

Abstract
TANK-binding kinase 1 (TBK1/NAK/T2K) and I-κB Kinase (IKK-i/IKK-ɛ) play important roles in the regulation of interferon (IFN)-inducible genes during the immune response to bacterial and viral infections. Cell stimulation with ssRNA virus, dsDNA virus or gram-negative bacteria leads to activation of TBK1 or IKK-i, which in turn phosphorylates the transcription factors, IFN-regulatory factor (IRF) 3 and IRF7, promoting their translocation in the nucleus. To understand the molecular basis of activation of TBK1, we analyzed the sequence of TBK1 and IKK-i and identified a ubiquitin-like domain (ULD) adjacent to their kinase domains. Deletion or mutations of the ULD in TBK1 or IKK-i impaired activation of respective kinases, failed to induce IRF3 phosphorylation and nuclear localization and to activate IFN-β or RANTES promoters. The importance of the ULD of TBK1 in LPS- or poly(I:C)-stimulated IFN-β production was demonstrated by reconstitution experiments in TBK1-IKK-i-deficient cells. We propose that the ULD is a regulatory component of the TBK1/IKK-i kinases involved in the control of the kinase activation, substrate presentation and downstream signaling pathways.
Keywords: innate immunity signal, interferon-inducible gene, TANK-binding kinase 1, ubiquitin, ubiquitin-like domain
Introduction
Toll-like receptors (TLRs) are type I receptor proteins, evolutionarily conserved from Caenorhabditis elegans to mammals (Janeway and Medzhitov, 2002; Lemaitre, 2002). The TLR family consists of at least 11 TLRs (Janeway and Medzhitov, 2002), which are expressed on various cells such as macrophages, dendritic cells, B-cells, T-cells and fibroblasts. Among TLR members, TLR3 and TLR4 recognize bacterial proteins LPS, envelope proteins of viruses, heat-shock protein 60 as well as dsRNA and mediate the downstream signal (Akira et al, 2006). In this signaling pathway, type I interferons (IFNs) are induced by the activation of transcription factors such as IFN-regulatory factor (IRF) 3 and IRF7 (Akira et al, 2006; Honda and Taniguchi, 2006). IRF3 and IRF7 are localized in the cytoplasm as inactive monomers (Hiscott et al, 2001; Taniguchi et al, 2001; Takeda and Akira, 2003; Honda and Taniguchi, 2006). Once activated by phosphorylation, IRFs form dimers, translocate into the nucleus and bind to the IFN-stimulated response element (ISRE) in the promoter region of its target genes, such as IFN-β and RANTES. This causes the upregulation of their transcriptional activities (Hiscott et al, 2001; Taniguchi et al, 2001; Takeda and Akira, 2003; Honda and Taniguchi, 2006). The absence of either IRF3 or IRF7 in mouse results in marked reduction of IFN production in response to virus infection, demonstrating the critical role of IRF3 and IRF7 in the IFN production in vivo (Sato et al, 2000; Honda et al, 2005).
Two IKK-related serine/threonine kinases, TBK1 and IKK-i, which directly phosphorylate IRF3 and IRF7, have been identified so far (Fitzgerald et al, 2003a; Sharma et al, 2003). TBK1 and IKK-i have a kinase domain in the N terminus and share 64% of similarities in amino-acid sequences (Pomerantz and Baltimore, 1999; Shimada et al, 1999). Although both kinases mediate similar signaling events downstream of TLR3 or TLR4 via the adaptor molecules TRAM and TRIF and activate the same substrates, it seems that TBK1 and IKK-i differ in their regulation of downstream signaling (Fitzgerald et al, 2003b; Hoebe et al, 2003; Yamamoto et al, 2003a, 2003b). IRF3 activation and IFN-β production by poly(I:C) are decreased in TBK1-deficient fibroblasts, whereas normal activation was observed in the IKK-i-deficient fibroblasts (Hemmi et al, 2004). However, in double-deficient fibroblasts, the activation of IRF3 is completely abolished, suggesting a partially redundant functions of TBK1 and IKK-i (Yamamoto et al, 2003a). Many studies have been carried out to clarify the mechanisms of IRF3 activation, such as determination of the critical phosphorylation sites for dimer formation and translocation (Qin et al, 2003; Takahasi et al, 2003). The adaptor protein TRIF is required for the activation of TBK1 and forms a complex with TBK1, TRAF6 and IRF3 (Sato et al, 2003). Nonetheless, the mechanism how TRIF recruits TBK1 in the complex upon stimulation is unclear.
Ubiquitin-like (UBL) proteins for example, SUMO, NEDD8, ATG12, ATG8, URM1, ISG15 and FAT10 share structural similarities with ubiquitin, are conjugated to substrates and are involved in regulation of different cellular responses (Kerscher et al, 2006). In addition, UBL may be found within a larger context of a protein, which then can represent UBL domains in these proteins (Hartmann-Petersen and Gordon, 2004). UBL domains in some of the type II proteins such as Parkin or Rad23 have been shown to play a role in recruiting the binding partners, resulting in the regulation of downstream signaling (Schauber et al, 1998; Walters et al, 2004).
In this study, we have identified a UBL domain (ULD) adjacent to the kinase domain in TBK1 and IKK-i. Unlike the UBL domain of Parkin, the ULD of TBK1 or IKK-i did not interact with known ubiquitin-binding domains (UBAs), but rather with its own kinase domain and its substrate IRF3. We have shown that an intact ULD in TBK1 is required for the full function of TBK1 kinase domain and interaction with kinase substrates. We have also identified the crucial amino acids in the ULD that control the kinase activity of TBK1 and are critical for the regulation of IFN-inducible gene transcription. Our findings shed light on the novel role of the ULD in the regulation TBK1 functions in vivo.
Results
Identification of ULD in IKK-related kinases
To analyze structural domains in TBK1 that might be important for its signaling properties, we initially performed a bioinformatical analysis of the TBK1 sequence. Profile-guided sequence analysis revealed that TBK1 (Figure 1A, lane 6), as well as other IKK-related kinases, such as IKKα (Figure 1A, lane 2), IKKβ (Figure 1A, lane 1,3) and IKK-i (Figure 1A, lane 4,5) contain an internal domain with distant similarity to ubiquitin fold proteins. Despite the high divergence, a statistically significant relationship to several UBL proteins could be established; the best human match (P<0.001) was the first UBL domain of the Fat10 protein (Figure 1A, lane 7). The evolutionary distance between the IKK proteins and the typical ULD, such as ubiquitin, Parkin and Rad23, is much larger than the distances between the typical UBLs. As a consequence, a larger degree of functional divergence is to be expected; we thus refer to the homology domain in the IKKs as a ULD. The ULD in mouse TBK1 consists of 79 amino acids, starts at amino acid 305 and ends at amino acid 383. The ULD in mouse IKK-i shares 65% similarity with the ULD in mouse TBK1. The amino-acid sequences of the hydrophobic patch of ubiquitin surrounding isoleucine 44 (Figure 1A, lane 8, arrowhead) are conserved between the ULDs in IKK-related proteins (Figure 1A, lane 1–6) and Fat10 (Figure 1A, lane 7).
Figure 1.
