

Abstract
Homologous recombination (HR) plays a critical role in the restart of blocked replication forks, but how this is achieved remains poorly understood. We show that mutants in the single Rad51 paralog in Caenorhabditis elegans, rfs-1, permit discrimination between HR substrates generated at DNA double-strand breaks (DSBs), or following replication fork collapse from HR substrates assembled at replication fork barriers (RFBs). Unexpectedly, RFS-1 is dispensable for RAD-51 recruitment to meiotic and ionizing radiation (IR)-induced DSBs and following replication fork collapse, yet, is essential for RAD-51 recruitment to RFBs formed by DNA crosslinking agents and other replication blocking lesions. Deletion of rfs-1 also suppresses the accumulation of toxic HR intermediates in him-6; top-3 mutants and accelerates deletion formation at presumed endogenous RFBs formed by poly G/C tracts in the absence of DOG-1. These data suggest that RFS-1 is not a general mediator of HR-dependent DSB repair, but acts specifically to promote HR at RFBs. HR substrates generated at conventional DSBs or following replication fork collapse are therefore intrinsically different from those produced during normal repair of blocked replication forks.
Keywords: C. elegans , homologous recombination, ICL repair, Rad51 paralogs, replication fork barriers
Introduction
The ability of cells to complete DNA replication is essential for the maintenance of genomic integrity and prevention of potentially carcinogenic rearrangements. In order to complete replication, cells must overcome replication fork barriers (RFBs). Endogenous RFBs include DNA secondary structures (e.g., quadruplex DNA), non-histone protein:DNA complexes (e.g., centromeres and replication termination sites), and the intersection of the replication and transcription machinery (e.g., tRNA genes). Treatment of cells with certain agents such as DNA crosslinking agents or camptothecin (CPT) also result in lesions that present a physical barrier to replication.
Several potential roles for homologous recombination (HR) repair (HRR) in responding to RFBs have been proposed, including fork stabilization, replication restart, and nascent strand protection (Courcelle and Hanawalt, 2001; Sogo et al, 2002; Lambert et al, 2005). HRR is an error-free DNA double-strand break (DSB) repair pathway that uses either an intact sister or homologous chromosome to repair the break. Following formation of a DSB, the break is resected, leaving a 3′ssDNA tail, which subsequently becomes coated with replication protein A (RPA) (Krogh and Symington, 2004). BRCA2 has been implicated in the nuclear targeting of RAD51, displacing or preventing RPA accumulation, and loading RAD51 at the resected DSB (Gudmundsdottir and Ashworth, 2006). RAD51, which is the eukaryotic homolog of the bacterial RecA recombinase, is then able to catalyze strand exchange between homologous sequences. In Escherichia coli, the RecFOR proteins are able to promote HRR in the absence of DSBs by promoting RecA loading on ssDNA gaps (Umezu et al, 1993; Umezu and Kolodner, 1994).
In mammalian cells, there are five paralogs of RAD51 (RAD51B, RAD51C, RAD51D, XRCC2, XRCC3), which exhibit 20–30% sequence identity to RAD51 (Thacker, 2005). Although they have been demonstrated to be required for HRR, their exact role has not yet been ascertained. They appear to be essential genes, as RAD51B, RAD51D, and XRCC2 mutations in mice cause embryonic lethality (Pittman et al, 1998; Shu et al, 1999; Deans et al, 2003). Knockout studies in chicken DT40 cells have demonstrated that mutation in any of the RAD51 paralogs renders cells acutely sensitive to DNA interstrand crosslinking agents, while only mildly sensitive to ionizing radiation (IR) (Takata et al, 2001) Furthermore, the same phenotype is observed for RAD51 paralog mutations in hamster CHO cells (Jones et al, 1987; Fuller and Painter, 1988; French et al, 2002). All of the paralogs influence the formation of IR- or interstrand crosslink (ICL)-induced RAD51 foci (Bishop et al, 1998; French et al, 2002; Godthelp et al, 2002).
The paralogs are found in two complexes in cells, a RAD51B–RAD51C–RAD51D–XRCC2 complex (BCDX2) and a RAD51C–XRCC3 complex (CX3) (Masson et al, 2001a, 2001b; Miller et al, 2002; Wiese et al, 2002). Biochemical studies have revealed that a subcomplex of RAD51B and RAD51C can alleviate the inhibitory effect of RPA in vitro and promote ATP-independent strand exchange, the CX3 complex is associated with Holliday junction resolution activity, and the BCDX2 complex preferentially binds Y-shaped DNA and Holliday junctions (Sigurdsson et al, 2001; Lio et al, 2003; Liu et al, 2004; Yokoyama et al, 2004). Data showing alleviation of RPA inhibition, strand exchange, and RAD51 focus formation argue that the paralogs play an early role in HRR, possibly as RAD51 cofactors, preferential binding of Y-shaped DNA and Holliday junction resolution activity associated with the CX3 complex suggest an additional later role in HRR.
Recent work in both Saccharomyces cerevisiae and Schizosaccharomyces pombe has discovered additional Rad51 paralogs to Rad55 and Rad57, a heterodimeric complex that promotes strand exchange by Rad51 in both yeasts (Krogh and Symington, 2004; Raji and Hartsuiker, 2006). Rlp1 and Rdl1 in S. pombe are believed to form a RAD51D–XRCC2-like complex, with the two proteins contributing a Walker A and Walker B ATPase motif, respectively (Khasanov et al, 2004; Martin et al, 2006). Sws1, a novel pro-recombinogenic factor conserved in humans, is also a member of this complex (Martin et al, 2006). It has also been suggested that three proteins identified in a genetic screen for top3 lethality suppressors in S. cerevisiae, Shu1, Shu2, and Psy3 form a complex analogous to the Sws1–Rlp1–Rdl1 complex (Shor et al, 2005; Martin et al, 2006).
Caenorhabditis elegans is increasingly being used as a model to study repair at both DSBs and RFBs. Not only are all the major metazoan DNA repair pathways in C. elegans conserved, but viable mutants are also available, including genes involved in NER, translesion synthesis, mismatch repair, Fanconi anemia, HRR, and non-homologous end joining. The germ line is an invaluable tool for dissecting DNA repair pathways, as it is both temporally and spatially polarized, with cells first progressing through mitosis before passing through meiotic prophase I. The restriction of SPO-11-induced DSBs to a specific region of the meiotic compartment allows separation of factors required for repair of meiotic DSBs from factors required for the repair of replication-induced DSBs. The mitotic compartment has been used to study repair at impeded forks arising from treatment with exogenous agents such as cisplatin, as well as more physiologically relevant endogenous lesions (Collis et al, 2006). A DEAH helicase, DOG-1, was implicated in the prevention of deletions at polyG/C tracts in the C. elegans genome, and is believed to prevent fork stalling by removing secondary structure formed by the G/C tracts (Cheung et al, 2002). Recent work has demonstrated that the HRR proteins RAD-51, BRD-1, and XPF, as well as the TLS polymerases POL eta and POL kappa, contribute to G/C tract stability in the absence of DOG-1 (Youds et al, 2006).
C. elegans possesses a single RAD-51 paralog, rfs-1, which has been previously demonstrated to interact with the C. elegans homologs of RAD51 and BRCA2 (CeBRC-2) (Boulton et al, 2002; Martin et al, 2005). We show here that RFS-1 performs a specialized role in promoting HR-mediated repair at lesions that block replication fork progression. Surprisingly, we demonstrate that RFS-1 does not respond to conventional DSBs and is dispensable for HR-mediated DSB repair. This suggests that RFS-1 is not a general mediator of HR-mediated repair, but rather performs specific roles in facilitating HRR of lesions encountered by the replication fork during S-phase. Our studies reveal striking differences in the generation of HR substrates at DSBs from those produced during the normal repair of replication blocking lesions. Indeed, repair of these replication blocking lesions does not appear to proceed via a conventional DSB intermediate.
