

Abstract
The low-density lipoprotein (LDL) receptor (LDLR) binds to and internalizes lipoproteins that contain apolipoproteinB100 (apoB100) or apolipoproteinE (apoE). Internalization of the apoB100 lipoprotein ligand, LDL, requires the FDNPVY807 sequence on the LDLR cytoplasmic domain, which binds to the endocytic machinery of coated pits. We show here that inactivation of the FDNPVY807 sequence by mutation of Y807 to cysteine prevented the uptake of LDL; however, this mutation did not prevent LDLR-dependent uptake of the apoE lipoprotein ligand, β-VLDL. Comparison of the surface localization of the LDLR-Y807C using LDLR-immunogold, LDL-gold and β-VLDL-gold probes revealed enrichment of LDLR-Y807C-bound β-VLDL in coated pits, suggesting that β-VLDL binding promoted the internalization of the LDLR-Y807C. Consistent with this possibility, treatment with monensin, which traps internalized LDLR in endosomes, resulted in the loss of surface LDLR-Y807C only when β-VLDL was present. Reconstitution experiments in which LDLR variants were introduced into LDLR-deficient cells showed that the HIC818 sequence is involved in β-VLDL uptake by the LDLR-Y807C. Together, these experiments demonstrate that the LDLR has a very low-density lipoprotein (VLDL)-induced, FDNPVY-independent internalization mechanism.
Keywords: ARH, fibroblasts, LDL, LDLR, VLDL
Introduction
The low-density lipoprotein (LDL) receptor (LDLR) plays a critical role in reducing the risk of atherosclerosis by binding to and internalizing atherogenic lipoproteins that contain apolipoproteinB100 (apoB100) or apolipoproteinE (apoE). Lipoprotein internalization involves first binding of the lipoprotein at the cell surface, followed by internalization of the LDLR–lipoprotein complex through clathrin-coated pits into clathrin-coated vesicles (Brown and Goldstein, 1986). These vesicles lose their clathrin coats and fuse with sorting endosomes, wherein the low pH of the endosomal lumen promotes the release of the lipoprotein from the LDLR (Maxfield and McGraw, 2004). The receptors recycle back to the cell surface, while the released lipoproteins remain in the endosomal lumen. Sorting endosomes mature into late endosomes through rab conversion and fuse with lysosomes, resulting in the degradation of the internalized lipoproteins (Goldstein et al, 2001; Bright et al, 2005; Rink et al, 2005).
The principal lipoproteins internalized by the LDLR are very low-density lipoprotein (VLDL), VLDL remnants and LDL (Goldstein and Brown, 1974; Kita et al, 1982). VLDL is synthesized in the liver and consists of a triglyceride-rich core surrounded by a shell of phospholipids, cholesterol and apolipoproteins. The apolipoprotein content includes a single copy of apoB100 and multiple copies of apoE (Shelness and Sellers, 2001). VLDL is converted to VLDL remnants in a process involving lipoprotein lipase, which releases fatty acids from the triglycerides carried by VLDL (Hahn, 1943). As compared to VLDL, VLDL remnants have a smaller size, an increased density and a decreased ratio of triglycerides to cholesterol esters (Berman et al, 1978; Innerarity et al, 1986). Internalization of VLDL and VLDL remnants by the LDLR requires apoE but not apoB100 (Weisgraber et al, 1983; Krul et al, 1985). In addition to uptake by the LDLR, VLDL remnants can also be eliminated by conversion to LDL, a pathway that accounts for most LDL production in vivo (Bilheimer et al, 1972; Phair et al, 1975). LDL consists of a cholesterol ester-rich core surrounded by a shell of phospholipids, cholesterol and a single copy of apoB100. The internalization of LDL by the LDLR requires the association of the LDLR with apoB100 (Shireman et al, 1977; Kita et al, 1981). LDL is the principal atherogenic lipoprotein in the circulation and the LDLR plays a central role in reducing the number of LDL both by increasing the rate of LDL clearance and by reducing the production of LDL via uptake of VLDL and VLDL remnants (Kita et al, 1982).
The importance of the LDLR in controlling LDL levels is revealed by loss-of-function mutations, which result in hypercholesterolemia and early onset of coronary artery disease. Among these loss-of-function mutations are variants that can support normal LDL binding, but fail to internalize LDL. The best studied of these internalization-defective alleles is the Y807C mutation, which has been extensively studied using fibroblasts from individual JD, who has one Y807C allele and one null allele for the LDLR (Brown and Goldstein, 1976; Goldstein et al, 1977). JD fibroblasts bind a normal amount of LDL; however, these fibroblasts are largely unable to degrade LDL, because the Y807C LDLR fails to target LDL to clathrin-coated pits for internalization (Anderson et al, 1977). This observation led to the hypothesis that the LDLR has an internalization determinant in the cytoplasmic domain that allows the LDLR to associate with the endocytic machinery in clathrin-coated pits. Characterization of additional internalization-defective mutations led to the identification of the FDNPVY807 sequence in the LDLR cytoplasmic domain as the internalization sequence that is required for LDL uptake by the LDLR (Davis et al, 1987; Chen et al, 1990; Hobbs et al, 1992).
The identification of the FDNPVY807 sequence initiated a search for clathrin-coated pit-associated proteins that could bind the FDNPVY sequence. Many candidate proteins have been identified, including clathrin heavy chain, adaptor protein-2 (AP2), β-arrestin, disabled protein-1 (dab1), disabled protein-2 (dab2) and the autosomal recessive hypercholesterolemia protein (ARH) (Kibbey et al, 1998; Howell et al, 1999; Morris and Cooper, 2001; Boll et al, 2002; He et al, 2002; Mishra et al, 2002; Wu et al, 2003). Of these proteins, only dab2 and ARH appear to play a significant role in targeting the LDLR to coated pits (Michaely et al, 2004; Garuti et al, 2005; Keyel et al, 2006; Maurer and Cooper, 2006). As its name implies, ARH is associated with a recessive form of hypercholesterolemia (Garcia et al, 2001) and individuals lacking ARH (ARH−/−) have LDL clearance rates that are as slow as those of individuals lacking the LDLR (LDLR−/−) (Zuliani et al, 1999; Jones et al, 2003). This observation indicates that liver hepatocytes, which are responsible for LDL clearance in vivo, require ARH for LDLR-dependent uptake of LDL. By contrast, fibroblasts from ARH−/− individuals can internalize and degrade LDL (Harada-Shiba et al, 1991; Zuliani et al, 1995), because fibroblasts express both ARH and dab2, while hepatocytes express only ARH (Keyel et al, 2006).
In contrast to LDL uptake, the role of the FDNPVY sequence on the uptake of apoE containing lipoproteins has not been examined. One reason to suspect that the FDNPVY sequence might be less important for LDLR-dependent uptake of apoE lipoprotein ligands is the observation that while the LDL clearance rates are similar in LDLR−/− and ARH−/− individuals, the circulating level of LDL is significantly lower in ARH−/− individuals as compared to LDLR−/− individuals (Arca et al, 2002; Pisciotta et al, 2006). This difference indicates that less LDL is produced in the absence of ARH than in the absence of the LDLR. A lower LDL production would be expected if ARH is not required for VLDL remnant uptake by the LDLR. Consistent with this possibility, Arh−/− mice have higher rates of clearance of β-VLDL and VLDL than Ldlr−/− mice (Jones et al, 2007). Here, we employed primary fibroblasts from normal, JD, ARH−/− and LDLR−/− individuals to test whether the FDNPVY sequence of the LDLR is required for uptake of β-VLDL, a VLDL remnant-like lipoprotein, that is internalized by the LDLR in an apoE-dependent manner (Mahley et al, 1980; Schneider et al, 1981). We show that inactivation of the FDNPVY807 sequence by the Y807C mutation prevents LDL but not β-VLDL uptake, and provide evidence that the LDLR has a ligand-induced, FDNPVY-independent internalization mechanism that can support uptake of the apoE lipoprotein ligand, β-VLDL.