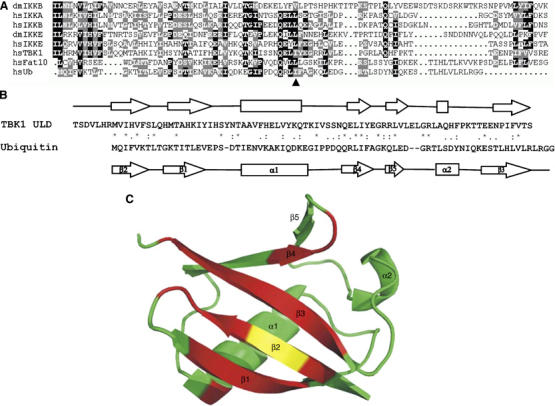
Identification and structural characterization of the ULD in TBK1 (A) An alignment of Fat10, ubiquitin and ULD domains of IKKβ, IKKα, IKK-i and TBK1. Conserved residues are shown on black or gray background. The hydrophobic patch at isoleucine 44 in ubiquitin (arrowhead) is conserved in ULD domain in TBK1. (dm: Drosophila, hs: Homo sapiens) (B) Primary and secondary structure alignment of mouse TBK1-ULD and human ubiquitin. Secondary structure elements (arrows for β-strands, boxes for helical regions) for TBK1-ULD represent the consensus of the chemical shifts analyses with the programs TALOS and CSI. Secondary structure elements as well as atomic coordinates for ubiquitin are taken from PDB entry 1UBI. (C) Dynamic behavior of TBK1-ULD. The ribbon diagram of the structure of human ubiquitin is colored according to the broadening and splitting of the TBK1-ULD resonances (red: significant, yellow: intermediate, green: no line broadening).
ULD of TBK1 adopts ubiquitin-like (β-grasp) fold
To characterize the identified ULD structurally, we have used heteronuclear high-resolution NMR spectroscopy. The [15N,1H]-TROSY spectrum (Supplementary Figure S1) of the isolated mouse TBK1-ULD (amino acids 302–383) shows a chemical shift dispersion that is indicative of a well-folded and globular domain. Analysis of the secondary structure elements based on the 13Cα, 13Cβ, 13CO and 1Hα chemical shifts identified one long α-helix and four β-strands that have the same sequential arrangement (ββαββ) as the corresponding secondary structure elements in ubiquitin (Figure 1B), demonstrating that the TBK1-ULD belongs to the UBL protein superfamily. Furthermore, the chemical shift analysis indicates the presence of an additional β-strand (corresponding to β5 in ubiquitin) and a short helical segment between residues 367 and 369 that corresponds to the 310-helix of ubiquitin.
Despite these structural similarities between the ULD and ubiquitin, differences exist in their dynamic behavior. Interestingly, the whole β-sheet and, in particular, the two β-strands that correspond to strand β1 and β3 in ubiquitin exhibit broad resonances with several maxima (Supplementary Figure S1, insert), which is characteristic for conformational exchange processes. Being plotted on the ubiquitin three-dimensional structure (Figure 1C), these residues form a well-defined surface.
The ULD in TBK1 plays a role in the regulation of the TBK1 kinase activity and downstream signaling
To understand the role of the ULDs in the regulation of downstream molecules, we generated deletion mutants of the ULDs in TBK1 (Δ-ULD) (Figure 2A) and IKK-i (Figure 2B). TBK1-Δ-ULD and IKK-i-Δ-ULD were neither misfolded nor degraded when expressed in HEK293T cells and was able to bind to the isolated GST-ULD of TBK1 (Figure 4C and D, Δ-ULD). As one of the important roles of TBK1 is to regulate IFN-inducible genes, the effects of TBK1-wild type (wt) and TBK1-Δ-ULD on the regulation of gene transcription were examined by a reporter gene assay. TBK1-wt induced both IFN-β (Figure 2C) and RANTES promoter activities (Figure 2D). On the other hand, Δ-ULD did not induce the gene transcriptions, comparable to the effect of a kinase dead mutant (KM) of TBK1 (Figure 2C and D). Similar effects on the gene transcription of IFN-β (Figure 2E) and RANTES (Figure 2F) were observed with IKK-i-Δ-ULD mutants. The hydrophobic patch of ubiquitin is known as the critical binding surface to UBA domains (Hicke et al, 2005). To test its functional relevance in the ULD of TBK1, we generated the corresponding ULD mutant of TBK1 (L352A I353A) (Figure 2A) and IKK-i (L353A F354A) (Figure 2B). The TBK1-L352A I353A mutant neither induced the gene transcriptions of IFN-β (Figure 2C) nor RANTES (Figure 2D), similar to TBK1-Δ-ULD. Mutation of either L352 or I353 alone did not affect the activities of IFN-β and RANTES promoters (data not shown). The introduction of mutations in the hydrophobic patch of IKK-i at L353 and F354 also abolished IFN-β (Figure 2E) and RANTES (Figure 2F) promoter activations. To clarify the mechanism by which IFN-inducible genes are regulated by the ULD, we examined the effects of TBK1-L352A I353A on IRF3 nuclear translocation (Figure 2G and H). IRF3 alone was not observed in the nucleus (Figure 2G, lower right, cont). Co-expression of TBK1-wt with IRF3 strongly induced the nuclear translocation of IRF3 (Figure 2G, upper left, lower right, 1 TBK1-wt). On the other hand, the translocation of IRF3 was not induced by Δ-ULD (Figure 2G, upper right, lower right, 2 TBK1-Δ-ULD) and by the L352A I353A mutant (Figure 2G, lower left, lower right, 3 TBK1 L352A I353A). The nuclear localization of IRF3 was also determined by immunoblot using nuclear fractions (Figure 2H). Similar results with immunofluorescence were obtained (Figure 2H). Next, we examined the dimer formation of IRF3 by native PAGE in the presence of wt versus different mutants of TBK1. The dimer formation of IRF3 was observed upon co-expression of TBK1-wt, but not when IRF3 was co-expressed together with TBK1-Δ-ULD, L352A I353A mutant or KM (Figure 2I). The dimerized IRF3 induced by TBK1-wt was detected by a specific antibody against phospho-Ser 386 IRF3 (Figure 2I), which is known to be important for the dimer formation of IRF3 (Takahasi et al, 2003). To examine the involvement of the ULD of TBK1 in the functional regulation of its own kinase activity, kinase activities of TBK1-wt and mutants were examined in vitro using MBP as an in vitro substrate (Figure 3A). Phosphorylation of MBP by TBK1 (Figure 3A) or IKK-i (Figure 3B) was completely abolished by the ULD deletion. The TBK1-L352A I353A mutant retained only 26% of the kinase activity of wt TBK1, determined by densitometric analysis (Figure 3A, L352A I353A). Moreover, the in vitro-measured autophosphorylation of TBK1 (Figure 3A) or IKK-i (Figure 3B) correlated with the results obtained for phosphorylation of an exogenous substrate. The effects of TBK1-wt and mutants on the phosphorylation of IRF3 were also determined by immunoblotting, using the specific antibody recognizing phospho-Ser 396 of IRF3 (Figure 3C). The effects of TBK1-wt, Δ-ULD and L352A I353A on the phosphorylation of IRF3 (Figure 3C) were similar to the results of kinase assay (Figure 3A). The introduction of mutations only at L352 or I353 did not affect the phosphorylation of IRF3 (Figure 3C, L352A and I353A). TBK1-KM did not phosphorylate IRF3 as expected (Figure 3C, KM). Identical observations on IRF3-phosphorylation were obtained with IKK-i Δ-ULD and IKK-i-L353A F354A mutants (Figure 3D). Effects of TBK1-wt and mutants on IκBα, which is another known substrate of TBK1 in vitro, were also examined (Figure 3E). Effects of TBK1-wt, TBK1-Δ-ULD and TBK1-KM on IκBα-phosphorylation were similar to IRF3 phosphorylation (Figure 3E). Interestingly, TBK1-L352A I353A mutant did not induce phosphorylation of IκBα (Figure 3E).