Results
Identification of RFS-1 through its interaction with RAD-51
RFS-1 was originally identified as a yeast two-hybrid interacting partner with RAD-51 in a screen to identify novel DNA damage response proteins (Boulton et al, 2002). Subsequent studies not only confirmed the RAD-51 interaction, but also found that RFS-1 interacts with the N-terminal domain of CeBRC-2 (Martin et al, 2005). PSI-BLAST sequence homology searches with RFS-1 reveal a RAD51/DMC1/RADA-like domain present in all Rad51 paralog proteins. Phylogenetic analysis suggests RFS-1 is most related to the RAD51D group of the RAD51 family, and sequence alignments demonstrate conservation of the Walker A and B ATPase motif (Supplementary Figures S1 and S2). To examine the contribution of RFS-1 in HR-mediated repair processes, we characterized rfs-1(ok1372), a deletion mutant that partially removes exon 1 and eliminates the remaining three exons entirely (Supplementary Figure S3A and B). The ok1372 deletion removes the Walker A and B boxes, which in the mammalian RAD51D protein are required for both repair of mitomycin C-induced DNA ICLs and interaction with XRCC2 and RAD51C (Wiese et al, 2006). Unexpectedly, rfs-1 mutants are homozygous viable, contrasting with Cebrc-2 and rad-51 that are essential for viability (Figure 1A). Interestingly, rfs-1 mutants display a high incidence of males (Him) phenotype, indicative of a defect in meiotic chromosome segregation (Figure 1A). The observation of 2.2% male progeny for rfs-1 mutants relative to the 0.1% observed for wild-type (Wt) animals is reminiscent of the weak Him phenotype observed following brc-1 and brd-1 knockdown by RNAi (2.54 and 2.88%, respectively) (Boulton et al, 2004). Like brc-1 and brd-1 mutants, rfs-1 mutants display a Him phenotype without accompanying embryonic lethality, indicating that the chromosome segregation defect is restricted to non-disjunction of the X-chromosome.
Figure 1.
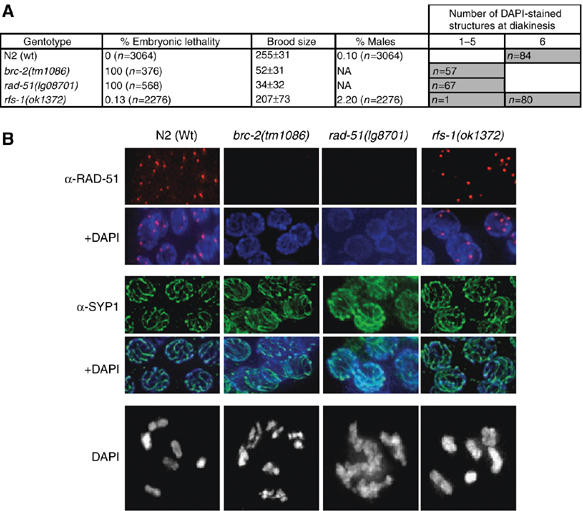
RFS-1 is dispensable for meiotic recombination and crossing over. (A) Table of embryonic lethality, broodsize (± s.d.), % males, and the number of DAPI-stained structures observed at diakinesis in animals of the indicated genotypes (n=number of embryos counted for embryonic lethality and animals scored for % males). (B) Representative images of germ lines of the indicated genotypes stained with either α-RAD-51 or the core SC component α-SYP-1 (MacQueen et al, 2002). Representative images of single oocyte nuclei of the indicated genotypes arrested at diakinesis, counterstained with DAPI.
RFS-1 is dispensable for repair of meiotic DSBs
To further examine the contribution of rfs-1 in meiosis, we performed a detailed cytological examination of the germ line. The C. elegans germ line is polarized in a distal-to-proximal manner, with respect to proliferation and meiotic prophase I. The distal portion of the germ line comprises a zone of mitotic proliferation, which is the only actively dividing cell population in the adult animal. Mitotic cells then enter the leptotene phase of meiotic prophase I, where homologous chromosomes align, synapse, and are held together along their entire length by the synaptonemal complex (SC). Meiotic recombination is initiated by the action of SPO-11 that induces the formation of DSBs that can be detected by the appearance of RAD-51 foci (Dernburg et al, 1998; Alpi et al, 2003; Colaiacovo et al, 2003; Martin et al, 2005). The completion of meiotic prophase in Wt animals produces six bivalents, pairs of homologs held together by a chiasmata, the result of successful crossing over. Analogous to Cebrc-2 and rad-51 mutants, the SC is unperturbed in rfs-1 mutants, as indicated by intact germ line immunostaining against a core component of the SC, SYP-1 (Figure 1B) (MacQueen et al, 2002; Martin et al, 2005). However in contrast to CeBRC-2 and RAD-51, RFS-1 is completely dispensable for repair of meiotic DSBs, as meiotic RAD-51 foci form in rfs-1 mutants and the normal complement of 6 DAPI stained bivalents are observable at diakinesis (Figure 1B).
RFS-1 is required for repair of lesions that block DNA replication
Since the C. elegans BRCA1 homolog (brc-1) is also superfluous for crossover formation in meiosis, but is crucial for repair of IR-induced DSBs, we next assayed whether rfs-1 mutations conferred enhanced sensitivity to IR (Boulton et al, 2004). While irradiated brc-1(tm1145) mutants have extensive embryonic lethality at the relatively low dose (for C. elegans) of 50 Gy irradiation, rfs-1(ok1372) mutants are only moderately sensitive to IR (Figure 2A). Since DT40 cells mutant for any of the RAD51 paralogs are also only mildly sensitive to IR, but are acutely sensitive to ICLs, we next tested the sensitivity of rfs-1 mutants to the crosslinking agents cisplatin (CDDP) and nitrogen mustard (HN2) (Takata et al, 2001). Both brc-1(tm1145) and rfs-1(ok1372) mutations significantly compromised progeny survival, relative to Wt animals (Figure 2B and C). We next assessed sensitivity of rfs-1 mutants to CPT, a topoisomerase I poison that inhibits the enzyme and prevents its release from DNA, creating capped single-ended DSBs (Strumberg et al, 2000) (Figure 2D). Both brc-1(tm1145) and rfs-1(ok1372) mutants exhibit severely reduced progeny survival rates relative to Wt animals following exposure to CPT.
Figure 2.
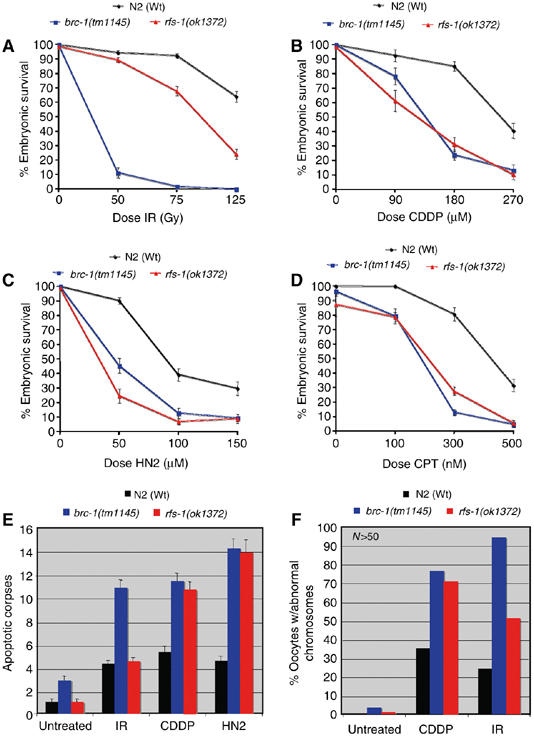
Lesions that impede replication forks cause increased progeny lethality, apoptosis, and chromosomal fragmentation in rfs-1 mutants. (A–D) Percentage progeny survival of N2 (Wt), brc-1(tm1145), and rfs-1(ok1372) animals treated with the indicated doses of ionizing radiation (IR; (A)), cisplatin (CDDP; (B)), nitrogen mustard (HN2; (C)), and CPT (D). Error bars indicate standard error of the mean (s.e.m.) from at least 24 adult worms over two independent experiments. (E) Number of apoptotic corpses scored by DIC microscopy in animals of the indicated genotype before (untreated), or 24 h post-treatment with 75 Gy IR, 180 μM CDDP, or 150 μM HN2. Error bars indicate s.e.m. from at least 20 adult animals. (F) Percentage of examined chromosomes at the diakinesis stage of meiosis I with abnormalities. Young adult animals were dissected and fixed before (untreated) or 24 h post-treatment with 75 Gy IR or 180 μM CDDP, and DNA was observed by counterstaining with DAPI. At least 50 oocyte nuclei at diakinesis were scored for chromosomal abnormalities (e.g., alteration in bivalent number or fragmentation) per strain for each condition. Representative images are shown in Supplementary Figure 5.