Results
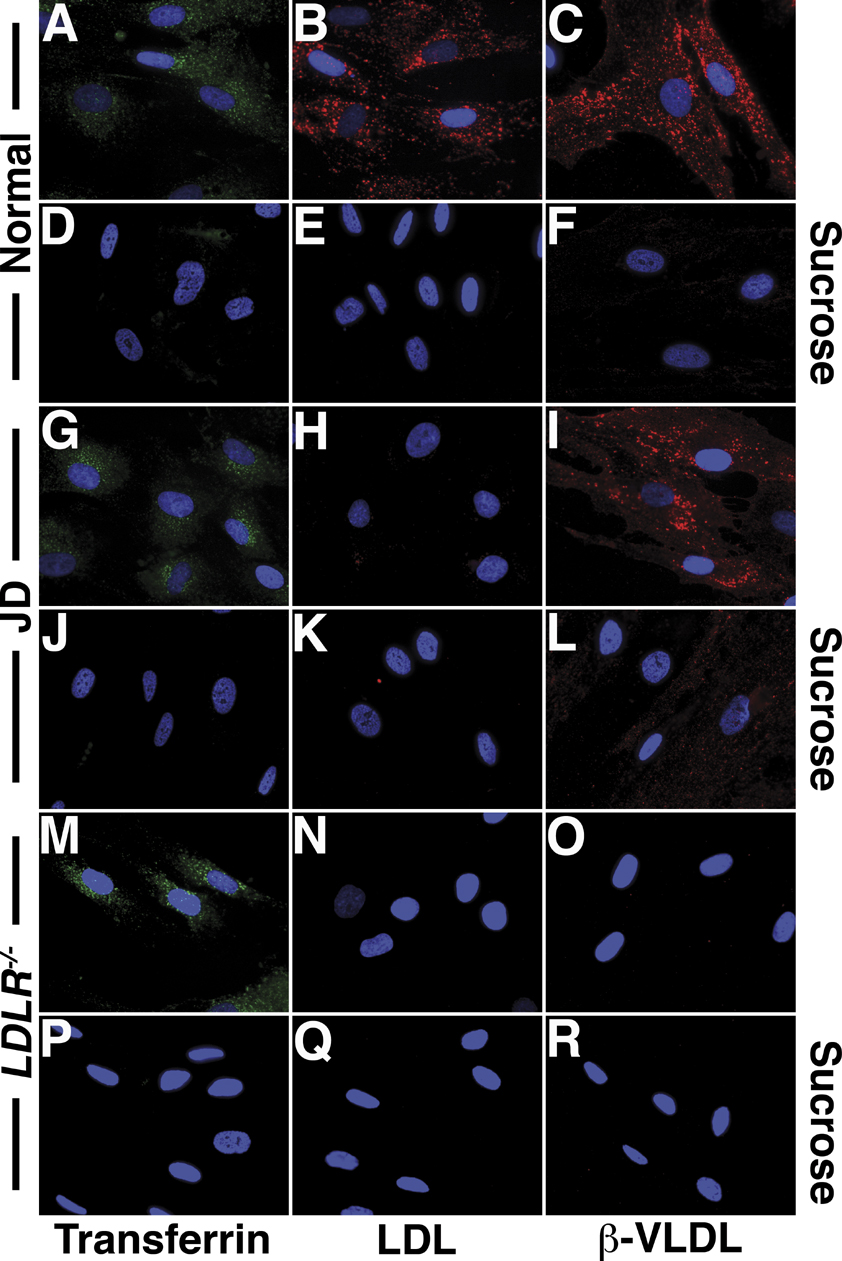
Internalization of the apoB100 lipoprotein ligand, LDL, requires the FDNPVY807 sequence of the LDLR (Davis et al, 1986; Chen et al, 1990). To determine if the FDNPVY807 sequence is required for LDLR-mediated internalization of apoE lipoprotein ligands, we compared uptake of fluorescently labeled LDL and β-VLDL in primary skin fibroblasts from normal, JD, ARH-null (ARH−/−) and LDLR-null (LDLR−/−) individuals (Figure 1). Normal fibroblasts supported robust uptake of both LDL and β-VLDL. JD fibroblasts, which express only the Y807C variant of the LDLR (LDLR-Y807C), were unable to support LDL uptake, but showed substantial internalization of β-VLDL. ARH−/− fibroblasts internalized both LDL and β-VLDL. The LDLR was required for uptake of both LDL and β-VLDL because LDLR−/− fibroblasts failed to internalize either lipoprotein. Thus, the integrity of the canonical FDNPVY807 internalization sequence of the LDLR is required for LDL but not β-VLDL internalization.
Figure 1.
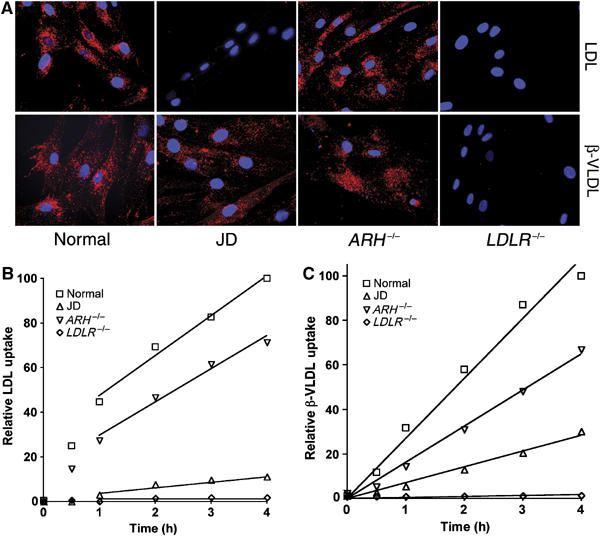
JD fibroblasts can internalize β-VLDL but not LDL. Normal, JD, ARH−/− and LDLR−/− fibroblasts were cultured in lipoprotein poor media for 48 h. (A) Cells were treated with 20 μg/ml Alexa546-labeled LDL or 10 μg/ml DiI-labeled β-VLDL for 2 h at 37°C and visualized by epifluorescent microscopy. (B, C), cells were treated with 10 μg/ml Alexa546-labeled LDL (B) or 5 μg/ml DiI-labeled β-VLDL (C) for 1 h at 4°C, and then shifted to 37°C for the times indicated. Cells were washed, suspended and fixed at 4°C. Mean fluorescent uptake of lipoproteins was determined by flow cytometry and is reported as a percentage of normal cell uptake at 4 h.
The FDNPVY807 sequence binds to the clathrin adaptor proteins, ARH and dab2, which facilitate the association of the LDLR and its bound ligands with clathrin-coated pits (Morris and Cooper, 2001; He et al, 2002; Mishra et al, 2002; Michaely et al, 2004). To determine whether the LDLR-Y807C employed clathrin-coated pits for β-VLDL uptake, β-VLDL uptake was assayed in the presence of hypertonic media, which disrupts clathrin-coated pits (Heuser and Anderson, 1989). As shown in Supplementary Figure S1, hypertonic sucrose blocked uptake of β-VLDL uptake in JD cells, indicating that the LDLR-Y807C employs clathrin-coated pits for β-VLDL uptake.
The finding that the Y807C mutation eliminated LDL but not β-VLDL uptake suggested that β-VLDL might promote the clustering of LDLRs into clathrin-coated pits. To determine the effect of LDL and β-VLDL on the cell surface distribution of the LDLR in the four fibroblasts, we labeled LDLR on the cell surfaces with LDLR-immunogold, LDL-gold or β-VLDL-gold and visualized the LDLR distribution using electron microscopy (Figure 2). For LDLR-immunogold labeling, cells were fixed before immunogold labeling to determine the localization of LDLR in the absence of lipoproteins. For the LDL-gold and β-VLDL-gold labelings, cells were incubated with the labeled lipoprotein for 90 min before fixation, to determine the localization of the LDLR in the presence of lipoproteins. In normal cells, the LDLR-immunogold, LDL-gold and β-VLDL-gold labels were all enriched in coated pits, indicating that the LDLR is localized in clathrin-coated pits in the absence of lipoproteins and in the presence of either LDL or β-VLDL. By contrast, coated pits of JD cells displayed little enrichment of either the LDLR-immunogold label (1.59-fold enrichment) or LDL-gold label (1.65-fold enrichment); however, coated pits of JD cells displayed significant enrichment of the β-VLDL-gold label (14.77-fold). These observations indicate that the Y807C mutation inhibits the ability of the LDLR to accumulate in coated pits in the absence of lipoproteins or in the presence of LDL, but does not prevent the LDLR from accumulating in coated pits in the presence of β-VLDL. The LDL-gold results with normal and JD cells are similar to previous results obtained with these fibroblasts using ferritin-labeled LDL (Anderson et al, 1977). In ARH−/− cells, all three gold labels were enriched in coated pits, indicating that the binding of ARH to the FDNPVY sequence of the LDLR is not required for the targeting of the LDLR to coated pits in fibroblasts, consistent with the ability of ARH−/− fibroblasts to internalize LDL (Harada-Shiba et al, 1991; Zuliani et al, 1995) and β-VLDL (Figure 1). The labeling of cells with LDLR-immunogold, LDL-gold and β-VLDL-gold was LDLR dependent because few gold particles were observed on LDLR−/− cells (Figure 2). Together, these observations indicate that the LDLR-Y807C supports targeting of β-VLDL but not LDL to clathrin-coated pits, and suggest that β-VLDL promotes the accumulation of the LDLR-Y807C in coated pits.
