EMBO J 21: 3728–3738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindsten T, June CH, Thompson CB, Leiden JM (1988) Regulation of 4F2 heavy-chain gene expression during normal human T-cell activation can be mediated by multiple distinct molecular mechanisms. Mol Cell Biol 8: 3820–3826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maillard I, Adler SH, Pear WS (2003) Notch and the immune system. Immunity 19: 781–791 [DOI] [PubMed] [Google Scholar]
- Manning BD, Tee AR, Logsdon MN, Blenis J, Cantley LC (2002) Identification of the tuberous sclerosis complex-2 tumor suppressor gene product tuberin as a target of the phosphoinositide 3-kinase/akt pathway. Mol Cell 10: 151–162 [DOI] [PubMed] [Google Scholar]
- Mao C, Tili EG, Dose M, Haks MC, Bear SE, Maroulakou I, Horie K, Gaitanaris GA, Fidanza V, Ludwig T, Wiest DL, Gounari F, Tsichlis PN (2007) Unequal contribution of akt isoforms in the double-negative to double-positive thymocyte transition. J Immunol 178: 5443–5453 [DOI] [PubMed] [Google Scholar]
- McManus EJ, Collins BJ, Ashby PR, Prescott AR, Murray-Tait V, Armit LJ, Arthur JS, Alessi DR (2004) The in vivo role of PtdIns(3,4,5)P3 binding to PDK1 PH domain defined by knock-in mutation. EMBO J 23: 2071–2082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michie AM, Zuniga-Pflucker JC (2002) Regulation of thymocyte differentiation: pre-TCR signals and beta-selection. Semin Immunol 14: 311–323 [DOI] [PubMed] [Google Scholar]
- Na SY, Patra A, Scheuring Y, Marx A, Tolaini M, Kioussis D, Hemmings BA, Hunig T, Bommhardt U (2003) Constitutively active protein kinase B enhances Lck and Erk activities and influences thymocyte selection and activation. J Immunol 171: 1285–1296 [DOI] [PubMed] [Google Scholar]
- Parmacek MS, Karpinski BA, Gottesdiener KM, Thompson CB, Leiden JM (1989) Structure, expression and regulation of the murine 4F2 heavy chain. Nucleic Acids Res 17: 1915–1931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parsons MJ, Jones RG, Tsao MS, Odermatt B, Ohashi PS, Woodgett JR (2001) Expression of active protein kinase B in T cells perturbs both T and B cell homeostasis and promotes inflammation. J Immunol 167: 42–48 [DOI] [PubMed] [Google Scholar]
- Patra AK, Na SY, Bommhardt U (2004) Active protein kinase B regulates TCR responsiveness by modulating cytoplasmic-nuclear localization of NFAT and NF-kappa B proteins. J Immunol 172: 4812–4820 [DOI] [PubMed] [Google Scholar]
- Pende M, Um SH, Mieulet V, Sticker M, Goss VL, Mestan J, Mueller M, Fumagalli S, Kozma SC, Thomas G (2004) S6K1(−/−)/S6K2(−/−) mice exhibit perinatal lethality and rapamycin-sensitive 5′-terminal oligopyrimidine mRNA translation and reveal a mitogen-activated protein kinase-dependent S6 kinase pathway. Mol Cell Biol 24: 3112–3124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rathmell JC, Elstrom RL, Cinalli RM, Thompson CB (2003) Activated Akt promotes increased resting T cell size, CD28-independent T cell growth, and development of autoimmunity and lymphoma. Eur J Immunol 33: 2223–2232 [DOI] [PubMed] [Google Scholar]
- Rathmell JC, Vander Heiden MG, Harris MH, Frauwirth KA, Thompson CB (2000) In the absence of extrinsic signals, nutrient utilization by lymphocytes is insufficient to maintain either cell size or viability. Mol Cell 6: 683–692 [DOI] [PubMed] [Google Scholar]