To further analyze the role of rfs-1 in DNA repair, we characterized the eDf25 deficiency that deletes rfs-1, in addition to a number of adjacent genes (Supplementary Figure S3C). The Egl phenotype of eDf25 precluded its analysis for sensitivity to DNA damaging agents, as this relies on hatching rates in a narrow window of time. However, an ok1372/eDf25 transheterozygote showed similar sensitivities to IR, CDDP, HN2, and CPT as rfs-1(ok1372) homozygotes, strongly suggesting that DNA damage sensitivity is caused by mutation in rfs-1 (Supplementary Figure S4A). Interestingly both ICLs and CPT form lesions that impede replication forks, causing fork stalling. Thus, it appears RFS-1 may have a specific role in the repair of lesions that physically block replication fork progression.
Since the RAD51 paralogs are implicated in promoting HRR rather than possessing a checkpoint or signal transduction role, it is likely that the sensitivity of rfs-1 mutants to crosslinking agents and CPT is a result of compromised repair. To further examine this possibility, we quantified apoptotic corpses, which are known to be induced by the presence of irreparable or persistent DNA damage, in CDDP- and HN2-treated animals (Boulton et al, 2002). While brc-1(tm1145) animals displayed elevated levels of apoptosis relative to Wt animals for all treatments tested (IR, CDDP, and HN2), rfs-1(ok1372) mutants only displayed increased germ cell death following treatment with crosslinking agents but not IR (Figure 2E).
Previous work analyzing the role of C. elegans FCD-2 in ICL repair had demonstrated that failure to repair trimethylpsoralen-UVA-induced ICLs in fcd-2 mutants leads to chromosomal abnormalities at diakinesis (Lee et al, 2007). We next examined whether the increase in apoptosis following ICL-induced damage was accompanied by chromosomal aberrations. Indeed, both CDDP-treated brc-1(tm1145) and rfs-1(ok1372) mutants showed increased levels of chromosomal abnormalities at diakinesis, relative to Wt animals (Figure 2F; Supplementary Figure S5). However, while irradiated rfs-1(ok1372) mutants have a moderate increase in abnormalities relative to Wt animals, following IR treatment, virtually all chromosomes at diakinesis examined in irradiated brc-1(tm1145) mutants were aberrant (Figure 2F; Supplementary Figure S5). Together, these data underline a severe defect in repairing DNA lesions that impede replication progression in the absence of rfs-1.
We next examined mitotic RAD-51 focus formation in rfs-1 mutants following treatment with IR, CDDP, and HN2. Cebrc-2 and rad-51 mutations eliminated RAD-51 focus formation under all conditions examined (Figure 3). Strikingly, rfs-1(ok1372), eDf25, and ok1372/eDf25 transheterozygotes are all compromised for RAD-51 focus formation following treatment with ICL-inducing agents, whereas IR-induced RAD-51 foci are unaffected (Figure 3; Supplementary Figure 4B and C). This appears to be a defect as opposed to a delay in RAD-51 recruitment, as the defect in rfs-1 mutants persists up to 32 h post-CDDP treatment (Supplementary Figure 6). Thus, it seems likely that the sensitivity, elevated apoptosis, and chromosome abnormalities observed in the absence of rfs-1 following treatment with CDDP and HN2 result from a failure to load RAD-51 specifically at lesions that block DNA replication but not at conventional DSBs (Figure 3).
Figure 3.
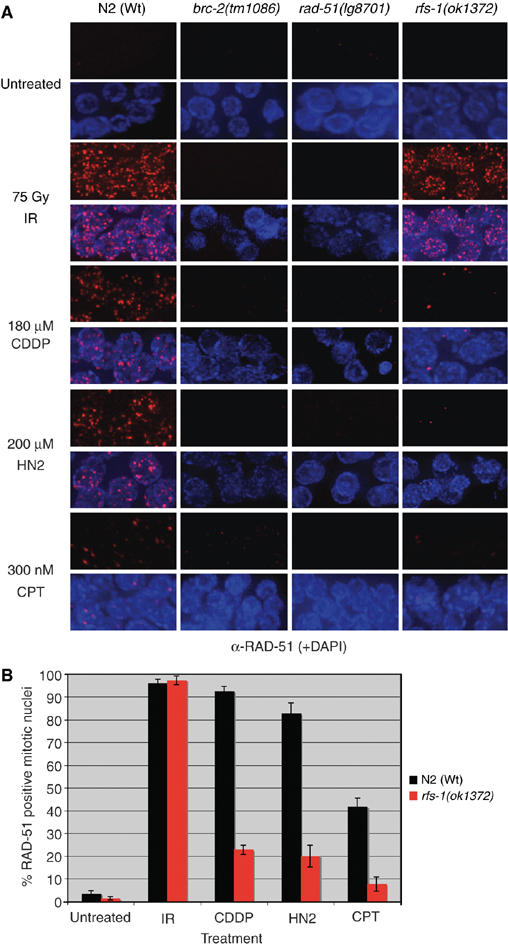
RFS-1 mutants are defective for RAD-51 focus formation specifically following ICL-induced DNA damage. (A) Representative images of RAD-51 staining in fixed mitotic nuclei either before (untreated), 4 h post treatment with 75 Gy IR, 18 h post-treatment with 180 μM CDDP, 16 h post-treatment with 200 μM HN2, or 7 h post-treatment with 300 nM CPT. (B) Quantification of RAD-51-positive N2 (Wt) and rfs-1(ok1372) mitotic nuclei as treated in panel A. Error bars indicate s.e.m. from 20 mitotic nuclei from 10–15 worms of each genotype from two independent experiments.
Since the CPT sensitivity and RAD-51 focus formation data argue that RFS-1 is promoting RAD-51 loading at single-ended DSBs, we wished to examine whether ssDNA gaps generated by UVC are also a substrate for RFS-1-dependent RAD-51 loading (Strumberg et al, 2000). While both Wt animals and mutants for the NER repair factor xpa-1 displayed extensive UVC-induced RAD-51 focus formation, both rfs-1 and rfs-1;xpa-1 mutants exhibited severe reduction in the percentage of RAD-51-positive mitotic cells (Figure 4). Interestingly, rfs-1 mutants are insensitive to UVC and rfs-1;xpa-1 double mutants are no more sensitive than xpa-1 single mutants, suggesting that while RFS-1 is required for RAD-51 loading at UVC induced lesions, it is not critical for repair (Figure 5). This suggests that RFS-1 is able to promote RAD-51 loading at ssDNA gaps as well as at one-ended DSBs.
Figure 4.
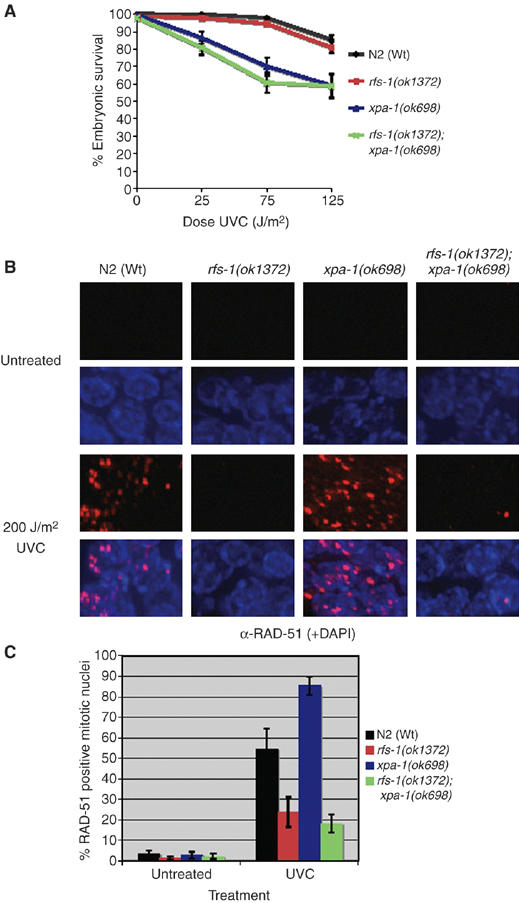
RFS-1 is required for RAD-51 loading at UVC-induced lesions, but not for repair. (A) Percentage progeny survival of animals of the indicated genotype treated with the indicated doses of UVC (254 nm). Error bars indicate standard error of the mean (s.e.m.) from at least 24 adult worms over two independent experiments. (B) Representative images of RAD-51 staining in fixed mitotic nuclei either before (untreated) or 2 h post-treatment with 200 J/m2 UVC. (C) Quantification of RAD-51-positive mitotic nuclei as treated in panel A. Error bars indicate s.e.m. from 20 mitotic nuclei from 10–15 worms of each genotype from two independent experiments.