Figure 2.
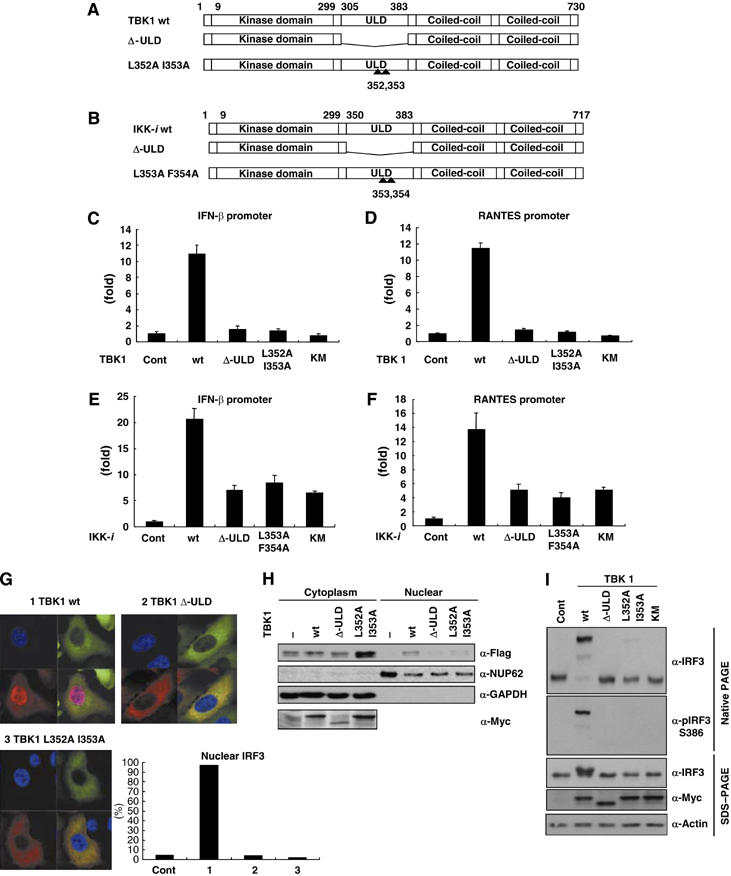
The ULD plays an important role in the regulation of downstream signaling. (A, B) Deletion-ULD (Δ-ULD) of TBK1 and IKK-i, and mutants with mutations at the hydrophobic patch of TBK1-L352A I353A and IKK-i-L353AF354A were generated as described previously. (C, D) The effects of TBK1-wt, Δ-ULD, L352A I353A and KM on promoter activities of IFN-inducible genes were examined. Luciferase construct driven by IFN-β promoter or RANTES promoter was co-expressed in HEK293T cells with TBK1-wt or mutants. After 48 h of transfection, cells were harvested for the luciferase assay. The internal control was measured by β-GAL activity. (E, F) The effects of IKK-i wt, Δ-ULD, L353A F354A and KM on promoter activities of IFN-inducible genes were examined. (G) Effects of TBK1 mutants on IRF3-nuclear localization were examined. Cellular localization of IRF3 was examined by immunofluorescence. Myc-IRF3 was co-expressed with GFP-TBK1-wt (upper left), GFP-TBK1-Δ-ULD (upper right) or GFP-TBK1-L352AI353A mutant (lower left). Nuclear was stained by DAPI (blue). GFP-TBK1-wt, GFP-TBK1-Δ-ULD and GFP-TBK1-L352AI353A mutants were localized in the cytoplasm (green). Nuclear translocation of Myc-IRF3 (red) was only induced by TBK1-wt (upper left). More than 100 cells in which both of GFP-TBK1 (wt, Δ-ULD or L352A I353A) and Myc-IRF3 were expressed were counted and percentage of the cells with nuclear IRF3 was shown (lower right). All of the panels were under the magnification of × 400. (H) HeLa cells were co-transfected with plasmids encoding Flag-IRF3 and Myc-TBK1. Nuclear and cytoplasmic fractions were prepared and analyzed by immunoblot. NUP62 and GAPDH were used for fractionation markers of nuclear and cytoplasm, respectively. (I) Dimer formation of IRF3 was examined by native PAGE. Flag-IRF3 was transfected with Myc-TBK1 wt or mutants into HEK293T cells. Total cell lysates were used for native PAGE and the membrane transferred was blotted with α-IRF3 or α-pIRF3 S386 antibody. Expressions of Flag-IRF3, Myc-TBK1 and loading control were determined by SDS–PAGE.
Figure 3.
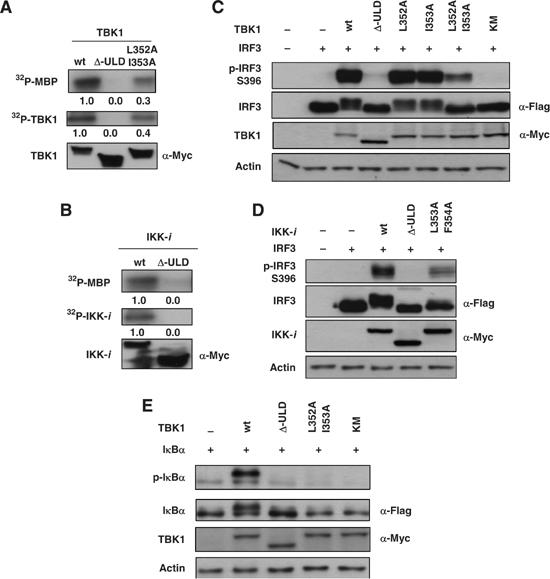
The ULD is required for the activation of downstream molecules and kinase activity of TBK1. (A) Various Myc-tagged TBK1 constructs were transfected into HEK293T cells and total cell lysates were subjected to immunoprecipitation by α-Myc antibody. Immunoprecipitated samples were subjected to in vitro kinase assay using MBP as substrate. The phosphorylation of MBP is shown in the upper panel, phosphorylation of TBK1 is shown in the middle panel and the expression level of TBK1 is shown in the lower panel. Densitometric analysis was performed using Scion Image software (Scion Corporations) and the numbers of the density are indicated. (B) Myc-IKK-i constructs were transfected into HEK293T cells and total cell lysates were subjected to immunoprecipitation by α-Myc antibody. Immunoprecipitated samples were subjected to in vitro kinase assay. (C, D) The effects of TBK1 and IKK-i-wt and mutants on phosphorylation of IRF3 were examined. Flag-IRF3 was co-expressed with Myc-TBK1 and IKK-i-wt or mutants in HEK293T cells. Total cell lysates were collected and examined by immunoblotting using α-phospho-IRF3 (Ser396) for the determination of IRF3-phosphorylation. The expression levels of Flag-IRF3, Myc-TBK1 and Myc-IKK-i were examined and are shown in the lower panels. Loading control was determined by using α-actin antibody. (E) The effects of TBK1-wt and mutants on phosphorylation of IκBα. Flag-IκBα was co-expressed with Myc-TBK1-wt or mutants in HEK293T cells. Total cell lysates were examined by immunoblotting using α-phospho-IκBα antibody for the determination of IκBα-phosphorylation. The expression levels of Flag-IκBα and Myc-TBK1 were examined and are shown in the lower panels. Loading control was determined by using α-actin antibody.