- Sapkota GP, Cummings L, Newell FS, Armstrong C, Bain J, Frodin M, Grauert M, Hoffmann M, Schnapp G, Steegmaier M, Cohen P, Alessi DR (2007) BI-D1870 is a specific inhibitor of the p90 RSK (ribosomal S6 kinase) isoforms in vitro and in vivo. Biochem J 401: 29–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarbassov DD, Ali SM, Sengupta S, Sheen JH, Hsu PP, Bagley AF, Markhard AL, Sabatini DM (2006) Prolonged rapamycin treatment inhibits mTORC2 assembly and Akt/PKB. Mol Cell 22: 159–168 [DOI] [PubMed] [Google Scholar]
- Schmitt TM, Zuniga-Pflucker JC (2002) Induction of T cell development from hematopoietic progenitor cells by delta-like-1 in vitro. Immunity 17: 749–756 [DOI] [PubMed] [Google Scholar]
- Schmitt TM, Zuniga-Pflucker JC (2005) Thymus-derived signals regulate early T-cell development. Crit Rev Immunol 25: 141–159 [DOI] [PubMed] [Google Scholar]
- Schmitt TM, Zuniga-Pflucker JC (2006) T-cell development, doing it in a dish. Immunol Rev 209: 95–102 [DOI] [PubMed] [Google Scholar]
- Schmitt TM, Ciofani M, Petrie HT, Zuniga-Pflucker JC (2004a) Maintenance of T cell specification and differentiation requires recurrent notch receptor-ligand interactions. J Exp Med 200: 469–479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitt TM, de Pooter RF, Gronski MA, Cho SK, Ohashi PS, Zuniga-Pflucker JC (2004b) Induction of T cell development and establishment of T cell competence from embryonic stem cells differentiated in vitro. Nat Immunol 5: 410–417 [DOI] [PubMed] [Google Scholar]
- Schneider YJ, Limet JN, Octave JN, Otte-Slachmuylder C, Crichton RR, Trouet A (1982) The role of receptor-mediated endocytosis in iron metabolism. Prog Clin Biol Res 91: 495–521 [PubMed] [Google Scholar]
- Takahama Y, Ohishi K, Tokoro Y, Sugawara T, Yoshimura Y, Okabe M, Kinoshita T, Takeda J (1998) Functional competence of T cells in the absence of glycosylphosphatidylinositol-anchored proteins caused by T cell-specific disruption of the Pig-a gene. Eur J Immunol 28: 2159–2166 [DOI] [PubMed] [Google Scholar]
- Tanigaki K, Tsuji M, Yamamoto N, Han H, Tsukada J, Inoue H, Kubo M, Honjo T (2004) Regulation of alphabeta/gammadelta T cell lineage commitment and peripheral T cell responses by Notch/RBP-J signaling. Immunity 20: 611–622 [DOI] [PubMed] [Google Scholar]
- Trowbridge IS, Shackelford DA (1986) Structure and function of transferrin receptors and their relationship to cell growth. Biochem Soc Symp 51: 117–129 [PubMed] [Google Scholar]
- Visan I, Yuan JS, Tan JB, Cretegny K, Guidos CJ (2006) Regulation of intrathymic T-cell development by Lunatic Fringe- Notch1 interactions. Immunol Rev 209: 76–94 [DOI] [PubMed] [Google Scholar]
- von Boehmer H, Aifantis I, Feinberg J, Lechner O, Saint-Ruf C, Walter U, Buer J, Azogui O (1999) Pleiotropic changes controlled by the pre-T-cell receptor. Curr Opin Immunol 11: 135–142 [DOI] [PubMed] [Google Scholar]
- Wolfer A, Wilson A, Nemir M, MacDonald HR, Radtke F (2002) Inactivation of Notch1 impairs VDJbeta rearrangement and allows pre-TCR-independent survival of early alpha beta Lineage Thymocytes. Immunity 16: 869–879 [DOI] [PubMed] [Google Scholar]
- Zuniga-Pflucker JC (2004) T-cell development made simple. Nat Rev Immunol 4: 67–72 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1
Supplementary Figure 2
Supplementary Figure 3
Supplementary Figure 4
Supplementary Figure Legends