Figure 2.
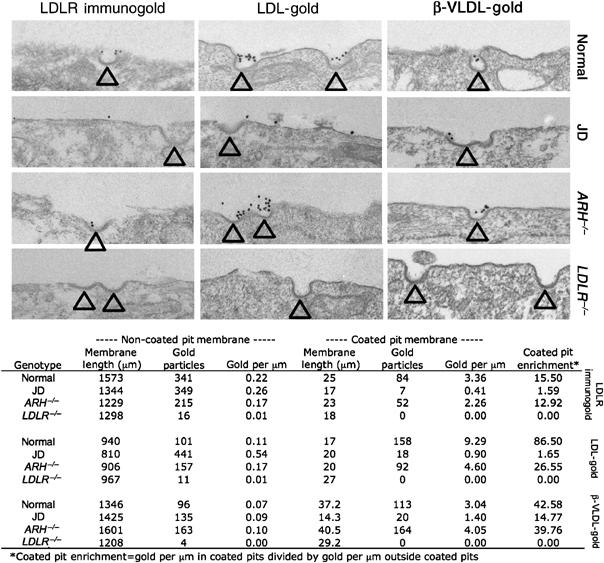
β-VLDL-gold but not LDLR-immunogold or LDL-gold is present in the clathrin-coated pits of JD fibroblasts. Normal, JD, ARH−/− and LDLR−/− fibroblasts were cultured in lipoprotein-poor media for 48 h and then processed for LDLR-immunogold, LDL-gold or β-VLDL-gold surface labeling as described in Materials and methods. Arrowheads indicate coated pits. Quantification of the gold particle distributions is presented below the electron micrographs.
To directly test whether β-VLDL induced the internalization of the LDLR-Y807C, we inhibited LDLR recycling using the proton ionophore, monensin, and examined the loss of surface LDLR over time (Basu et al, 1981; Michaely et al, 2004). LDLRs on the cell surface were labeled with biotin using a non-cell-permeable biotinylation reagent. The biotinylated proteins were isolated using neutravidin–agarose and then immunoblotted to assess the relative amount of surface LDLR (Michaely et al, 2004). Treatment of normal cells with monensin alone, monensin plus LDL or monensin plus β-VLDL significantly reduced the number of LDLRs on the cell surface. By contrast, treatment of JD fibroblasts with monensin alone or monensin plus LDL did not change the number of surface LDLR-Y807C; however, treatment of JD fibroblasts with monensin plus β-VLDL sharply reduced the number of surface LDLR-Y807C (Figure 3A). Treatment with monensin and lipoproteins did not cause a global loss of surface proteins, because no change was observed in the cell surface level of CD44 (Figure 3A), a hyaluronic acid receptor that is not internalized through clathrin-coated pits (Tammi et al, 2001). The amount of LDLR in the cell lysates before separation of the biotinylated material was similar under all conditions, indicating that the loss of surface LDLR was not due to a loss of total cellular LDLR content (Figure 3B). These observations show that β-VLDL promotes internalization of LDLR-Y807C.
Figure 3.
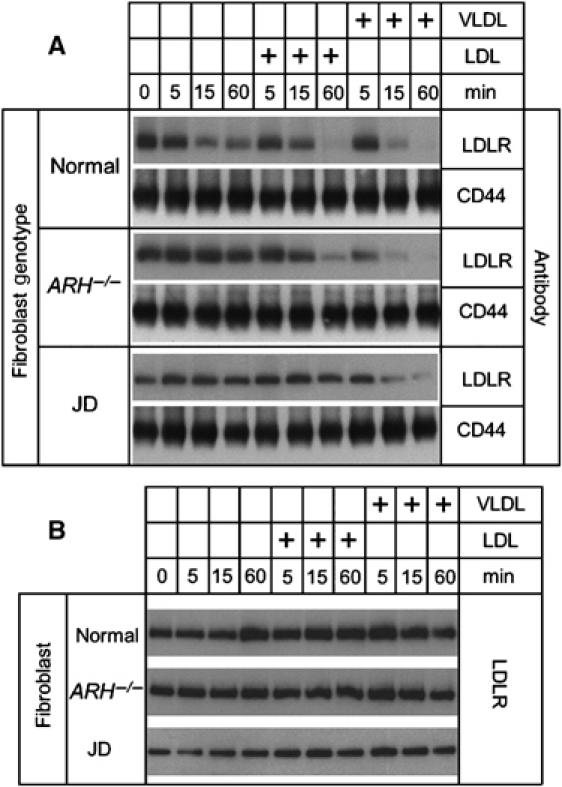
β-VLDL can drive uptake of the LDLR-Y807C of JD fibroblasts. Normal, JD and ARH−/− fibroblasts were cultured in lipoprotein-poor media for 48 h and then treated with monensin alone for 0, 5, 15 or 60 min, or in combination with 20 μg/ml LDL for 5, 15 or 60 min, or in combination with 10 μg/ml βVLDL for 5, 15 or 60 min. Cells were then surface biotinylated and lysed. The biotinylated proteins were isolated from the whole-cell lysates by neutravidin agarose precipitation. (A) Biotinylated proteins were run on 5–17% SDS–PAGE gels, transferred to nylon membranes and immunoblotted for the presence of LDLR or for CD44. (B) The whole-cell lysates were run on 5–17% SDS–PAGE gels, transferred to nylon membranes and immunoblotted for the presence of LDLR.
We next compared the fate of the internalized lipoproteins by incubating the fibroblasts with LDL-gold or β-VLDL-gold at 37°C. LDL-gold accumulated in late endosomes and lysosomes of normal and ARH−/− cells, but not JD cells. In contrast, treatment of fibroblasts with β-VLDL-gold at 37°C resulted in accumulation of β-VLDL-gold in late endosomes and lysosomes of normal, ARH−/− and JD cells (Figure 4). The greater accumulation of LDL-gold in endosomes of normal and ARH−/− cells as compared to β-VLDL-gold is likely due to the more efficient trafficking of LDL to late endosomes (Lombardi et al, 1993; Jones et al, 2000). The relative ability of the four fibroblasts to transport LDL-gold and β-VLDL-gold to late endosomes and lysosomes correlated with their ability to degrade these lipoproteins (Supplementary Figure S2). Because LDLR−/− cells failed to internalize or degraded either LDL or β-VLDL, the uptake and degradation of both LDL and β-VLDL required the presence of the LDLR. Taken together, these results show that the LDLR-Y807C has a ligand-induced, FDNPVY-independent internalization mechanism that can support the coated pit targeting, internalization and degradation of β-VLDL but not LDL.
Figure 4.
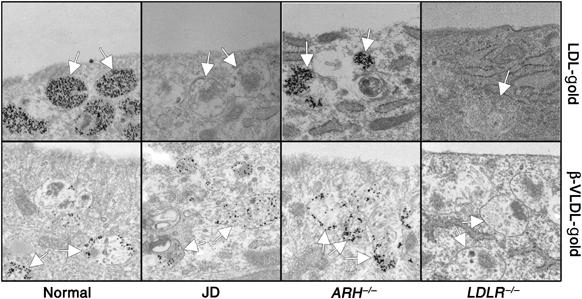
JD fibroblasts can target β-VLDL but not LDL to late endosomes. Normal, JD, ARH−/− and LDLR−/− fibroblasts were cultured in lipoprotein-poor media for 48 h and then treated with LDL-gold or β-VLDL-gold for 90 min at 37°C. Arrows indicate multivesicular bodies, a morphologically distinctive late endosome.