Figure 5.
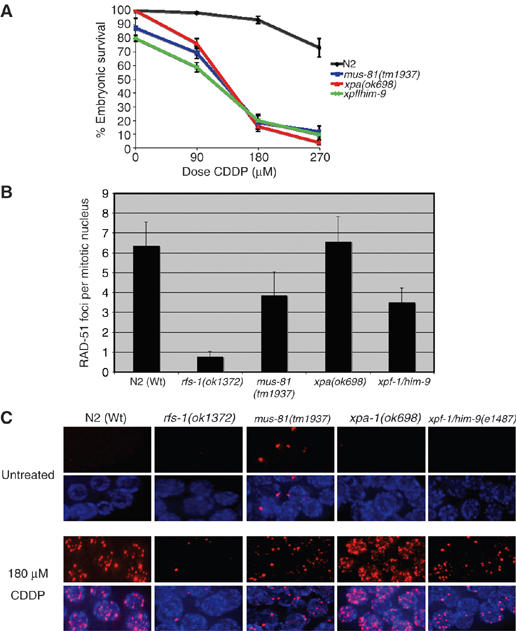
MUS-81 and XPF-1 are involved in HR substrate generation at CDDP-induced lesions. (A) Percentage progeny survival of N2 (Wt), mus-81(tm1937), xpa-1(ok698), xpf-1/him-9(el487) animals following treatment with the indicated doses of CDDP. Error bars indicate s.e.m. from at least 24 adult worms over two independent experiments. (B) Quantification of the number of RAD-51 foci per mitotic nucleus in animals of the indicated genotype dissected 18 h post-treatment with 180 μM CDDP. Error bars indicate s.e.m. from at least 10 nuclei from 20–35 animals of each genotype from three independent experiments. (C) Representative images of RAD-51 staining in fixed mitotic nuclei either before (untreated) or 18 hours post-treatment with 180 μM CDDP.
mus-81 and xpf-1 both contribute to the generation of an HR substrate at ICL lesions
The specific requirement for RFS-1 in repair at replication forks raises two questions; first, how is the HR substrate generated, and second, what is the nature of this HR substrate. Studies in yeast and mammalian cells have demonstrated that nucleolytic incision is required to convert an ICL lesion into a suitable substrate for HRR; however, the identity of the nuclease responsible for this remains debatable (Jachymczyk et al, 1981; Dardalhon and Averbeck, 1995; De Silva et al, 2000; McHugh et al, 2000). Work in yeast has implicated the NER pathways in processing ICLs (Jachymczyk et al, 1981). Two recent studies in mammalian cells have suggested that Mus81 or XPF may be the nucleases responsible for ICL incision (Hanada et al, 2006; Mogi and Oh, 2006). To determine the contribution of Mus81 and NER homologs in the generation of an HR substrate at ICL lesions in C. elegans, we obtained a mutant in mus-81(tm1937) that removes the first two exons and 210 bp of sequence upstream of the translation start site (Supplementary Figure S7A and B), and deletion mutants in both xpf-1/him-9 and xpa-1(ok698) (Park et al, 2002; Denver et al, 2006; O'Neil, 2006, unpublished data). Surprisingly, mus-81, xpa-1, and xpf-1 mutants were all exquisitely sensitive to CDDP (Figure 5A). However, xpa-1 mutants had Wt levels of RAD-51 foci following CDDP treatment, while mus-81 and xpf-1 mutants had reduced, but not abolished levels of RAD-51 foci (Figure 5B and C). This suggests that while MUS-81 and XPF-1 may contribute to HR substrate generation at ICLs in C. elegans, redundancy between these two nucleases exists.
RFS-1 is dispensable for promoting HRR at collapsed replication forks
To further examine the nature of the HR substrate generated at blocked replication forks we examined the role of rfs-1 under different replication stress conditions. To determine if RFS-1 is required to promote RAD-51 loading onto free DNA ends produced following the collapse of stalled replication forks, we utilized an S-phase checkpoint mutant, atl-1 (C. elegans ATR), that exhibits spontaneous RAD-51 foci in the mitotic compartment of the germ line as a result of replication fork collapse (Garcia-Muse and Boulton, 2005). Surprisingly, rfs-1;atl-1 double mutants display similar levels of spontaneous RAD-51 foci to that observed in atl-1 mutants, indicating that RFS-1 is dispensable for promoting HRR at collapsed replication forks (Figure 6A and C). It is known that stalled replication forks frequently collapse to generate DSBs at high doses of hydroxyurea (HU) that inhibits ribonucleotide reductase (Lundin et al, 2002). While Cebrc-2 and rad-51 mutations eradicate HU-induced RAD-51 foci, surprisingly rfs-1 mutants resemble Wt animals with respect to RAD-51 focus formation (Figure 6B and C). The enlargement and reduction in number of mitotic nuclei, indicative of an S-phase arrest, demonstrate that RFS-1 plays no detectable role in the S-phase checkpoint (Figure 6B; Supplementary Figure S8). The specific HRR defects in rfs-1 mutants imply that inherent differences exist between HRR at collapsed forks versus impeded forks.
Figure 6.
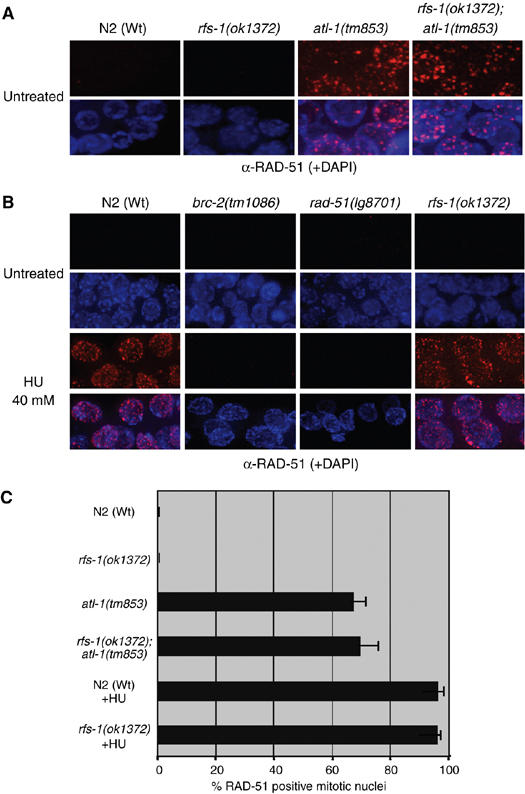
RFS-1 is not required for RAD-51 loading at collapsed replication forks. (A) Representative images of RAD-51 staining in fixed mitotic nuclei in untreated animals of the indicated genotypes. (B) Representative images of RAD-51 staining in fixed mitotic nuclei of the indicated genotypes either before (untreated) or 16 h post-treatment with 40 mM HU. (C) Quantification of RAD-51-positive N2 (Wt) and rfs-1(ok1372) mitotic nuclei as treated in panels A and B. Error bars indicate s.e.m. from 20 mitotic nuclei from 10–15 worms of each genotype from two independent experiments. For HU-treated animals, only 10 mitotic nuclei were scored, due to the reduction in mitotic nuclei number induced by the S-phase arrest.