The hydrophobic patch of the ULD in TBK1 plays a role in the interactions of TBK1
As ubiquitin and UBL molecules are involved in the inter- and intramolecular folding of many proteins (Hoeller et al, 2006), we examined whether the ULD in TBK1 is engaged in similar functions. We purified a GST-fusion protein of the ULD of TBK1 (GST-ULD) and performed pull-down assays, using sequential deletion mutants of TBK1, as shown in Figure 4A. A relatively weak binding between GST-ULD and TBK1-wt was observed by immunoblotting (Figure 4B). GST-ULD weakly bound the TBK1 1–383, which contains the kinase domain and the ULD (Figure 4B). On the other hand, a more potent binding of GST-ULD to its own kinase domain (Figure 4A, 1–301 (wt)) was detected (Figure 4B). Similarly, the inactive kinase domain of TBK1 (Figure 4A, TBK1 1–301 (KM)) was also able to bind strongly to GST-ULD (Figure 4B). We confirmed the direct binding between the isolated TBK1-ULD and the TBK1 kinase domain by one to one transformation in the yeast two-hybrid system (data not shown). These results suggest that the major binding site of ULD is located within the kinase domain of TBK1. Hence, in full-length TBK1 or the TBK1 1–383 mutant, which contains both the kinase domain and the ULD, the binding site for exogenous GST-ULD is blocked. To further examine whether the hydrophobic patch of TBK1-ULD is involved in binding to the kinase domain, TBK1-wt, Δ-ULD and L352A I353A (Figure 4A) were subjected to GST-pull-down assays. GST-ULD weakly bound to TBK1-wt (Figure 4C, wt). In contrast, it bound more potently to TBK1-L352A I353A (Figure 4C, L352A I353A) and Δ-ULD (Figure 4C, Δ-ULD). Similar results were observed for GST-ULD binding to IKK-i-Δ-ULD and IKK-i-L353A F354A mutant (Figure 4D). To characterize more precisely the involvement of the ULD in the molecular folding, we purified a GST-fusion protein, in which the hydrophobic patch was mutated (GST-ULD-L352AI353A). The binding between ULD and TBK1-wt (Figure 4E), TBK1-L352A I353A (Supplementary Figure S2A, L352A I353A) or the TBK1-Δ-ULD mutant (Supplementary Figure S2B, Δ-ULD) was almost completely abolished by introduction of mutations of the hydrophobic patch in GST-ULD. These results show that the ULD binds to the kinase domain of TBK1 via the binding surface containing the hydrophobic patch at L352 I353 in the ULD. As no known UBA domain (Bienko et al, 2005; Hicke et al, 2005; Hurley et al, 2006) was found in TBK1 or IKK-i, we examined whether TBK1-ULD behaves similarly to ubiquitin or the UBL from another type II UBL molecules, in terms of binding to its partner proteins. GST-pull-down assays using purified ubiquitin (GST-Ub) and the UBL of Parkin (GST-UBL-Parkin) showed that the ubiquitin-binding proteins, Sts-1 and -2, bound potently to both ubiquitin and the Parkin-UBL, but not to TBK1-ULD (Figure 4F). Moreover, GST-Ub did not interact with any of the TBK1 mutants or wt (data not shown).
Figure 4.
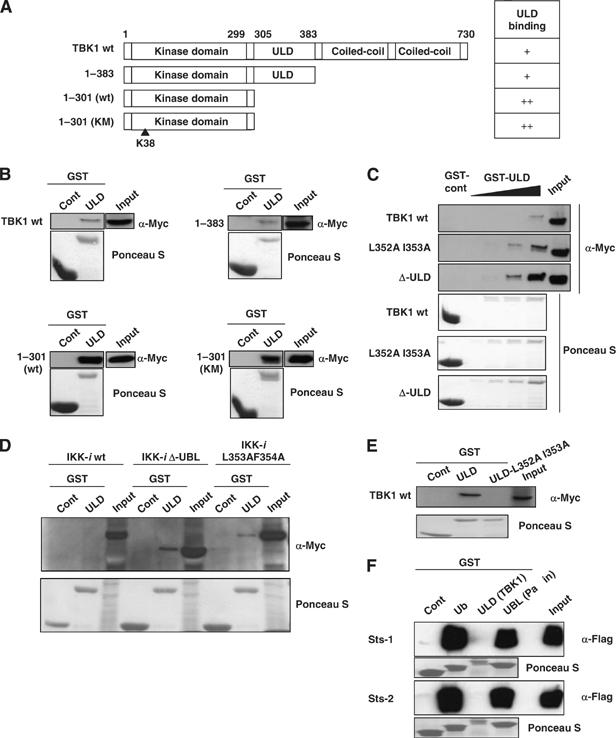
The ULD binds to kinase domain of TBK1 via hydrophobic patch L352 I353. (A) Deletion mutants of TBK1 were created as described. TBK1 only containing kinase domain with the ULD (1–383), kinase domain (1–301), kinase domain with mutations at ATP-binding site (1–301 KM) were generated as shown. (B) Myc-tagged TBK1-wt, 1–383, 1–301 (wt) and 1–301 (KM) were introduced into HEK293T cells and total cell lysates were used for GST-pull-down assay. After incubation of total cell lysates with GST-control or GST-ULD proteins, the binding was determined by immunoblotting using α-Myc antibody. (C) Myc-tagged TBK1-wt, L352A I353A and Δ-ULD were introduced into HEK293T cells and total cell lysates were used for GST-pull-down assay. (D) Myc-tagged IKK-i-wt, Δ-ULD and L353F F354A were introduced into HEK293T cells and total cell lysates were used for GST-pull-down assay. (E) GST-ULD and GST-ULD with L352A I353A-mutation (ULD-L352A I353A) were purified and used to examine the binding to Myc-tagged TBK1-wt. The amount of GST-fusion proteins was determined by Ponceau S staining, shown in the lower panels (B–E). (F) Sts-1 and -2, which have a UBA domain, do not bind the ULD of TBK1. Flag-Sts-1 and -2 were transfected into HEK293T cells and cell were harvested for the GST-pull-down assay. Total cell lysates were incubated with GST-fusion proteins of ubiquitin, TBK1-ULD or Parkin-UBL and immunoblotting using α-Flag antibody was performed. The amount of GST-fusion proteins was checked by Ponceau S staining as shown in the lower panels.