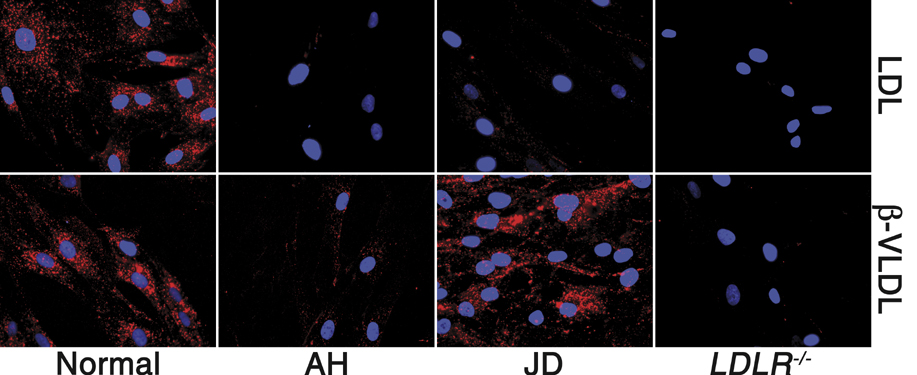
To identify the sequences in the LDLR-Y807C that are required for β-VLDL uptake, we generated a series of GFP-tagged LDLR constructs containing truncations or amino-acid substitutions and assayed their ability to support uptake of fluorescently labeled LDL and β-VLDL in transiently transfected LDLR−/− fibroblasts. Introduction of a stop codon at W792 (W792X) sharply reduced both LDL and β-VLDL uptake, indicating that the cytoplasmic domain is required for uptake of both LDL and β-VLDL (Figure 5 and Supplementary Figure S3). Consistent with this conclusion, neither LDL nor β-VLDL was internalized by fibroblasts from the hypercholesterolemic individual AH (Supplementary Figure S4), who is homozygous for the W792X allele of the LDLR (Lehrman et al, 1985). Introduction of a stop codon at residue 823 or 829 in the LDLR-Y807C had little effect on the ability of the LDLR to support β-VLDL uptake, whereas introduction of a stop codon at residue 812 or 817 of the LDLR-Y807C markedly reduced uptake of β-VLDL (Figure 5). These data suggest that the residues between 817 and 823 are involved in β-VLDL uptake by the LDLR-Y807C. Alanine substitution of amino acids 816–818 (but not 812–814, 814–816 or 818–820) markedly reduced internalization of β-VLDL by the LDLR-Y807C (Figure 5). β-VLDL uptake was not observed in cells not expressing the GFP-LDLR constructs, indicating that uptake required expression of the LDLR. Uptake was specific for β-VLDL because little uptake of LDL was observed in cells expressing GFP-LDLR constructs with the Y807C mutation (Supplementary Figure S3). These observations suggest that β-VLDL uptake by the LDLR-Y807C involves the HIC818 sequence.
Figure 5.
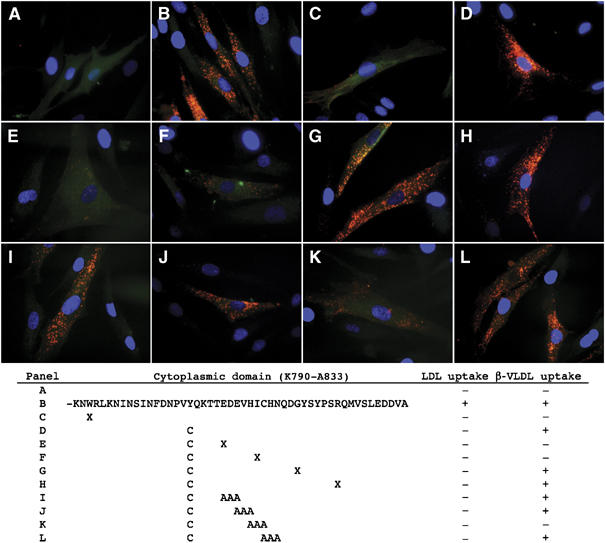
The HIC818 sequence is required for β-VLDL uptake by the LDLR-Y807C. LDLR−/− fibroblasts were transfected with plasmids that direct the expression of GFP (A), GFP-LDLR (B), GFP-LDLR-W792X (C), GFP-LDLR-Y807C (D), GFP-LDLR-Y807C+E812X (E), GFP-LDLR-Y807C+I817X (F), GFP-LDLR-Y807C+G823X (G), GFP-LDLR-Y807C+R829X (H), GFP-LDLR-Y807C+EDE814>AAA (I), GFP-LDLR-Y807C+EVH816>AAA (J), GFP-LDLR-Y807C+HIC818>AAA (K) or GFP-LDLR-Y807C+CHN820>AAA (L). Cells were then plated on coverslips, treated with lipoprotein poor media, incubated at 37°C with DiI-β-VLDL for 2 h and visualized for the presence of β-VLDL (red), GFP- (green) and DAPI-stained nuclei (blue). Shown below the fluorescent images is a diagram detailing the mutations and their effect on lipoprotein uptake.
To better assess the relative importance of the HIC818 sequence on β-VLDL uptake, we introduced the normal, HIC818>AAA, Y807C, Y807C+HIC818>AAA and W792X variants of the LDLR into LDLR−/− fibroblasts, using retroviruses. To avoid the possibility that fusion of GFP to the LDLR altered uptake, we generated retroviruses that allowed bicistronic expression of the LDLR and GFP through the presence of an internal ribosomal entry site, which separated the two coding sequences. Thus, because both the LDLR and GFP were produced from the same message, cells with GFP expression (green fluorescence) also expressed an LDLR and the amount of GFP expression is proportional to the amount of LDLR expression (Liu et al, 1997). Comparison of LDL uptake showed that cells expressing LDLR variants with the Y807C mutation were unable to internalize LDL, and that the HIC818>AAA mutation had little impact on LDL uptake (Supplementary Figure S5A). By contrast, comparison of β-VLDL uptake showed that the LDLR-Y807C was able to support approximately half the β-VLDL uptake of the normal LDLR, and that the HIC818>AAA mutation reduced β-VLDL uptake of the LDLR-Y807C by half (Supplementary Figure S5B). These observations indicate that approximately half of the β-VLDL uptake activity of the LDLR-Y807C depends on the HIC818 sequence.
Uptake at steady state involves endocytosis, release of bound lipoprotein in the endosome and recycling of unbound receptors back to the cell surface. To determine whether the reduced β-VLDL uptake observed with the LDLR-Y807C+HIC818>AAA was caused by a reduced rate of β-VLDL endocytosis, we measured initial rates of β-VLDL endocytosis. For these experiments, cells infected with viruses that direct expression of normal, HIC818>AAA, Y807C, Y807C+HIC818>AAA LDLR variants were sorted by fluorescence-activated cell sorting (FACS) to produce cell populations in which >95% of the cells expressed GFP and had LDLR on their cell surfaces (data not shown). The total LDLR expression by the infected fibroblasts was similar to the endogenous level expressed by normal human fibroblasts (Figure 6A). Comparison of the initial rates of uptake of 125I-β-VLDL showed that the HIC818>AAA mutation reduced by half the rate of 125I-β-VLDL endocytosis supported by the LDLR-Y807C (Figure 6B and C). These observations indicate that the reduced uptake of β-VLDL by the LDLR-Y807C+HIC818>AAA results from a reduced ability to internalize β-VLDL.
Figure 6.
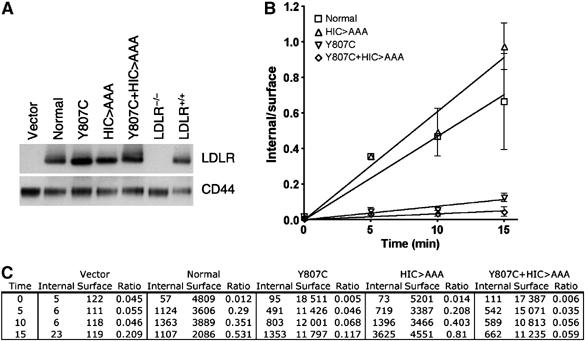
Mutation of the HIC818 sequence reduces the rate of β-VLDL uptake by the Y807C LDLR by half—LDLR−/− fibroblasts were infected with bicistronic retroviruses encoding just GFP (vector) or the normal, Y807C, HIC818>AAA or Y807C+HIC818>AAA variants of the LDLR, together with GFP. Cells were sorted based upon green fluorescence to >95% homogeneity. (A) A 5 μg weight of total cell lysate from each fibroblast was run on SDS–PAGE gels, transferred to nylon membranes and immunoblotted for the LDLR or for CD44. (B, C) Cells were incubated with 10 μg/ml 125I-β-VLDL for 1 h at 4°C, and then shifted to 37°C for 0, 5, 10 or 15 min. Internalized and surface-bound 125I-β-VLDL were separated as described in Materials and methods, and the ratio is presented in panel B Individual values for internalized and surface-bound β-VLDL are presented in panel C Each point is the mean of four determinations. Error bars show the standard deviation. The experiment shown is representative of three independent experiments.