RFS-1 is required for maintenance of polyG/C tracts in the absence of DOG-1
Similar to RFBs formed by ICL-inducing agents, endogenous polyG/C tracts are believed to form DNA secondary structures that hinder replication fork progression (Arthanari and Bolton, 2001). Data suggests that the DOG-1 (Deletion of G-tracts) helicase prevents deletion formation at polyG/C tracts by unwinding DNA secondary structures formed by these sequences (Cheung et al, 2002). DNA repair proteins including rad-51, brc-1, xpf-1, and him-6 (C. elegans BLM) have been implicated in the maintenance of poly G/C tract integrity in dog-1 mutants (Youds et al, 2006). Similar to previous studies, we observed deletion rates of 11.2 and 37.7% in dog-1 and rad-51;dog-1 animals, respectively (Table I). Correspondingly, rfs-1;dog-1 mutants exhibit a 32.4% deletion rate at the vab-1 poly G/C tract, a 2.9-fold increase relative to dog-1 mutants (Table I; Supplementary Figure S9). Thus rfs-1 is also involved in promoting HRR to maintain endogenous poly G/C tract stability in dog-1 mutants.
Table 1.
Deletion rate in the vab-1 G/C tract
| Genotype | Animals assayed | % animals with deletions | P-value in t-test with dog-1 | Fold increase relative to dog-1 |
|---|---|---|---|---|
| N2 | 95 | 0 | ||
| dog-1 | 428 | 11.2 | 1.0 | |
| dpy-13 rad-51 | 94 | 0 | ||
| dpy-13 rad-51;dog-l | 69 | 37.7 | 0.00085 | 3.4 |
| rfs-1 | 96 | 0 | ||
| rfs-1; dog-1 | 333 | 32.4 | 0.00045 | 2.9 |
| The percentage of animals with deletions is the number of individual animals that showed one or more deletions in the vab-1 locus polyG/C tract (as determined by PCR) divided by the total number of animals assayed. | ||||
rfs-1 mutations suppress mitotic catastrophe in the absence of HIM-6 and TOP-3
Previous work has demonstrated that toxic recombination intermediates that accumulate in the absence of him-6 and top-3 (C. elegans Topoisomerase IIIα) lead to mitotic catastrophe and spontaneous RAD-51 foci in the mitotic compartment of the germ line (Wicky et al, 2004). Given the observation that him-6 and rfs-1 are both required for polyG/C tract stability in the absence of dog-1 we created an rfs-1;him-6 double mutant to determine if the rfs-1 mutation could suppress the mitotic catastrophe phenotype and accumulation of recombination intermediates. As previously described, injection of top-3 dsRNA into Wt animals resulted in spontaneous mitotic RAD-51 foci (Figure 7A and B). Injection of top-3 dsRNA into him-6 animals resulted in further accumulation of mitotic RAD-51 foci and subsequent mitotic catastrophe (Figure 7). Strikingly, rfs-1; top-3 and rfs-1; top-3; him-6 mutants do not accumulate spontaneous mitotic RAD-51 foci and as a consequence mitotic catastrophe is averted (Figure 7). These data suggest that top-3 and him-6 act predominantly on recombination intermediates formed in an rfs-1-dependent manner at impeded replication forks.
Figure 7.
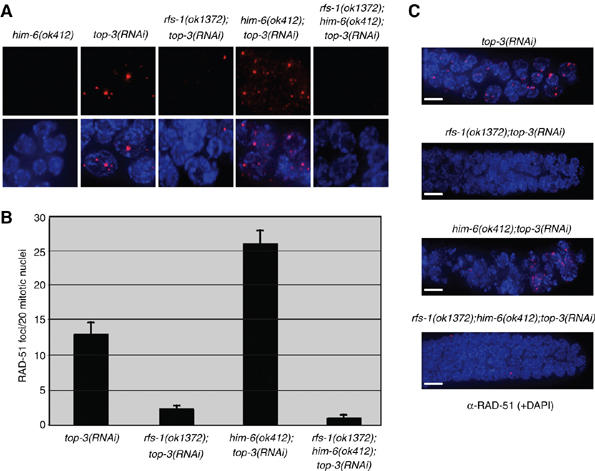
rfs-1 mutants suppress RAD-51 focus formation and mitotic catastrophe caused by the combined loss of HIM-6 and TOP-3. (A) Representative images of RAD-51 staining in fixed mitotic nuclei in untreated animals of the indicated genotypes. (B) Quantification of RAD-51 foci in the first 20 mitotic nuclei in animals of the indicated genotypes. Error bars indicate s.e.m. from at least 22 animals of each genotype from two independent experiments. (C) Representative images of RAD-51 staining in the fixed mitotic zone in animals of the indicated genotype. The severe chromosomal abnormalities in him-6(ok412);top-3(RNAi) animals are suppressed by rfs-1(ok1372). Scale bars, 5 μm.
Discussion
DNA lesions encountered during DNA replication are a major threat to genome integrity. It is known that HRR plays a critical role in maintenance of genome stability through its participation in regeneration of active replication forks at replication blocking lesions. Our study has revealed unexpected differences in the nature of HR substrates at impeded replication forks versus conventional DSBs that support the idea that repair of blocked replication forks does not proceed through a conventional DSB intermediate.
Rad51 paralogs are believed to be general mediators of HRR that act in concert with BRCA2 and Rad51 to promote all HR-mediated repair events. However, our analysis of the single C. elegans Rad51 paralog (RFS-1) suggests that this may not be the case. In contrast to Cebrc-2 and rad-51 mutants, rfs-1 mutants are viable, load RAD-51 onto SPO-11-induced meiotic DSBs, and complete meiotic recombination as normal (Figure 1). This observation, coupled with the fact that rfs-1 is also dispensable for RAD-51 loading and subsequent repair of IR-induced DSBs, suggests that RFS-1 is not a general mediator of HRR (Figures 2 and 3). Although rfs-1 is dispensable for meiotic and IR-induced DSB repair, rfs-1 mutants are profoundly sensitive to agents that impact on replication fork progression (Figure 2). Strikingly, the underlying cause of the sensitivity of rfs-1 mutants to these agents is a severe defect in RAD-51 loading (Figure 3). Rather than acting as a general HRR mediator our data suggests that RFS-1 performs a specialized role in promoting RAD-51 loading onto a substrate unique to blocked replication forks. Initially, an attractive candidate substrate was the free DNA ends produced following the processing/collapse of a blocked replication fork. However, rfs-1 is surprisingly dispensable for RAD-51 focus formation at forks that collapse in the absence of the S-phase checkpoint or following nucleotide depletion via HU treatment (Figure 6). This argues that collapsed forks resemble conventional DSBs similar to those formed by SPO-11 in meiosis or following IR-treatment. The defect in RAD-51 focus formation at both CPT and UVC-induced lesions suggest that the substrate(s) specific to impeded replication forks could be a one-ended DSB and/or a ssDNA gap (Strumberg et al, 2000).
An important issue raised by our findings is how HR substrates are generated at blocked forks. Current models of ICL repair predict that nucleolytic processing of an ICL lesion generates a DSB that creates a substrate for HRR and subsequent repair of the lesion (Dronkert and Kanaar, 2001; Niedernhofer et al, 2005). In yeast, the NER proteins have been implicated in generation of the HR substrate, while in mammalian cells both Mus81 and Xpf have been implicated in substrate generation (Jachymczyk et al, 1981; Hanada et al, 2006; Mogi and Oh, 2006). Our data show that C. elegans xpa-1 mutants are Wt for RAD-51 focus formation after CDDP treatment, indicating that the NER pathway is not generally involved in generation of HR substrates at ICLs in C. elegans (Figure 5). However, while both mus-81 and xpf-1 mutants have reduced RAD-51 foci following CDDP treatment, they fail to phenocopy rfs-1 mutants, indicating that neither is solely responsible for generating an HR substrate at ICL lesions (Figure 5). Although unlikely, an alternative possibility is that RFS-1 could be involved in the generation of a DSB at an ICL during S-phase. This is difficult to address in C. elegans, as only 3–5% of the cells in the adult animal are actively dividing, making assays such as pulsed field gel electrophoresis extremely difficult. Measuring lesions using the comet assay is also hampered by the fact that the germ line is a syncytium, which prevents resolution of individual nuclei. The reduction, but not complete attenuation of RAD-51 foci at ICL lesions, could indicate that functional redundancy exists between mus-81 and xpf-1, with respect to HR substrate generation. Future studies involving the generation of double mutants between mus-81 and xpf-1, as well as other endonucleases implicated in mammalian ICL repair, could further refine the genetic requirements of HR substrate generation at blocked forks.