The ULD in TBK1 interacts with IRF3 transcription factor
Next, we raised the question on how the active form of TBK1 can interact with its substrates. We hypothesized that the ULD might be involved in a dual binding to the kinase domain and to exogenous substrates, thus promoting activation of the former and phosphorylation of the latter. To address this issue, the direct interaction between TBK1-ULD and IRF3, a known substrate of TBK1, was examined in yeast (Supplementary Figure S3A). Yeast, in which both TBK1-ULD and IRF3 were introduced, gave a positive signal on Gal-substrate-containing plate (Supplementary Figure S3A, lane 1), whereas yeast in which TBK1-ULD with empty prey (Supplementary Figure S3A, lane 2) or empty prey with IRF3 (Supplementary Figure S3A, lane 3) was introduced gave a negative signal. Next, we examined whether the ULD and the ULD-L352A I353A bind to IRF3 (Figure 5A) or IRF7 (Supplementary Figure S3B) by GST-pull-down assay, using the proteins expressed in mammalian cells. Both GST-ULD and GST-ULD-L352A I353A bound to IRF3 (Figure 5A) and IRF7 (Supplementary Figure S3B), whereas GST-ubiquitin did not bind to IRF3 or IRF7 (Figure 5A and Supplementary Figure S3B). The binding site of TBK1-ULD in IRF3 was mapped by generating deletion mutants of IRF3 as shown in Figure 5B. The binding between GST-ULD and IRF3 was lost when IRF association domain (IAD) was deleted (Figure 5C, IRF3 1–189). IRF3 (190–427), which contains IAD and SRR, as well as IRF3 (190–384) that contains only IAD, bound to GST-TBK1-ULD (Figure 5C). The deletion of SRR in IRF7 did not affect the binding to GST-ULD (Supplementary Figure S3C, IRF7 1–470). The deletion mutant of IAD and SRR in IRF7 abolished the binding to GST-ULD (Supplementary Figure S3C, IRF7 1–270). Moreover, the binding between GST-ULD and IRF3 was completely abolished when IRF3 is phosphorylated by TBK1, but not when co-expressed with the kinase inactive TBK1 (TBK1-KM, Figure 5D). This result supports the model in which phosphorylation of IRF3 might release it from the TBK1 complex, leading to the nuclear translocation of phosphorylated IRF3 (Figure 2G).
Figure 5.
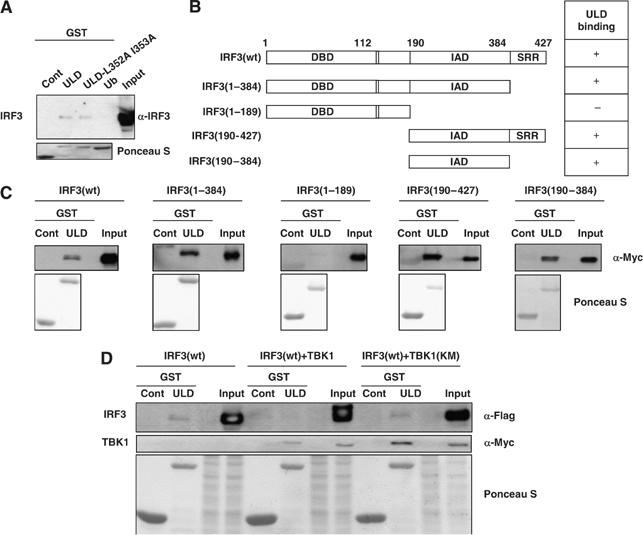
The ULD binds to IRF3. (A) Myc-IRF3 was introduced into HEK293T cells and total cell lysates were used to examine the binding to GST-ULD, GST-ULD-L352A I353A and GST-Ub. (B) The deletion mutants were generated for the experiments as indicated: IRF3 lacking serine-rich region (SRR, 1–384), lacking SRR and IAD (1–189) containing IAD domain and SRR (190–427) and containing IAD domain (190–384) were created. The positive binding between each mutant and GST-ULD is indicated as +, and negative binding is indicated as – on the right panel. (C) Each of the Myc-tagged IRF3 mutants was introduced into the HEK293T cells and total cell lysates were subjected to GST-pull-down assay. The binding between GST-ULD and Myc-IRF3 was examined by immunoblotting. (D) Flag-IRF3 with or without Myc-TBK1-wt or KM was transfected into HEK293T cells. Total cell lysates were incubated with GST-ULD. The precipitates were subjected to immunoblotting using α-Flag antibody or α-Myc antibody.
TBK1-Δ-ULD fails to restore the target gene induction by LPS and poly(I:C)
To further investigate the functional involvement of TBK1-ULD in the regulation of signaling by upstream stimuli in vivo, we examined the role of TBK1-ULD in the IFN-inducible gene production by LPS and poly(I:C) in TBK1−/−IKK-i−/− MEF cells. We introduced TBK1-wt, Δ-ULD, L352A I353A and KM into MEF cells using a retroviral system (Figure 6A). The level of the protein expression of exogenous TBK1 was comparable with the level of endogenous TBK1 in control MEF cells, as shown by immunoblotting using α-TBK1 antibody (Figure 6A, left). The expression level of IRF3 was not altered by introduction of TBK1-wt or mutants in control and TBK1−/−IKK-i−/− MEF cells (Figure 6A). We examined the gene transcription activity using an IFN-β-promoter-driven luciferase construct in TBK1−/−IKK-i−/− MEF cells (Figure 6B). TBK1−/−IKK-i−/− MEF cells were transfected with an IFN-β-promoter-driven luciferase construct and TBK1-wt or TBK1 mutants were reconstituted by retrovirus infection. The gene promoter activities of IFN-β by LPS were restored by the presence of TBK1-wt (Figure 6B), whereas TBK1-Δ-ULD, L352A I353A and KM failed to rescue the response to LPS (Figure 6B). Similar effects of poly(I:C) transfection on IFN-β-promoter-driven luciferase activation were observed in reconstituted TBK1−/−IKK-i−/− MEF cells (Figure 6C). Poly(I:C)-induced IFN-β production was also examined by ELISA (Figure 6D). IFN-β production was not fully restored by TBK1-Δ-ULD or TBK1-L352A I353A mutant (Figure 6D). These data strongly suggest that the activation of TBK1 is regulated by upstream stimuli and that the kinase domain and the ULD need to interact to be functionally competent. However, we could not observe any change of the exogenous-ULD binding to TBK1 in poly(I:C)- or LPS-stimulated or nonstimulated conditions (data not shown). These data indicate that TBK1-ULD is required for the regulation of TBK1 kinase activity in LPS- and dsRNA-induced TLR signaling pathways in vivo (Figure 7A and B).
Figure 6.
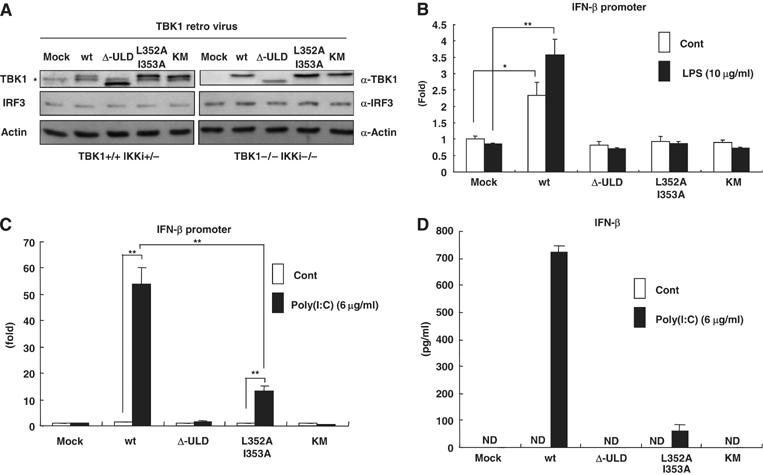
The ULD of TBK1 plays a role in LPS- and poly(I:C)-induced signaling pathway. (A) Mock, TBK1-wt, Δ-ULD, L352A I353A or KM retrovirus was infected into control (TBK1+/+IKK-i+/−) and TBK1−/−IKK-i−/− MEF cells. After 48 h of infection, cells were harvested and expression of TBK1 and IRF3 in total cell lysates was analyzed by immunoblotting using α-TBK1 antibody or α-IRF3 antibody. The endogenous TBK1 is indicated with *. The expression of IRF3 was not affected by retrovirus infection. The loading control was examined using α-actin antibody. (B, C) TBK1-wt, TBK1-Δ-ULD, TBK1-L352A I353A and TBK1-KM were introduced in TBK1−/−IKK-i−/− MEF cells by retrovirus. IFN-β promoter-driven luciferase construct was transfected together with β-GAL construct. The cells were treated or untreated with LPS (10 μg/ml) or poly(I:C) (6 μg/ml) for 16 h and harvested for the luciferase assay. The internal control was measured by β-GAL. (D) IFN-β production of infected TBK1−/−IKK-i−/− MEF cells after 24 h of poly(I:C) (6 μg/ml) treatment was examined by ELISA. (nd, not detected) (*P<0.05, **P<0.01).