Discussion
The central finding of this study is that the Y807C mutation in the LDLR (LDLR-Y807C), which disrupts the canonical internalization motif (FDNPVY807), prevented internalization of LDL but not β-VLDL. JD fibroblasts, which express only the LDLR-Y807C, were able to internalize β-VLDL in a clathrin-coated pit-dependent manner, but were unable to internalize LDL (Figure 1 and Supplementary Figure S1). When β-VLDL-gold was added to JD fibroblasts, the label accumulated in coated pits, which is consistent with uptake occurring through clathrin-coated pits (Figure 2). Uptake was specific for β-VLDL because no coated pit enrichment was seen when the LDLR-Y807C was labeled with LDLR-immunogold or LDL-gold (Figure 2), and because β-VLDL but not LDL promoted the internalization of the LDLR-Y807C (Figure 3). The internalized β-VLDL trafficked to late endosomes and lysosomes (Figure 4) and was degraded (Supplementary Figure S2) by the JD fibroblasts. The inability of LDLR−/− fibroblasts to bind (Figure 2), internalize (Figures 1 and 4) or degrade (Supplementary Figure S2) β-VLDL indicated that β-VLDL uptake by JD fibroblasts required the LDLR-Y807C. These findings suggest that the LDLR has an ancillary mechanism, independent of the FDNPVY sequence, which allows ligand-dependent uptake of β-VLDL in JD cells. This novel mechanism appears to involve the HIC818 sequence in the LDLR cytoplasmic domain, because mutation of these residues to alanine reduced the ability of the LDLR-Y807C to support β-VLDL uptake (Figures 5 and 6 and Supplementary Figure S5).
Together, these observations show that the LDLR is more than a simple, constitutive, endocytic receptor. Endocytic receptors can be classified into two categories: constitutive and ligand dependent (Hopkins et al, 1985). Ligand-dependent endocytic receptors, such as the EGF receptor, are efficiently targeted to coated pits and internalized only in the presence of their ligands (Gorden et al, 1978; Wiley et al, 1991). By contrast, constitutive endocytic receptors, such as the transferrin receptor, are targeted to coated pits and internalized in both the presence and absence of ligand (Willingham et al, 1984; Ajioka and Kaplan, 1986). The LDLR qualifies as a constitutive receptor because the it is present in coated pits in the absence of LDL (Anderson et al, 1982; Figure 2), and because the LDLR can be internalized in the absence of LDL (Basu et al, 1981; Figure 3); however, the results of this study indicate that the LDLR can also function as a ligand-dependent endocytic receptor for the apoE-dependent uptake of the VLDL remnant-like lipoprotein, β-VLDL. Thus, the LDLR functions as a hybrid endocytic receptor with both constitutive and ligand-induced properties.
Uptake by endocytic receptors involves internalization determinants that function by associating with components of clathrin-coated pits. The NPXY and YXXΦ motifs are examples of constitutive determinants that bind to coated pit components in the presence or absence of bound ligands (Bonifacino and Traub, 2003). Ligand-induced determinants frequently involve post-translational modifications that are triggered by ligand binding. Common examples include phosphorylation and ubiquitination events (Bonifacino and Traub, 2003; Lefkowitz and Whalen, 2004). The LDLR can be phosphorylated (Kishimoto et al, 1987), however, the phosphorylation site at S833 does not appear to be involved in β-VLDL uptake by the LDLR-Y807C, because truncation of the LDLR-Y807C at R829 did not appear to reduce β-VLDL uptake (Figure 5). Ubiquitination of the LDLR is also unlikely because ubiquitination of receptors frequently results in their degradation (Hurley and Emr, 2006), yet JD cells showed no loss in their ability to internalize β-VLDL over time (Figure 1). Ubiquitination may play a role through clathrin adaptor proteins that can be ubiquitinated (Lefkowitz and Whalen, 2004), and future experiments will address this possibility. Ligand-induced endocytosis can also involve receptor dimerization (Bonifacino and Traub, 2003). The LDLR can form dimmers, and interestingly, C818 is likely in or near the dimerization surface because dimerization requires residues 812–839 and is sensitive to the sulfhydryl alkylating agent, N-ethylmaleimide (van Driel et al, 1987). β-VLDL binding may promote LDLR dimerization because β-VLDL, like other VLDL species, has multiple copies of apoE and can engage multiple LDLR simultaneously (Windler et al, 1980). VLDL-induced dimerization may generate a novel binding site for a coated pit component. Future work with cells stably expressing LDLR variants will address this possibility.
A second question is why the LDLR has a separate, ligand-induced mechanism for the internalization of VLDL. One possibility suggested by our data is that the VLDL-induced mechanism augments VLDL uptake by the normal receptor. β-VLDL-gold labeling of the normal LDLR showed higher coated pit enrichment (44-fold) than the immunogold labeling (15-fold) in the absence of lipoproteins (Figure 2), suggesting that the binding of β-VLDL-gold promoted the accumulation of the LDLR in coated pits. In support of this possibility, co-treatment of normal fibroblasts with monensin and β-VLDL resulted in a more complete loss of LDLR from the cell surface than was observed with monensin alone (Figure 3).
The presence of a VLDL-specific mechanism for the uptake of the LDLR suggests that the LDLR can function solely as an endocytic receptor for VLDL. The degree to which the LDLR functions in LDL or VLDL uptake may be modulated by the relative expression or activity of adaptor proteins, which bind to the FDNPVY sequence. Perhaps by modulating the expression of these adaptors, the function of the LDLR can be altered from a dual specificity apoB100/apoE receptor to a ligand-induced, apoE-specific receptor. This change may play a role in maintaining lipoprotein homeostasis under different nutritional states, or allow uptake of VLDL but not LDL in specific tissues. Lung, for example, expresses moderate levels of the LDLR, but does not express ARH or dab2 (Fazili et al, 1999; Garcia et al, 2001), and organ perfusion studies have shown that lung will absorb VLDL and VLDL remnants but not LDL (Pietra et al, 1976). Liver may also employ the FDNPVY-independent mechanism because Arh−/− mice are able to clear VLDL and β-VLDL faster than Ldlr−/− mice (Jones et al, 2007). Interestingly, ARH deficiency in humans is characterized by the formation of large xanthomas, which are cholesterol ester-rich deposits formed by macrophages (Khachadurian and Uthman, 1973; Harada-Shiba et al, 2003). VLDL remnants such as β-VLDL are potent activators of cholesterol ester production in macrophages (Goldstein et al, 1980; Mahley et al, 1980), and an FDNPVY-independent mechanism of VLDL remnant uptake may contribute to xanthoma formation in ARH deficiency. Consistent with this possibility, previous studies suggest that macrophages internalize β-VLDL via a mechanism that is distinct from that of LDL uptake (Tabas et al, 1990; Myers et al, 1993).
In summary, we have shown that the LDLR has a ligand-induced, FDNPVY-independent internalization mechanism that can support uptake of β-VLDL but not LDL when the FDNPVY sequence is inactivated by the Y807C mutation. This novel internalization mechanism appears to involve the HIC818 sequence in the LDLR cytoplasmic domain.
Materials and methods
Materials
All cell culture reagents were purchased from Gibco (Carlsbad, CA). The LDLR−/− and JD fibroblasts were a gift from Michael Brown and Joseph Goldstein (Department of Molecular Genetics, UT Southwestern Medical Center, Dallas, TX). The Normal and ARH−/− fibroblasts were a gift from Sebastiano Calandra and Roberta Tiozzo (Department of Biomedical Sciences, University of Modena, Italy). Rabbit anti-LDLR IgG used for electron microscopy was from Maine Biotechnology Services (Portland, ME). Rabbit anti-LDLR IgG used for immunoblotting was a gift from Joachim Herz (Department of Molecular Genetics, UT Southwestern Medical Center, Dallas, TX). Goat anti-rabbit IgG (10 nm gold labeled) and PD-10 columns were from Amersham (Piscataway, NJ). Formaldehyde was from Fluka (Buchs, Switzerland). Alexa546 succinimidyl ester was from Molecular Probes (Carlsbad, CA). All other chemicals were from Sigma (St Louis, MO). LDL was prepared from freshly drawn human plasma, as previously described (Goldstein et al, 1983). Beta migrating VLDL (β-VLDL) was prepared from the serum of cholesterol fed rabbits, as previously described (Kovanen et al, 1981).
Cell culture
Fibroblasts, isolated from normal, ARH-deficient and LDLR-deficient subjects, and from JD, were cultured in media A (DMEM media supplemented with 10% (v/v) fetal bovine serum, 20 mM HEPES pH 7.5, 100 U/ml penicillin G and 100 μg/ml streptomycin). LDLR expression was upregulated by replacing media A with media B (DMEM media supplemented with 10% (v/v) lipoprotein-poor serum, 20 mM HEPES pH 7.5, penicillin G (100 U/ml) and streptomycin (100 μg/ml)) for 48 h.
Electron microscopy
Colloidal gold-conjugated LDL (LDL-gold) and colloidal gold-conjugated β-VLDL (β-VLDL-gold) were produced using methods established for the production of LDL-gold (Handley et al, 1981; Michaely et al, 2004). Cell surface immunolabeling was performed as previously described (Michaely et al, 2004). Surface labeling and uptake with LDL-gold and β-VLDL-gold were performed by incubation at either 4 or 37°C, in the presence of 10 μg/ml of LDL-gold or β-VLDL-gold for 90 min. The cells were washed three times using buffer A (PBS plus 2 mg/ml BSA, pH 7.4) and fixed in buffer B (3% (w/v) paraformaldehyde in PBS). The cells were then embedded, sectioned and visualized as previously described, using a JEOL 1200 electron microscope operated at 80 kV (Michaely et al, 2004).