PolyG/C tracts are believed to form secondary DNA structures that hinder replication fork progression and are expected to be removed by the action of a specialized helicase, DOG-1 (Cheung et al, 2002). A recent study has shown that in the absence of DOG-1, HRR proteins are required to prevent polyG/C tract instability and deletion (Youds et al, 2006). The fact that RFS-1 is also required for stability of polyG/C tracts in dog-1 mutants suggests that these sequences can indeed form endogenous replication blocking lesions that represent a source of spontaneous DNA damage in S-phase (Table I). Interestingly, HIM-6 (C. elegans BLM helicase) is also required for polyG/C tract stability in dog-1 mutants. Combined depletion of HIM-6 and TOP-3 leads to spontaneous RAD-51 foci and mitotic catastrophe (Wicky et al, 2004) analogous to that observed in S-phase checkpoint mutants (Garcia-Muse and Boulton, 2005). The him-6; top-3 phenotype is believed to be produced by the accumulation of toxic recombination intermediates, as rad-51 mutations can suppress these phenotypes, and similarly rad51, rad54, rad55, and rad57 mutations suppress the top3 growth defect in S. cerevisiae (Wicky et al, 2004; Shor et al, 2005). The ability of rfs-1 to suppress the him-6;top-3 phenotype is similar to the ability of mutations in the S. cerevisiae Rad51 paralogs shu1, shu2 and psy3 to suppress top-3 lethality (Shor et al, 2005). In contrast to the inability of rfs-1 to suppress spontaneous RAD-51 focus formation in atl-1 mutants (C. elegans ATR) at collapsed replication forks, the rfs-1 mutation suppresses both RAD-51 focus formation and mitotic catastrophe following him-6 and top-3 depletion (Figures 6 and 7). These data argue that HIM-6 and TOP-3 are predominantly acting on RFS-1 dependent recombination intermediates formed by naturally occurring RFBs. Interestingly, it has been proposed that the Sgs1/Top3 and Mus81/Eme1 function in S. cerevisiae to prevent accumulation of toxic recombination intermediates at ssDNA exposed by stalled replication forks as opposed to DSBs (Fabre et al, 2002). This would be consistent with the observed dependence on RFS-1 for RAD-51 focus formation at UV-induced ssDNA gaps (Figure 4). Further studies of the role of rfs-1 in unperturbed mitosis should illuminate the nature of the endogenous replication blocking lesions RFS-1, HIM-6, and TOP-3 respond to in vivo.
A specialized role for RFS-1 in promoting HRR at blocked replication forks rather than conventional DSBs could be unique to the C. elegans RAD51 paralog, but existing data suggest that this is unlikely. RAD51 paralog knockouts in DT40 cells, whilst acutely sensitive to DNA crosslinking agents, are only mildly sensitive to IR (Takata et al, 2001). Furthermore, RAD51 paralog mutants in CHO cells are also extremely sensitive to DNA crosslinking agents, and only mildly sensitive to IR (Jones et al, 1987; Fuller and Painter, 1988; French et al, 2002). We were surprised that IR-induced RAD-51 foci formed in rfs-1 mutants, given the role of the vertebrate Rad51 paralogs in promoting RAD-51 loading at both IR and ICL-induced lesions and repair at I-SceI-induced DSBs (Bishop et al, 1998; Johnson et al, 1999; Pierce et al, 1999; Takata et al, 2001; French et al, 2002; Godthelp et al, 2002). However, if Rad51 paralogs were general mediators of HRR, one would predict that paralog-deficient cells should phenocopy RAD51 knockout cell lines, which are inviable (Sonoda et al, 1998). This is clearly not the case, and does not appear to be due to functional redundancy between the paralogs, as DT40 double mutants in the same complex (rad51B/rad51d) or both complexes (rad51d/xrcc3) are viable and have similar mild sensitivities to IR as single mutants (Yonetani et al, 2005). It is possible that the decreased RAD51 recruitment to IR-induced lesions in vertebrate cells reflects a defect in loading RAD51 at replication blocking lesions caused by the high doses of IR (8–12 Gy) used in these studies. Consistent with this hypothesis, at doses of 1–3 Gy in XRCC3 defective CHO cells, the defect in RAD-51 loading is much less pronounced (Bishop et al, 1998). Interestingly, while RAD51 and RAD51C levels are highly enriched in S-G2 cell cycle phases (10.2- and 7.5-fold, respectively) at sequences adjacent to an induced I-SceI DSB in human cells, RAD51D, XRCC2, and XRCC3 levels are only enriched 1.62-, 1.56-, and 1.65-fold, respectively (Rodrigue et al, 2006). The presence of RAD51C at DSBs could reflect the postulated late role for RAD51C in DSB repair in mammalian cells (Liu et al, 2004).
Competitive binding studies found that the human BCDX2 complex preferentially binds to branched DNA structures such as Y-shaped DNA and synthetic Holliday junctions that resemble structures believed to form at blocked replication forks (Yokoyama et al, 2004). The preferential binding of BCDX2 to branched DNA over ssDNA, dsDNA, 3′ and 5′ tailed duplexes and nicked DNA is consistent with our proposed replication-specific role for RFS-1 and its vertebrate counterparts (Yokoyama et al, 2004; Rodrigue et al, 2006). RFS-1 could be involved in targeting RAD-51 and CeBRC-2 to impeded forks, or could bind and stabilize impeded forks to facilitate HR substrate generation. Additionally, RFS-1 could promote RAD-51 loading at impeded forks, in order to protect the newly synthesized nascent strands from nucleolytic degradation. This is consistent with the proposed role for the RecFOR proteins in E. coli in loading RecA onto ssDNA at exposed replication forks to prevent degradation by the RecJ nuclease (Umezu et al, 1993; Umezu and Kolodner, 1994; Chow and Courcelle, 2004). RAD51 loading to protect nascent strands rather than active repair has also been proposed to occur at an inducible RFB in S. pombe, as HRR leads to gross chromosomal rearrangements at impeded forks (Lambert et al, 2005). It was proposed that RAD51 loading could promote fork stabilization until specialized helicases/nucleases removed the blocking lesion. It is therefore possible that the role of RFS-1 in promoting HRR at impeded forks may be one of stabilization rather than active participation in repair.
In summary, our study of the single C. elegans Rad51 paralog has revealed that HR substrates generated at impeded replication forks are intrinsically different from substrates generated following replication fork collapse or at conventional DSBs. Our data would suggest that RFS-1 plays a specialized role in promoting RAD-51 loading onto ssDNA gaps generated at stalled replications forks and/or one ended DSBs, potentially formed following replication fork regression. It is likely that further study will allow us to refine the role of the Rad51 paralogs in HRR and gain insight into the nature and function of the HR substrate generated during the normal repair of impeded replication forks.
Materials and methods
Stains and culture conditions
C. elegans strains were cultured and maintained as described previously (Brenner, 1974). The rfs-1(ok1372) strain was generated and kindly provided by the C. elegans Gene Knockout Project at Oklahoma Medical Research Foundation, a part of the International C. elegans Gene Knockout Consortium. rfs-1(ok1372) was backcrossed six times with the Wt Bristol N2 strain. The following strains were kindly provided by the Caenorhabditis Genetics Centre (University of Minnesota, St Paul, MN): Wt Bristol N2, rad-51(lg08701), brc-1(tm1145), him-6(e1423), him-6(ok412), eDf25, and him-9/xpf-1(e1487) (Alpi et al, 2003; Boulton et al, 2004; Wicky et al, 2004). mus-81(tm1937) was kindly provided by Shohei Mitani of the National Bioresource Project for the Nematode, Department of Physiology, School of Medicine, Tokyo Women's Medical University, Tokyo, Japan. atl-1(tm853) and brc-2(tm1086) were described previously (Garcia-Muse and Boulton, 2005; Martin et al, 2005).
For additional methods see Supplementary data.
Supplementary Material
Supplementary Figure 1
{kind=link}
Supplementary Figure 2
{kind=link}
Supplementary Figure 3
{kind=link}
Supplementary Figure 4
{kind=link}
Supplementary Figure 5
{kind=link}
Supplementary Figure 6
{kind=link}
Supplementary Figure 7
{kind=link}
Supplementary Figure 8
{kind=link}
Supplementary Figure 9
{kind=link}
Supplementary Methods
Acknowledgments
We thank the following: Anton Gartner for kindly providing the top-3 cDNA and RAD-51 antibodies, Ann Villeneuve and Monica Colaiacovo for kindly providing SYP-1 antibodies, the National Bioresource Project for the Nematode (Shohei Mitani) and the C. elegans Gene Knockout Consortium (R Barstead) for providing mus-81(tm1937) and rfs-1(ok1372), respectively, Anne Rose for providing dog-1(gk10);rad-51(lg8701), to Nigel O'Neil for providing unpublished information on the nature of the xpf-1/him-9(e1487) allele, and Jillian Youds and members of the Boulton laboratory for technical advice and comments on the manuscript. This work was funded by Cancer Research UK.