Figure 7.
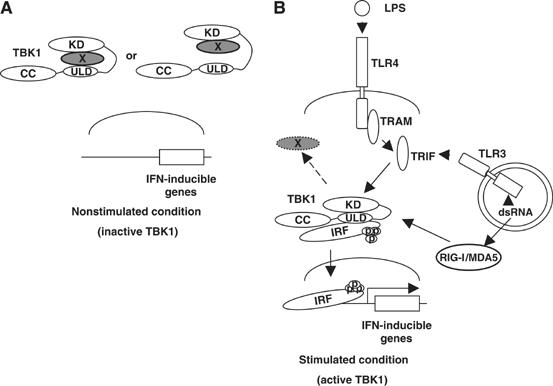
Active and inactive form of TBK1. (A) Under the nonstimulated condition, the domain interfaces of the ULD and kinase domain of TBK1 are not in the close proximity, failing to be fully functional. (B) TBK1 is active when the ULD interacts with the kinase domain. LPS- or poly(I:C)-induced active form of TBK1 interacts with IAD of IRF3, at least partially, via the ULD and phosphorylates it. Phosphorylated IRF3 dissociates from TBK1 and translocates into the nucleus, binds to the ISRE motif in the promoter region of the target genes and regulates their transcription. In the scheme, we draw interactions between single TBK1 and IRF molecules due to the clarity of presentation. It is, however, likely that these interactions are taking place in larger oligomeric TBK1/IRF complexes in vivo.
Discussion
In this study, we identified and characterized ubiquitin fold domains in two IKK-related kinases, TBK1 and IKK-i, and demonstrated their importance for initiation of downstream signaling pathways. Analogous ULDs were also found in other IKK-related kinases, IKKβ and IKKα. Our analyses suggest that the ULDs of the IKK-related kinases are most closely related to that of Fat10, although they have diverged quite far from the typical family of ULD. As a consequence, the interaction partners of the IKK-ULDs appear to be different from those of ubiquitin and its closer relatives (Figures 4F and 5A). In addition, a recent study demonstrated that the ULD of IKKɛ was shown to be important for the IKKɛ-Drosophila IAP1 complex formation, which appears to be important for IKKɛ-mediated phosphorylation and subsequent degradation of IAP1 (Kuranaga et al, 2006). This pathway is essential for the proneural clusters of the wing imaginal disc in which nonapoptotic caspase activity is required for proper sensory organ precursor development (Kuranaga et al, 2006). Taken together, the ULDs of TBK1 and IKK-i may function as protein–protein interaction domains important for the recruitment and phosphorylation of the binding partners, thereby regulating dowstream signaling.
An interesting feature of the ULD in TBK1 is its involvement in the regulation of the kinase activity of TBK1. There are kinases, like Aurora-A and -B and MARK2, which compromise C-terminal extensions of the catalytic core, which wrap around and terminate in a subdomain that binds N lobe (Panneerselvam et al, 2006). It is suggested that the C-terminal extensions are involved in regulation of kinase activity in these kinases (Bayliss et al, 2003; Sessa et al, 2005; Panneerselvam et al, 2006). In MARK2, there is a UBA domain, which corresponds to the C-terminal extension and binds to the catalytic cleft (Panneerselvam et al, 2006). Although the structural analysis of TBK1 is required for the conclusion, a similar conformational regulation of kinase activity in TBK1 is suggested. This is so far the first study, which demonstrates the positive role of the ULD domain in the control of the adjacent kinase domain. Moreover, the important role of ULD of IKKβ in the regulation of NF-κB signaling was shown previously (May et al, 2004). They have found that the ULD of IKKβ is required for the dissociation from p65; however, the precise mechanism how the ULD of IKKβ regulates its function has been obscure. We found that purified TBK1-ULD binds to other IKK-related serine-threonine kinases, IKKα, IKKβ (data not shown) or IKK-i, which share a high similarity with the kinase domain in TBK1 in amino-acid sequences. On the other hand, GST-ULD did not interact with PAK1, which is a serine-threonine kinase unrelated to IKK (data not shown). At present, we have not been able to map the binding surface on the kinase domain and it is thus still unclear how interactions between the ULD and the kinase domain may lead to the activation of TBK1. We also found that autophoshorylations of TBK1 and IKK-i were abolished by deletion of the ULD. It has been shown that S172 in the kinase domain of IKK-i is a critical phosphorylation site for the kinase activity (Shimada et al, 1999). These observations further support the important role of the ULD in making a formation of kinase-active TBK1 and IKK-i. An alternative scenario may involve a more favorable dimerization ability of the closed conformation of TBK1, upon overexpression or following interactions with upstream adaptor proteins involved in the TBK1 activation process, such as TRIF. However, we did not observe any changes of bindings of GST-ULD and TBK1 in the LPS- or poly(I:C)-stimulated conditions. As we observed a strong kinase activity of TBK1 even without any upstream stimuli in vitro, we speculate that there are negative regulatory mechanisms in the nonstimulated condition, which hold TBK1 in an inactive status by blocking the interaction between the kinase domain and the ULD (Figure 7A). Whether the interaction between the ULD and the kinase domain occurs within a single molecule or in an oligomer remains unclear.
Interestingly, we found that the TBK1-L352A I353A mutant did not lose the kinase activity completely, although the nuclear translocation of IRF3 and IFN-β and RANTES promoter activities were completely abolished. Moreover, we could detect only a very weak dimer formation of IRF3 by TBK1-L352A I353A mutant. These data raised one possibility that TBK1-L352A I353A does not modify the specific phosphorylation site of IRF3, which is important for the dimer formation. Indeed, phosphorylation of IRF3 at S386, which is a critical phosphorylation site for dimerization, by TBK1-L352A I353A mutant was barely detectable (Figure 2I). Interestingly, we found that the phosphorylation of IκBα by TBK1 was completely abolished by mutations at L352I353 (Figure 3E). It is well known that an NF-κB-responsive element exists in the promoter region of IFN-β and that p65 is involved in the regulation of RANTES promoter activation (Honda et al, 2006). These observations suggest that the mutation at L352A I353A abolishes phosphorylations of the targets in a substrate-specific manner. It is also suggested that the promoter activations of IFN-β and RANTES were abolished by the mutation at L352A I353A due to the incapability of IκBα phosphorylation. Another possibility is that IRF3 gets an extra modification required for proper nuclear translocation, such as ubiquitylation, in addition to the phosphorylation by TBK1. Previous studies revealed that ubiquitylations of Smad2 and Bcl-3 are required for their nuclear translocations (Lo and Massague, 1999; Massoumi et al, 2006). Moreover, IRF7 is polyubiquitylated by TRAF6 coexpression and the ubiquitylation of IRF7 was required for the TRAF6-induced IFN-β promoter activation (Kawai et al, 2005).