Quantification of gold labeling
Electron microscope images were obtained by taking 50 micrographs of each cell type (normal, ARH−/−, LDLR−/− and JD) that had been labeled with LDLR-immunogold, LDL-gold or β-VLDL-gold. Micrographs were coded and the length of the non-coated pit membranes, the diameter of the coated pits and the number of gold particles associated with each of these two regions were determined by a naive observer. The labeling intensity was expressed as the number of gold particles per micrometer length of the different regions of the plasma membrane. Clusters of tightly associated gold particles were assumed to be aggregates and were counted as one particle. Gold particles separated by a gap greater than twice their diameter were counted as single particles.
Lipoprotein degradation assays
Human 125I-LDL and rabbit 125I-β-VLDL degradation assays were performed in triplicate at 37°C for 5 h using 15 μg/ml 125I-LDL or 15 μg/ml 125I-β-VLDL (Goldstein et al, 1983).
Monensin treatment and analysis of surface biotinylated proteins
Fibroblasts were treated with 25 μM monensin alone or in combination with 20 μg/ml LDL or 10 μg/ml β-VLDL for the times indicated. Cells were washed with ice-cold PBS and incubated with buffer C (1 mg/ml Sulfo-NHS-(LC)-biotin (Pierce) in PBS, pH 8.0) for 30 min at 4°C. Cells were then washed with ice-cold PBS and incubated with buffer D (100 mM NaCl and 20 mM Tris pH 8.0) for 30 min at 4°C. Cells were lysed in 1 ml buffer E (1% Triton X-100, 4 mM EGTA, 10 mM Tris pH 8.0) and lysates clarified by centrifugation for 15 min at 20 000 g at 4°C. A 100 μl volume of the lysate was set aside to compare total LDLR content. Biotinylated proteins from 800 μl of the lysate were precipitated by addition of 100 μl of a 50% slurry of neutravidin agarose (Pierce), followed by end over end mixing for 1 h at 4°C. Biotinylated proteins bound to the agarose were washed three times with buffer F (15 mM Tris pH 8.0, 500 mM NaCl, 4 mM EGTA and 0.5% Triton X-100) and once with buffer G (15 mM Tris pH 8.0, 4 mM EGTA and 0.5% Triton X-100). Biotinylated proteins were eluted from neutravidin agarose by the addition of an equal volume (50 μl) of 5 × SDS–PAGE sample buffer and heating to 100°C for 10 min. Samples were loaded on 5–17% SDS–PAGE gels, transferred to PVDF membranes and immunoblotted for LDLR or for CD44 (Chemicon).
Introduction of LDLR variants into LDLR−/− fibroblasts
For the GFP-LDLR fusions, the signal sequence of the human LDLR was inserted into the AgeI site at the 5′ end of GFP in the pEGFP-C2 vector. The remainder of the human LDLR was cloned into the HindIII/KpnI sites at the 3′ end of GFP. Mutations were introduced into the LDLR by site-directed mutagenesis (Quikchange, Stratagene). GFP-LDLR expression plasmids were introduced into LDLR−/− fibroblasts by electroporation (AMAXA). For the LDLR retroviruses, LDLR variants were cloned into the pMX-IRES-GFP bicistronic retroviral vector (Liu et al, 1997). Retroviral vectors were cotransfected with the pAmpho vector (Clontech) into 293T cells to produce infectious, replication-defective retroviruses. Retroviruses were added to LDLR−/− fibroblasts in the presence of hexadimethrine bromide to promote viral entry.
Flow cytometry
Cells were incubated with either 10 μg/ml Alexa546-labeled LDL or 5 μg/ml DiI-labeled β-VLDL in media C (bicarbonate-free DMEM supplemented with 20 mM HEPES pH 7.5, and 10% HLPPS) for 1 h at 4°C. The media was then replaced with warm media B containing 10 μg/ml 546-LDL or 5 μg/ml DiI-β-VLDL for the times indicated. Cells were washed with ice-cold PBS, suspended by scraping in PBS and fixed in the presence of 3% paraformaldehyde. Cells were washed with PBS and analyzed by flow cytometry on a FACS Calibur (BD Biosciences). For the determination of LDL and β-VLDL uptake by endogenous LDLRs, the mean fluorescence intensities were recorded for 10 000 events. For the determination of LDL and β-VLDL uptake by LDLRs introduced by retroviruses, intact GFP-positive cells were gated and the mean green (FL1) and red (FL2) fluorescence values were recorded. The FL2 fluorescence values were divided by the FL1 fluorescence values and are reported as a percentage of the uptake supported by the normal LDLR.
FACS
Cells infected with retroviruses were sorted twice based on green (GFP) fluorescence, using a MoFlo High Performance Cell Sorter (Dako). Purity was 90% after the first sorting and >95% after the second sorting.
Initial endocytic rates
Initial endocytic rates were determined as previously described (Lombardi et al, 1993). Briefly, cells were incubated with 10 μg/ml 125I-β-VLDL for 1 h at 4°C in media C. Media was replaced for the times indicated with warm media B, also containing 10 μg/ml 125I-β-VLDL. Cells were extensively washed with ice-cold PBS and incubated with 1 mg/ml Protease K in buffer H (PBS+1 mM EDTA) for 2 h at 4°C. The cell suspension was then centrifugated at 5000 g for 10 min over a cushion of 10% sucrose in PBS. The tubes were frozen in liquid nitrogen, cut to separate the cells (internal) from the solution (surface-bound material released by protease K) and counted on a gamma counter. Nonspecific activity was assessed in parallel experiments in the presence of 250 μg/ml unlabeled β-VLDL. Nonspecific activities were subtracted from mean values for each data point.
Supplementary Material
Supplementary Figure 1
{kind=link}
Supplementary Figure 2
{kind=link}
Supplementary Figure 3
{kind=link}
Supplementary Figure 4
{kind=link}
Supplementary Figure 5
{kind=link}
Acknowledgments
We thank Richard Anderson for many helpful discussions, Joachim Herz for the anti-LDLR antibody, Michael Brown and Joseph Goldstein for the LDLR−/− and JD fibroblasts, and Sebastiano Calandra and Roberta Tiozzo for the normal and ARH−/− fibroblasts. This work was supported by NIH grants HL085218-01 (PM) and HL20948 (HH).