References
- Alpi A, Pasierbek P, Gartner A, Loidl J (2003) Genetic and cytological characterization of the recombination protein RAD-51 in Caenorhabditis elegans. Chromosoma 112: 6–16 [DOI] [PubMed] [Google Scholar]
- Arthanari H, Bolton PH (2001) Functional and dysfunctional roles of quadruplex DNA in cells. Chem Biol 8: 221–230 [DOI] [PubMed] [Google Scholar]
- Bishop DK, Ear U, Bhattacharyya A, Calderone C, Beckett M, Weichselbaum RR, Shinohara A (1998) Xrcc3 is required for assembly of Rad51 complexes in vivo. J Biol Chem 273: 21482–21488 [DOI] [PubMed] [Google Scholar]
- Boulton SJ, Gartner A, Reboul J, Vaglio P, Dyson N, Hill DE, Vidal M (2002) Combined functional genomic maps of the C. elegans DNA damage response. Science 295: 127–131 [DOI] [PubMed] [Google Scholar]
- Boulton SJ, Martin JS, Polanowska J, Hill DE, Gartner A, Vidal M (2004) BRCA1/BARD1 orthologs required for DNA repair in Caenorhabditis elegans. Curr Biol 14: 33–39 [DOI] [PubMed] [Google Scholar]
- Brenner S (1974) The genetics of Caenorhabditis elegans. Genetics 77: 71–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung I, Schertzer M, Rose A, Lansdorp PM (2002) Disruption of dog-1 in Caenorhabditis elegans triggers deletions upstream of guanine-rich DNA. Nat Genet 31: 405–409 [DOI] [PubMed] [Google Scholar]
- Chow KH, Courcelle J (2004) RecO acts with RecF and RecR to protect and maintain replication forks blocked by UV-induced DNA damage in Escherichia coli. J Biol Chem 279: 3492–3496 [DOI] [PubMed] [Google Scholar]
- Colaiacovo MP, MacQueen AJ, Martinez-Perez E, McDonald K, Adamo A, La Volpe A, Villeneuve AM (2003) Synaptonemal complex assembly in C. elegans is dispensable for loading strand-exchange proteins but critical for proper completion of recombination. Dev Cell 5: 463–474 [DOI] [PubMed] [Google Scholar]
- Collis SJ, Barber LJ, Ward JD, Martin JS, Boulton SJ (2006) C. elegans FANCD2 responds to replication stress and functions in interstrand cross-link repair. DNA Repair (Amst) 5: 1398–1406 [DOI] [PubMed] [Google Scholar]
- Courcelle J, Hanawalt PC (2001) Participation of recombination proteins in rescue of arrested replication forks in UV-irradiated Escherichia coli need not involve recombination. Proc Natl Acad Sci USA 98: 8196–8202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dardalhon M, Averbeck D (1995) Pulsed-field gel electrophoresis analysis of the repair of psoralen plus UVA induced DNA photoadducts in Saccharomyces cerevisiae. Mutat Res 336: 49–60 [DOI] [PubMed] [Google Scholar]
- De Silva IU, McHugh PJ, Clingen PH, Hartley JA (2000) Defining the roles of nucleotide excision repair and recombination in the repair of DNA interstrand cross-links in mammalian cells. Mol Cell Biol 20: 7980–7990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deans B, Griffin CS, O'Regan P, Jasin M, Thacker J (2003) Homologous recombination deficiency leads to profound genetic instability in cells derived from Xrcc2-knockout mice. Cancer Res 63: 8181–8187 [PubMed] [Google Scholar]
- Denver DR, Feinberg S, Steding C, Durbin M, Lynch M (2006) The relative roles of three DNA repair pathways in preventing Caenorhabditis elegans mutation accumulation. Genetics 174: 57–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dernburg AF, McDonald K, Moulder G, Barstead R, Dresser M, Villeneuve AM (1998) Meiotic recombination in C. elegans initiates by a conserved mechanism and is dispensable for homologous chromosome synapsis. Cell 94: 387–398 [DOI] [PubMed] [Google Scholar]
- Dronkert ML, Kanaar R (2001) Repair of DNA interstrand cross-links. Mutat Res 486: 217–247 [DOI] [PubMed] [Google Scholar]
- Fabre F, Chan A, Heyer WD, Gangloff S (2002) Alternate pathways involving Sgs1/Top3, Mus81/ Mms4, and Srs2 prevent formation of toxic recombination intermediates from single-stranded gaps created by DNA replication. Proc Natl Acad Sci USA 99: 16887–16892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- French CA, Masson JY, Griffin CS, O'Regan P, West SC, Thacker J (2002) Role of mammalian RAD51L2 (RAD51C) in recombination and genetic stability. J Biol Chem 277: 19322–19330 [DOI] [PubMed] [Google Scholar]
- Fuller LF, Painter RB (1988) A Chinese hamster ovary cell line hypersensitive to ionizing radiation and deficient in repair replication. Mutat Res 193: 109–121 [DOI] [PubMed] [Google Scholar]
- Garcia-Muse T, Boulton SJ (2005) Distinct modes of ATR activation after replication stress and DNA double-strand breaks in Caenorhabditis elegans. EMBO J 24: 4345–4355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Godthelp BC, Artwert F, Joenje H, Zdzienicka MZ (2002) Impaired DNA damage-induced nuclear Rad51 foci formation uniquely characterizes Fanconi anemia group D1. Oncogene 21: 5002–5005 [DOI] [PubMed] [Google Scholar]
- Gudmundsdottir K, Ashworth A (2006) The roles of BRCA1 and BRCA2 and associated proteins in the maintenance of genomic stability. Oncogene 25: 5864–5874 [DOI] [PubMed] [Google Scholar]
- Hanada K, Budzowska M, Modesti M, Maas A, Wyman C, Essers J, Kanaar R (2006) The structure-specific endonuclease Mus81-Eme1 promotes conversion of interstrand DNA crosslinks into double-strands breaks. EMBO J 25: 4921–4932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jachymczyk WJ, von Borstel RC, Mowat MR, Hastings PJ (1981) Repair of interstrand cross-links in DNA of Saccharomyces cerevisiae requires two systems for DNA repair: the RAD3 system and the RAD51 system. Mol Gen Genet 182: 196–205 [DOI] [PubMed] [Google Scholar]
- Johnson RD, Liu N, Jasin M (1999) Mammalian XRCC2 promotes the repair of DNA double-strand breaks by homologous recombination. Nature 401: 397–399 [DOI] [PubMed] [Google Scholar]
- Jones NJ, Cox R, Thacker J (1987) Isolation and cross-sensitivity of X-ray-sensitive mutants of V79-4 hamster cells. Mutat Res 183: 279–286 [DOI] [PubMed] [Google Scholar]
- Khasanov FK, Salakhova AF, Chepurnaja OV, Korolev VG, Bashkirov VI (2004) Identification and characterization of the rlp1+, the novel Rad51 paralog in the fission yeast Schizosaccharomyces pombe. DNA Repair (Amst) 3: 1363–1374 [DOI] [PubMed] [Google Scholar]
- Krogh BO, Symington LS (2004) Recombination proteins in yeast. Annu Rev Genet 38: 233–271 [DOI] [PubMed] [Google Scholar]
- Lambert S, Watson A, Sheedy DM, Martin B, Carr AM (2005) Gross chromosomal rearrangements and elevated recombination at an inducible site-specific replication fork barrier. Cell 121: 689–702 [DOI] [PubMed] [Google Scholar]
- Lee KY, Yang I, Park JE, Baek OR, Chung KY, Koo HS (2007) Developmental stage- and DNA damage-specific functions of C. elegans FANCD2. Biochem Biophys Res Commun 352: 479–485 [DOI] [PubMed] [Google Scholar]
- Lio YC, Mazin AV, Kowalczykowski SC, Chen DJ (2003) Complex formation by the human Rad51B and Rad51C DNA repair proteins and their activities in vitro. J Biol Chem 278: 2469–2478 [DOI] [PubMed] [Google Scholar]
- Liu Y, Masson JY, Shah R, O'Regan P, West SC (2004) RAD51C is required for Holliday junction processing in mammalian cells. Science 303: 243–246 [DOI] [PubMed] [Google Scholar]