An additional function of the ULD is its interaction with the TBK1 substrates, IRF3 and IRF7. Interestingly, the binding between the ULD and IRFs was not blocked by mutations in the hydrophobic patch of the ULD, which is required for the binding to kinase domain. Therefore, the binding surfaces on the ULD to kinase domain or to IRFs are different from one another. This makes sense in the view of a positive regulation of this signal, because active TBK1, in which the binding surface of ULD to the kinase domain is occupied, can still access its substrates via the ULD and mediate their phosphorylation. However, the binding of the isolated ULD of TBK1 to IRFs was weak (Figure 5A and Supplementary Figure S3B) and the ULD binding to IRF3 was abolished when IRF3 was phosphorylated by TBK1 (Figure 5D), suggesting that in vivo interactions between TBK1 and IRFs might be more complex. It is also interesting that the binding region of the ULD in IRF3 is IAD (Figure 5B and C), which is responsible for the dimer formation of IRF3. These observations suggest a model of interactions between TBK1 and IRFs, whereby a transient interaction between TBK1-ULD and IRF3-IAD is regulated by phosphorylation of IRFs, leading to their dissociation from TBK1, subsequent dimerization via IADs and translocation of phosphorylated IRFs into the nucleus to regulate LPS-mediated IFN responses.
TBK1/IKK-i kinases are involved in different pathways mediating immune responses against exogenous agents such as bacteria and viruses. For example, bacterial polysaccharides LPS stimulate TLR to activate TBK1/IKK-i and IFN-inducible genes, whereas poly(I:C) stimulates TBK1 via TLR3 and also via RIG-I/MDA5 pathway independently of TLRs (Andrejeva et al, 2004; Yoneyama et al, 2004). Using in vivo model of TBK1-reconstituted MEF cells, we observed similar effects of both LPS and poly(I:C) on promoter activation and production of IFN-β. These observations collectively indicate that the TBK1-ULD functions in the more general aspects of the innate immune responses. We thus propose that the ULD in TBK1 and IKK-i play important roles in the regulation of their activities and downstream signaling pathway.
Materials and methods
Reporter gene assay
HEK293T cells or MEF cells were seeded on 48-well plates (2 × 104) 24 h before the transfection. Cells were transiently transfected with 40 ng of luciferase construct and 40 ng of β-Gal construct with various constructs. A total of 0.5 μg plasmid was introduced by FuGene6 (Roche) or FuGeneHD (Roche), following the manufacturer's protocol. After 24 h of transfection, MEF cells were treated by 10 μg/ml LPS or 6 μg/ml poly(I:C) for 16 h. Poly(I:C) was denatured at 55°C for 30 min, allowed to anneal to room temperature before use and complexed with cationic lipids, Lipofectamine 2000 (Invitrogen) for cell stimulations. Luciferase activity was measured with the luciferase reporter gene assay system (Roche). The internal control was measured by β-GAL reporter gene assay (Roche), following the manufacturer's protocol.
Immunofluorescence
Transfected HeLa cells were grown on poly-L-lysine-coated coverslips, washed twice in phosphate-buffered saline (PBS) and fixed 8 min with 4% paraformaldehyde in PBS. Cells were then permeabilized with 0.5% Triton-X-100 for 10 min. After washing with PBS, cells were blocked for 15 min in 5% fish gelatin in PBS containing 0.02% NaN3 and incubated 1 h with primary antibody (α-Myc), diluted 1:2000 in blocking solution. Coverslips were washed with blocking solution three times and incubated 45 min with Alexa Fluor 546-conjugated secondary antibody (Molecular Probes), diluted 1:500 in blocking solution. Unbound antibodies were washed and cells were incubated in 100 ng/ml 4′,6-diamidino-2 phenylindole HCl (DAPI) for 5 min to stain DNA and mounted in Fluoromount-G mounting solution (Southern Biotechnology Associates). Fluorescence microscopy was performed on an LSM510 META confocal microscope.
Immunoblotting and GST-pull-down assay
Immunoblotting and GST-pull-down assay were performed as described previously (Hoeller et al, 2006). Briefly, GST-ULD (TBK1 and IKK-i) and GST-Parkin-UBL were generated by amplifying the ULD or the UBL domain with PCR and subcloned into pGEX-4T1, using EcoRI-XhoI, BamHI-EcoRI and EcoRI-XhoI sites, respectably. GST-ULD-L352AI353A was generated by site-directed mutagenesis. GST-constructs were transformed into the bacterial strain BL21 and protein productions were induced with IPTG at 37°C. GST-proteins were purified with Sepharose 4B (Amersham).
Subcellular fractionation
To obtain nuclear and cytoplasmic fractions, cells were washed and sampled in 500 μl of cold PBS. Pelleted cells (500 g) were resuspended in hypotonic buffer (10 mM Tris–HCl (pH 7.4), 10 mM sodium iodide, 5 mM MgCl2, 1 mM PMSF) and homogenized with 20 strokes in a tissue grinder. The supernatant (600 g) was designated the cytoplasmic fraction. The pellet, representing the nuclear fraction, was washed with hypotonic buffer containing 0.1% NP-40 (Calbiochem).
Native PAGE
Native PAGE was performed as described previously (Iwamura et al, 2001) by using 7.5% Acrylamide gel. The gel was pre-run with 25 mM Tris and 192 mM glycine, pH 8.4 with or without 0.2% deoxycholate in the cathode and anode chamber, respectively, for 30 min at 40 mM. Samples in the native sample buffer (62.5 mM Tris–HCL (pH 6.8), 15% glycerol) were applied to the gel and electrophoresed for 50 min at 25 mM. Immunoblotting was performed.
Kinase assay
Kinase assay was performed as described previously (tenOever et al, 2004). Briefly, immunoprecipitated samples were prepared from total cell lysates of HEK293T cells in which Myc-TBK1 or Myc-IKK-i constructs were transfected using α-Myc antibody. γ-32P ATP (5 μCi), MBP (1 μg) and immunoprecipitated samples were incubated in the kinase buffer (20 mM HEPES, 10 mM MgCl2, 0.1 mM sodium orthovanadate, 20 mM β-glycerophosphate, 10 mM p-nitrophenylphosphate, 1 mM DTT and 50 mM NaCl) at 30°C for 30 min. The 50% of the precipitated samples were used for the kinase assay and the rest for the immunoblotting.
Retrovirus infection
Ecotropic retrovirus constructs were generated by amplifying the TBK1-wt, Δ-ULD, L352AI353A or KM by PCR and subcloned into pBabe-vector, using EcoRI-SalI sites. These plasmids were transfected into Phoenix cells and the supernatant was collected. After centrifuging and filtering the supernatants, they were used as viruses and were infected into 3T3 cells or MEF cells with polybrane (4 μg/ml). The medium was changed 6 h after infection and cells were harvested 36–48 h later for the further experiments.