References
- Ajioka RS, Kaplan J (1986) Intracellular pools of transferrin receptors result from constitutive internalization of unoccupied receptors. Proc Natl Acad Sci USA 83: 6445–6449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson RG, Brown MS, Beisiegel U, Goldstein JL (1982) Surface distribution and recycling of the low density lipoprotein receptor as visualized with antireceptor antibodies. J Cell Biol 93: 523–531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson RG, Goldstein JL, Brown MS (1977) A mutation that impairs the ability of lipoprotein receptors to localise in coated pits on the cell surface of human fibroblasts. Nature 270: 695–699 [DOI] [PubMed] [Google Scholar]
- Arca M, Zuliani G, Wilund K, Campagna F, Fellin R, Bertolini S, Calandra S, Ricci G, Glorioso N, Maioli M, Pintus P, Carru C, Cossu F, Cohen J, Hobbs HH (2002) Autosomal recessive hypercholesterolaemia in Sardinia, Italy, and mutations in ARH: a clinical and molecular genetic analysis. Lancet 359: 841–847 [DOI] [PubMed] [Google Scholar]
- Basu SK, Goldstein JL, Anderson RG, Brown MS (1981) Monensin interrupts the recycling of low density lipoprotein receptors in human fibroblasts. Cell 24: 493–502 [DOI] [PubMed] [Google Scholar]
- Berman M, Hall MR, Levy RI, Eisenberg S, Bilheimer DW, Phair RD, Goebel RH (1978) Metabolsim of apoB and apoC lipoproteins in man: kinetic studies in normal and hyperlipoproteininemic subjects. J Lipid Res 19: 38–56 [PubMed] [Google Scholar]
- Bilheimer DW, Eisenberg S, Levy RI (1972) The metabolism of very low density lipoprotein proteins. I. Preliminary in vitro and in vivo observations. Biochim Biophys Acta 260: 212–221 [DOI] [PubMed] [Google Scholar]
- Boll W, Rapoport I, Brunner C, Modis Y, Prehn S, Kirchhausen T (2002) The mu2 subunit of the clathrin adaptor AP-2 binds to FDNPVY and YppO sorting signals at distinct sites. Traffic 3: 590–600 [DOI] [PubMed] [Google Scholar]
- Bonifacino JS, Traub LM (2003) Signals for sorting of transmembrane proteins to endosomes and lysosomes. Annu Rev Biochem 72: 395–447 [DOI] [PubMed] [Google Scholar]
- Bright NA, Gratian MJ, Luzio JP (2005) Endocytic delivery to lysosomes mediated by concurrent fusion and kissing events in living cells. Curr Biol 15: 360–365 [DOI] [PubMed] [Google Scholar]
- Brown MS, Goldstein JL (1976) Analysis of a mutant strain of human fibroblasts with a defect in the internalization of receptor-bound low density lipoprotein. Cell 9: 663–674 [DOI] [PubMed] [Google Scholar]
- Brown MS, Goldstein JL (1986) A receptor-mediated pathway for cholesterol homeostasis. Science 232: 34–47 [DOI] [PubMed] [Google Scholar]
- Chen WJ, Goldstein JL, Brown MS (1990) NPXY, a sequence often found in cytoplasmic tails, is required for coated pit-mediated internalization of the low density lipoprotein receptor. J Biol Chem 265: 3116–3123 [PubMed] [Google Scholar]
- Davis CG, Lehrman MA, Russell DW, Anderson RG, Brown MS, Goldstein JL (1986) The J.D. mutation in familial hypercholesterolemia: amino acid substitution in cytoplasmic domain impedes internalization of LDL receptors. Cell 45: 15–24 [DOI] [PubMed] [Google Scholar]
- Davis CG, van Driel IR, Russell DW, Brown MS, Goldstein JL (1987) The low density lipoprotein receptor. Identification of amino acids in cytoplasmic domain required for rapid endocytosis. J Biol Chem 262: 4075–4082 [PubMed] [Google Scholar]
- Fazili Z, Sun W, Mittelstaedt S, Cohen C, Xu XX (1999) Disabled-2 inactivation is an early step in ovarian tumorigenicity. Oncogene 18: 3104–3113 [DOI] [PubMed] [Google Scholar]
- Garcia CK, Wilund K, Arca M, Zuliani G, Fellin R, Maioli M, Calandra S, Bertolini S, Cossu F, Grishin N, Barnes R, Cohen JC, Hobbs HH (2001) Autosomal recessive hypercholesterolemia caused by mutations in a putative LDL receptor adaptor protein. Science 292: 1394–1398 [DOI] [PubMed] [Google Scholar]
- Garuti R, Jones C, Li WP, Michaely P, Herz J, Gerard RD, Cohen JC, Hobbs HH (2005) The modular adaptor protein autosomal recessive hypercholesterolemia (ARH) promotes low density lipoprotein receptor clustering into clathrin-coated pits. J Biol Chem 280: 40996–41004 [DOI] [PubMed] [Google Scholar]
- Goldstein JL, Basu SK, Brown MS (1983) Receptor-mediated endocytosis of low-density lipoprotein in cultured cells. Methods Enzymol 98: 241–260 [DOI] [PubMed] [Google Scholar]
- Goldstein JL, Brown MS (1974) Binding and degradation of low density lipoproteins by cultured human fibroblasts. Comparison of cells from a normal subject and from a patient with homozygous familial hypercholesterolemia. J Biol Chem 249: 5153–5162 [PubMed] [Google Scholar]
- Goldstein JL, Brown MS, Stone NJ (1977) Genetics of the LDL receptor: evidence that the mutations affecting binding and internalization are allelic. Cell 12: 629–641 [DOI] [PubMed] [Google Scholar]
- Goldstein JL, Ho YK, Brown MS, Innerarity TL, Mahley RW (1980) Cholesteryl ester accumulation in macrophages resulting from receptor-mediated uptake and degradation of hypercholesterolemic canine beta-very low density lipoproteins. J Biol Chem 255: 1839–1848 [PubMed] [Google Scholar]
- Goldstein JL, Hobbs HH, Brown MS (2001) Familial hypercholesterolemia. In The Metabolic and Molecular Bases of Inherited Disease, Scriver CR (ed) pp 2863–2913. New York, USA: McGraw-Hill New York [Google Scholar]
- Gorden P, Carpentier JL, Cohen S, Orci L (1978) Epidermal growth factor: morphological demonstration of binding, internalization, and lysosomal association in human fibroblasts. Proc Natl Acad Sci USA 75: 5025–5029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hahn PF (1943) Abolishment of alimentary lipemia following injection of heparin. Science 98: 19–20 [DOI] [PubMed] [Google Scholar]
- Handley DA, Arbeeny CM, Witte LD, Chien S (1981) Colloidal gold—low density lipoprotein conjugates as membrane receptor probes. Proc Natl Acad Sci USA 78: 368–371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harada-Shiba M, Tajima S, Miyake Y, Kojima S, Tsushima M, Yamamoto A (1991) Severe hypercholesterolemia patients with normal LDL receptor. J Jpn Atheroscler Soc 19: 227–242 [DOI] [PubMed] [Google Scholar]
- Harada-Shiba M, Takagi A, Miyamoto Y, Tsushima M, Ikeda Y, Yokoyama S, Yamamoto A (2003) Clinical features and genetic analysis of autosomal recessive hypercholesterolemia. J Clin Endocrinol Metab 88: 2541–2547 [DOI] [PubMed] [Google Scholar]
- He G, Gupta S, Yi M, Michaely P, Hobbs HH, Cohen JC (2002) ARH is a modular adaptor protein that interacts with the LDL receptor, clathrin, and AP-2. J Biol Chem 277: 44044–44049 [DOI] [PubMed] [Google Scholar]
- Heuser JE, Anderson RG (1989) Hypertonic media inhibit receptor-mediated endocytosis by blocking clathrin-coated pit formation. J Cell Biol 108: 389–400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hobbs HH, Brown MS, Goldstein JL (1992) Molecular genetics of the LDL receptor gene in familial hypercholesterolemia. Hum Mutat 1: 445–466 [DOI] [PubMed] [Google Scholar]
- Hopkins CR, Miller K, Beardmore JM (1985) Receptor-mediated endocytosis of transferrin and epidermal growth factor receptors: a comparison of constitutive and ligand-induced uptake. J Cell Sci Suppl 3: 173–186 [DOI] [PubMed] [Google Scholar]
- Howell BW, Lanier LM, Frank R, Gertler FB, Cooper JA (1999) The disabled 1 phosphotyrosine-binding domain binds to the internalization signals of transmembrane glycoproteins and to phospholipids. Mol Cell Biol 19: 5179–5188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurley JH, Emr SD (2006) The ESCRT complexes: structure and mechanism of a membrane-trafficking network. Annu Rev Biophys Biomol Struct 35: 277–298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Innerarity TL, Pitas RE, Mahley RW (1986) Lipoprotein–receptor interactions. Methods Enzymol 129: 542–565 [DOI] [PubMed] [Google Scholar]
- Jones C, Garuti R, Michaely P, Li WP, Maeda N, Cohen JC, Herz J, Hobbs HH (2007) Disruption of LDL but not VLDL clearance in autosomal recessive hypercholesterolemia. J Clin Invest 117: 165–174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones C, Hammer RE, Li WP, Cohen JC, Hobbs HH, Herz J (2003) Normal sorting but defective endocytosis of the low density lipoprotein receptor in mice with autosomal recessive hypercholesterolemia. J Biol Chem 278: 29024–29030 [DOI] [PubMed] [Google Scholar]
- Jones NL, Saunders JA, Mallory RR (2000) Intracellular trafficking of pigeon beta-very low density lipoprotein and low density lipoprotein at low and high concentrations in pigeon macrophages. J Lipid Res 41: 1823–1831 [PubMed] [Google Scholar]