- Lundin C, Erixon K, Arnaudeau C, Schultz N, Jenssen D, Meuth M, Helleday T (2002) Different roles for nonhomologous end joining and homologous recombination following replication arrest in mammalian cells. Mol Cell Biol 22: 5869–5878 [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacQueen AJ, Colaiacovo MP, McDonald K, Villeneuve AM (2002) Synapsis-dependent and -independent mechanisms stabilize homolog pairing during meiotic prophase in C. elegans. Genes Dev 16: 2428–2442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin JS, Winkelmann N, Petalcorin MI, McIlwraith MJ, Boulton SJ (2005) RAD-51-dependent and -independent roles of a Caenorhabditis elegans BRCA2-related protein during DNA double-strand break repair. Mol Cell Biol 25: 3127–3139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin V, Chahwan C, Gao H, Blais V, Wohlschlegel J, Yates JR III, McGowan CH, Russell P (2006) Sws1 is a conserved regulator of homologous recombination in eukaryotic cells. EMBO J 25: 2564–2574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masson JY, Stasiak AZ, Stasiak A, Benson FE, West SC (2001a) Complex formation by the human RAD51C and XRCC3 recombination repair proteins. Proc Natl Acad Sci USA 98: 8440–8446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masson JY, Tarsounas MC, Stasiak AZ, Stasiak A, Shah R, McIlwraith MJ, Benson FE, West SC (2001b) Identification and purification of two distinct complexes containing the five RAD51 paralogs. Genes Dev 15: 3296–3307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McHugh PJ, Sones WR, Hartley JA (2000) Repair of intermediate structures produced at DNA interstrand cross-links in Saccharomyces cerevisiae. Mol Cell Biol 20: 3425–3433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller KA, Yoshikawa DM, McConnell IR, Clark R, Schild D, Albala JS (2002) RAD51C interacts with RAD51B and is central to a larger protein complex in vivo exclusive of RAD51. J Biol Chem 277: 8406–8411 [DOI] [PubMed] [Google Scholar]
- Mogi S, Oh DH (2006) gamma-H2AX formation in response to interstrand crosslinks requires XPF in human cells. DNA Repair (Amst) 5: 731–740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niedernhofer LJ, Lalai AS, Hoeijmakers JH (2005) Fanconi anemia (cross)linked to DNA repair. Cell 123: 1191–1198 [DOI] [PubMed] [Google Scholar]
- Park HK, Yook JS, Koo HS, Choi IS, Ahn B (2002) The Caenorhabditis elegans XPA homolog of human XPA. Mol Cell 14: 50–55 [PubMed] [Google Scholar]
- Pierce AJ, Johnson RD, Thompson LH, Jasin M (1999) XRCC3 promotes homology-directed repair of DNA damage in mammalian cells. Genes Dev 13: 2633–2638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pittman DL, Weinberg LR, Schimenti JC (1998) Identification, characterization, and genetic mapping of Rad51d, a new mouse and human RAD51/RecA-related gene. Genomics 49: 103–111 [DOI] [PubMed] [Google Scholar]
- Raji H, Hartsuiker E (2006) Double-strand break repair and homologous recombination in Schizosaccharomyces pombe. Yeast 23: 963–976 [DOI] [PubMed] [Google Scholar]
- Rodrigue A, Lafrance M, Gauthier MC, McDonald D, Hendzel M, West SC, Jasin M, Masson JY (2006) Interplay between human DNA repair proteins at a unique double-strand break in vivo. EMBO J 25: 222–231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shor E, Weinstein J, Rothstein R (2005) A genetic screen for top3 suppressors in Saccharomyces cerevisiae identifies SHU1, SHU2, PSY3 and CSM2: four genes involved in error-free DNA repair. Genetics 169: 1275–1289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shu Z, Smith S, Wang L, Rice MC, Kmiec EB (1999) Disruption of muREC2/RAD51L1 in mice results in early embryonic lethality which can Be partially rescued in a p53(−/−) background. Mol Cell Biol 19: 8686–8693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sigurdsson S, Van Komen S, Bussen W, Schild D, Albala JS, Sung P (2001) Mediator function of the human Rad51B–Rad51C complex in Rad51/RPA-catalyzed DNA strand exchange. Genes Dev 15: 3308–3318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sogo JM, Lopes M, Foiani M (2002) Fork reversal and ssDNA accumulation at stalled replication forks owing to checkpoint defects. Science 297: 599–602 [DOI] [PubMed] [Google Scholar]
- Sonoda E, Sasaki MS, Buerstedde JM, Bezzubova O, Shinohara A, Ogawa H, Takata M, Yamaguchi-Iwai Y, Takeda S (1998) Rad51-deficient vertebrate cells accumulate chromosomal breaks prior to cell death. EMBO J 17: 598–608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strumberg D, Pilon AA, Smith M, Hickey R, Malkas L, Pommier Y (2000) Conversion of topoisomerase I cleavage complexes on the leading strand of ribosomal DNA into 5′-phosphorylated DNA double-strand breaks by replication runoff. Mol Cell Biol 20: 3977–3987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takata M, Sasaki MS, Tachiiri S, Fukushima T, Sonoda E, Schild D, Thompson LH, Takeda S (2001) Chromosome instability and defective recombinational repair in knockout mutants of the five Rad51 paralogs. Mol Cell Biol 21: 2858–2866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thacker J (2005) The RAD51 gene family, genetic instability and cancer. Cancer Lett 219: 125–135 [DOI] [PubMed] [Google Scholar]
- Umezu K, Chi NW, Kolodner RD (1993) Biochemical interaction of the Escherichia coli RecF, RecO, and RecR proteins with RecA protein and single-stranded DNA binding protein. Proc Natl Acad Sci USA 90: 3875–3879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Umezu K, Kolodner RD (1994) Protein interactions in genetic recombination in Escherichia coli. Interactions involving RecO and RecR overcome the inhibition of RecA by single-stranded DNA-binding protein. J Biol Chem 269: 30005–30013 [PubMed] [Google Scholar]
- Wicky C, Alpi A, Passannante M, Rose A, Gartner A, Muller F (2004) Multiple genetic pathways involving the Caenorhabditis elegans Bloom's syndrome genes him-6, rad-51, and top-3 are needed to maintain genome stability in the germ line. Mol Cell Biol 24: 5016–5027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiese C, Collins DW, Albala JS, Thompson LH, Kronenberg A, Schild D (2002) Interactions involving the Rad51 paralogs Rad51C and XRCC3 in human cells. Nucleic Acids Res 30: 1001–1008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiese C, Hinz JM, Tebbs RS, Nham PB, Urbin SS, Collins DW, Thompson LH, Schild D (2006) Disparate requirements for the Walker A and B ATPase motifs of human RAD51D in homologous recombination. Nucleic Acids Res 34: 2833–2843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yokoyama H, Sarai N, Kagawa W, Enomoto R, Shibata T, Kurumizaka H, Yokoyama S (2004) Preferential binding to branched DNA strands and strand-annealing activity of the human Rad51B, Rad51C, Rad51D and Xrcc2 protein complex. Nucleic Acids Res 32: 2556–2565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yonetani Y, Hochegger H, Sonoda E, Shinya S, Yoshikawa H, Takeda S, Yamazoe M (2005) Differential and collaborative actions of Rad51 paralog proteins in cellular response to DNA damage. Nucleic Acids Res 33: 4544–4552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Youds JL, O'Neil NJ, Rose AM (2006) Homologous recombination is required for genome stability in the absence of DOG-1 in Caenorhabditis elegans. Genetics 173: 697–708 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1
Supplementary Figure 2
Supplementary Figure 3
Supplementary Figure 4
Supplementary Figure 5
Supplementary Figure 6
Supplementary Figure 7
Supplementary Figure 8
Supplementary Figure 9
Supplementary Methods