- Keyel PA, Mishra SK, Roth R, Heuser JE, Watkins SC, Traub LM (2006) A single common portal for clathrin-mediated endocytosis of distinct cargo governed by cargo-selective adaptors. Mol Biol Cell 17: 4300–4317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khachadurian AK, Uthman SM (1973) Experiences with the homozygous cases of familial hypercholesterolemia. A report of 52 patients. Nutr Metab 15: 132–140 [DOI] [PubMed] [Google Scholar]
- Kibbey RG, Rizo J, Gierasch LM, Anderson RG (1998) The LDL receptor clustering motif interacts with the clathrin terminal domain in a reverse turn conformation. J Cell Biol 142: 59–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kishimoto A, Brown MS, Slaughter CA, Goldstein JL (1987) Phosphorylation of serine 833 in cytoplasmic domain of low density lipoprotein receptor by a high molecular weight enzyme resembling casein kinase II. J Biol Chem 262: 1344–1351 [PubMed] [Google Scholar]
- Kita T, Beisiegel U, Goldstein JL, Schneider WJ, Brown MS (1981) Antibody against low density lipoprotein receptor blocks uptake of low density lipoprotein (but not high density lipoprotein) by the adrenal gland of the mouse in vivo. J Biol Chem 256: 4701–4703 [PubMed] [Google Scholar]
- Kita T, Brown MS, Bilheimer DW, Goldstein JL (1982) Delayed clearance of very low density and intermediate density lipoproteins with enhanced conversion to low density lipoprotein in WHHL rabbits. Proc Natl Acad Sci USA 79: 5693–5697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovanen PT, Brown MS, Basu SK, Bilheimer DW, Goldstein JL (1981) Saturation and suppression of hepatic lipoprotein receptors: a mechanism for the hypercholesterolemia of cholesterol-fed rabbits. Proc Natl Acad Sci USA 78: 1396–1400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krul ES, Tikkanen MJ, Cole TG, Davie JM, Schonfeld G (1985) Roles of apolipoproteins B and E in the cellular binding of very low density lipoproteins. J Clin Invest 75: 361–369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lefkowitz RJ, Whalen EJ (2004) beta-arrestins: traffic cops of cell signaling. Curr Opin Cell Biol 16: 162–168 [DOI] [PubMed] [Google Scholar]
- Lehrman MA, Goldstein JL, Brown MS, Russell DW, Schneider WJ (1985) Internalization-defective LDL receptors produced by genes with nonsense and frameshift mutations that truncate the cytoplasmic domain. Cell 41: 735–743 [DOI] [PubMed] [Google Scholar]
- Liu X, Sun Y, Constantinescu SN, Karam E, Weinberg RA, Lodish HF (1997) Transforming growth factor beta-induced phosphorylation of Smad3 is required for growth inhibition and transcriptional induction in epithelial cells. Proc Natl Acad Sci USA 94: 10669–10674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lombardi P, Mulder M, van der Boom H, Frants RR, Havekes LM (1993) Inefficient degradation of triglyceride-rich lipoprotein by HepG2 cells is due to a retarded transport to the lysosomal compartment. J Biol Chem 268: 26113–26119 [PubMed] [Google Scholar]
- Mahley RW, Innerarity TL, Brown MS, Ho YK, Goldstein JL (1980) Cholesteryl ester synthesis in macrophages: stimulation by beta-very low density lipoproteins from cholesterol-fed animals of several species. J Lipid Res 21: 970–980 [PubMed] [Google Scholar]
- Maurer ME, Cooper JA (2006) The adaptor protein Dab2 sorts LDL receptors into coated pits independently of AP-2 and ARH. J Cell Sci 119: 4235–4246 [DOI] [PubMed] [Google Scholar]
- Maxfield FR, McGraw TE (2004) Endocytic recycling. Nat Rev Mol Cell Biol 5: 121–132 [DOI] [PubMed] [Google Scholar]
- Michaely P, Li WP, Anderson RG, Cohen JC, Hobbs HH (2004) The modular adaptor protein ARH is required for low density lipoprotein (LDL) binding and internalization but not for LDL receptor clustering in coated pits. J Biol Chem 279: 34023–34031 [DOI] [PubMed] [Google Scholar]
- Mishra SK, Watkins SC, Traub LM (2002) The autosomal recessive hypercholesterolemia (ARH) protein interfaces directly with the clathrin-coat machinery. Proc Natl Acad Sci USA 99: 16099–16104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris SM, Cooper JA (2001) Disabled-2 colocalizes with the LDLR in clathrin-coated pits and interacts with AP-2. Traffic 2: 111–123 [DOI] [PubMed] [Google Scholar]
- Myers JN, Tabas I, Jones NL, Maxfield FR (1993) Beta-very low density lipoprotein is sequestered in surface-connected tubules in mouse peritoneal macrophages. J Cell Biol 123: 1389–1402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phair RD, Hammond MG, Bowden JA, Fried M, Fisher WR, Berman M (1975) Preliminary model for human lipoprotein metabolism in hyperlipoproteinemia. Fed Proc 34: 2263–2270 [PubMed] [Google Scholar]
- Pietra GG, Spagnoli LG, Capuzzi DM, Sparks CE, Fishman AP, Marsh JB (1976) Metabolism of 125I-labeled lipoproteins by the isolated rat lung. J Cell Biol 70: 33–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pisciotta L, Oliva CP, Pes GM, Di Scala L, Bellocchio A, Fresa R, Cantafora A, Arca M, Calandra S, Bertolini S (2006) Autosomal recessive hypercholesterolemia (ARH) and homozygous familial hypercholesterolemia (FH): a phenotypic comparison. Atherosclerosis 188: 398–405 [DOI] [PubMed] [Google Scholar]
- Rink J, Ghigo E, Kalaidzidis Y, Zerial M (2005) Rab conversion as a mechanism of progression from early to late endosomes. Cell 122: 735–749 [DOI] [PubMed] [Google Scholar]
- Schneider WJ, Kovanen PT, Brown MS, Goldstein JL, Utermann G, Weber W, Havel RJ, Kotite L, Kane JP, Innerarity TL, Mahley RW (1981) Familial dysbetalipoproteinemia. Abnormal binding of mutant apoprotein E to low density lipoprotein receptors of human fibroblasts and membranes from liver and adrenal of rats, rabbits, and cows. J Clin Invest 68: 1075–1085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shelness GS, Sellers JA (2001) Very-low-density lipoprotein assembly and secretion. Curr Opin Lipidol 12: 151–157 [DOI] [PubMed] [Google Scholar]
- Shireman R, Kilgore LL, Fisher WR (1977) Solubilization of apolipoprotein B and its specific binding by the cellular receptor for low density lipoprotein. Proc Natl Acad Sci USA 74: 5150–5154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabas I, Lim S, Xu XX, Maxfield FR (1990) Endocytosed beta-VLDL and LDL are delivered to different intracellular vesicles in mouse peritoneal macrophages. J Cell Biol 111: 929–940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tammi R, Rilla K, Pienimaki JP, MacCallum DK, Hogg M, Luukkonen M, Hascall VC, Tammi M (2001) Hyaluronan enters keratinocytes by a novel endocytic route for catabolism. J Biol Chem 276: 35111–35122 [DOI] [PubMed] [Google Scholar]
- van Driel IR, Davis CG, Goldstein JL, Brown MS (1987) Self-association of the low density lipoprotein receptor mediated by the cytoplasmic domain. J Biol Chem 262: 16127–16134 [PubMed] [Google Scholar]
- Weisgraber KH, Innerarity TL, Harder KJ, Mahley RW, Milne RW, Marcel YL, Sparrow JT (1983) The receptor-binding domain of human apolipoprotein E. Monoclonal antibody inhibition of binding. J Biol Chem 258: 12348–12354 [PubMed] [Google Scholar]
- Wiley HS, Herbst JJ, Walsh BJ, Lauffenburger DA, Rosenfeld MG, Gill GN (1991) The role of tyrosine kinase activity in endocytosis, compartmentation, and down-regulation of the epidermal growth factor receptor. J Biol Chem 266: 11083–11094 [PubMed] [Google Scholar]
- Willingham MC, Hanover JA, Dickson RB, Pastan I (1984) Morphologic characterization of the pathway of transferrin endocytosis and recycling in human KB cells. Proc Natl Acad Sci USA 81: 175–179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Windler EE, Kovanen PT, Chao YS, Brown MS, Havel RJ, Goldstein JL (1980) The estradiol-stimulated lipoprotein receptor of rat liver. A binding site that membrane mediates the uptake of rat lipoproteins containing apoproteins B and E. J Biol Chem 255: 10464–10471 [PubMed] [Google Scholar]
- Wu JH, Peppel K, Nelson CD, Lin FT, Kohout TA, Miller WE, Exum ST, Freedman NJ (2003) The adaptor protein beta-arrestin2 enhances endocytosis of the low density lipoprotein receptor. J Biol Chem 278: 44238–44245 [DOI] [PubMed] [Google Scholar]
- Zuliani G, Arca M, Signore A, Bader G, Fazio S, Chianelli M, Bellosta S, Campagna F, Montali A, Maioli M, Pacifico A, Ricci G, Fellin R (1999) Characterization of a new form of inherited hypercholesterolemia: familial recessive hypercholesterolemia. Arterioscler Thromb Vasc Biol 19: 802–809 [DOI] [PubMed] [Google Scholar]
- Zuliani G, Vigna GB, Corsini A, Maioli M, Romagnoni F, Fellin R (1995) Severe hypercholesterolaemia: unusual inheritance in an Italian pedigree. Eur J Clin Invest 25: 322–331 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1
Supplementary Figure 2
Supplementary Figure 3
Supplementary Figure 4
Supplementary Figure 5