ELISA
IFN-β production was measured by using Mouse-IFN-β ELISA Kit (PBL), following the manufacturer's protocol. Briefly, MEF cells were seeded on 48-well plates (2 × 104) and assay was performed using the cultured medium after 24 h of 6 μg/ml poly(I:C) transfection with Lipofectamine 2000.
Yeast two-hybrid assay
One to one transformation in yeast was performed as described previously (Bienko et al, 2005). Briefly, sequence corresponding to TBK1-ULD was subcloned into pYTH9 vector, created fusion proteins with the Gal4 DNA-binding domain. The vector was introduced into Y190 yeast strain, as well as the prey plasmid containing TBK1 (wt, 1–383, 1–301) or IRF3 fused to Gal4 activation domain. Experiments were performed as described previously (Bienko et al, 2005).
NMR measurements
Double-labeled samples of mouse TBK1 ULD (amino acids 302–383) were expressed in BL21 bacteria as a GST-fusion protein in standard minimal medium. The protein was purified with glutathione affinity chromatography, followed after cleavage with thrombin by size-exclusion chromatography. The purified protein contains the amino-acid sequence GSPEF at the N terminus and LERPHRD at the C terminus due to cloning into the pGEX4T1 vector. Purified samples were concentrated to ∼0.2 mM. A sample selectively 15N labeled on Lys was produced by expression in BL21 bacteria, samples selectively 15N labeled on Phe, Tyr, Leu or Ile were expressed in the auxotrophic strain DL39, following standard protocols. Selectively labeled samples were concentrated to 0.1 mM. For the sequential assignment, an HNCA, an HNCACB, a 15N-separated 3D-NOESY, a 15N-separated 3D-TOCSY, as well as homonuclear 2D-TOCSY and 2D-NOESY spectra were measured on Bruker Avance spectrometers operating at proton frequencies between 500 and 800 MHz. The identification of secondary structure elements was based on the analysis of the 1H and 13C chemical shifts with the programs CSI (Wishart and Sykes, 1994) and TALOS (Cornilescu et al, 1999).
Supplementary Material
Supplementary Figures
Supplementary Materials and Methods
Acknowledgments
We thank Drs Nobuhiko Kayagaki, Osamu Takeuchi, Stefan Knapp, Daniela Hoeller, Mirko Schmidt and Kaisa Haglund for discussions, constructive comments and critical reading of the manuscript. We also thank Ingrid Konrad, Masuda Sader, Natascha Rogova and Frank Löhr for the technical assistance. This work was supported by grants from the Deutsche Forschungsgemeinschaft, the German-Israeli Foundation to ID and by the Center for Biomolecular Magnetic Resonance. FI received a postdoctoral fellowship from The Uehara Memorial Foundation and the Alexander von Humboldt Foundation and CH received a fellowship from the Hessian Ministry for Science and Arts.
References
- Akira S, Uematsu S, Takeuchi O (2006) Pathogen recognition and innate immunity. Cell 124: 783–801 [DOI] [PubMed] [Google Scholar]
- Andrejeva J, Childs KS, Young DF, Carlos TS, Stock N, Goodbourn S, Randall RE (2004) The V proteins of paramyxoviruses bind the IFN-inducible RNA helicase, mda-5, and inhibit its activation of the IFN-beta promoter. Proc Natl Acad Sci USA 101: 17264–17269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bayliss R, Sardon T, Vernos I, Conti E (2003) Structural basis of Aurora-A activation by TPX2 at the mitotic spindle. Mol Cell 12: 851–862 [DOI] [PubMed] [Google Scholar]
- Bienko M, Green CM, Crosetto N, Rudolf F, Zapart G, Coull B, Kannouche P, Wider G, Peter M, Lehmann AR, Hofmann K, Dikic I (2005) Ubiquitin-binding domains in Y-family polymerases regulate translesion synthesis. Science 310: 1821–1824 [DOI] [PubMed] [Google Scholar]
- Cornilescu G, Delaglio F, Bax A (1999) Protein backbone angle restraints from searching a database for chemical shift and sequence homology. J Biomol NMR 13: 289–302 [DOI] [PubMed] [Google Scholar]
- Fitzgerald KA, McWhirter SM, Faia KL, Rowe DC, Latz E, Golenbock DT, Coyle AJ, Liao SM, Maniatis T (2003a) IKKepsilon and TBK1 are essential components of the IRF3 signaling pathway. Nat Immunol 4: 491–496 [DOI] [PubMed] [Google Scholar]
- Fitzgerald KA, Rowe DC, Barnes BJ, Caffrey DR, Visintin A, Latz E, Monks B, Pitha PM, Golenbock DT (2003b) LPS-TLR4 signaling to IRF-3/7 and NF-kappaB involves the toll adapters TRAM and TRIF. J Exp Med 424: 1043–1055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartmann-Petersen R, Gordon C (2004) Integral UBL domain proteins: a family of proteasome interacting proteins. Semin Cell Dev Biol 15: 247–259 [DOI] [PubMed] [Google Scholar]
- Hemmi H, Takeuchi O, Sato S, Yamamoto M, Kaisho T, Sanjo H, Kawai T, Hoshino K, Takeda K, Akira S (2004) The roles of two IkappaB kinase-related kinases in lipopolysaccharide and double stranded RNA signaling and viral infection. J Exp Med 199: 1641–1650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hicke L, Schubert HL, Hill CP (2005) Ubiquitin-binding domains. Nat Rev Mol Cell Biol 6: 610–621 [DOI] [PubMed] [Google Scholar]
- Hiscott J, Grandvaux N, Sharma S, Tenoever BR, Servant MJ, Lin R (2001) Convergence of the NF-kappaB and interferon signaling pathways in the regulation of antiviral defense and apoptosis. Ann N Y Acad Sci 19: 237–248 [DOI] [PubMed] [Google Scholar]
- Hoebe K, Du X, Georgel P, Janssen E, Tabeta K, Kim SO, Goode J, Lin P, Mann N, Mudd S, Crozat K, Sovath S, Han J, Beutler B (2003) Identification of Lps2 as a key transducer of MyD88-independent TIR signalling. Nature 301: 743–748 [DOI] [PubMed] [Google Scholar]
- Hoeller D, Crosetto N, Blagoev B, Raiborg C, Tikkanen R, Wagner S, Kowanetz K, Breitling R, Mann M, Stenmark H, Dikic I (2006) Regulation of ubiquitin-binding proteins by monoubiquitination. Nat Cell Biol 8: 163–169 [DOI] [PubMed] [Google Scholar]
- Honda K, Taniguchi T (2006) IRFs: master regulators of signalling by Toll-like receptors and cytosolic pattern-recognition receptors. Nat Rev Immunol 6: 644–658 [DOI] [PubMed] [Google Scholar]
- Honda K, Yanai H, Negishi H, Asagiri M, Sato M, Mizutani T, Shimada N, Ohba Y, Takaoka A, Yoshida N, Taniguchi T (2005) IRF-7 is the master regulator of type-I interferon-dependent immune responses. Nature 434: 772–777 [DOI] [PubMed] [Google Scholar]
- Hurley JH, Lee S, Prag G (2006) Ubiquitin-binding domains. Biochem J 399: 361–372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwamura T, Yoneyama M, Yamaguchi K, Suhara W, Mori W, Shiota K, Okabe Y, Namiki H, Fujita T (2001) Induction of IRF-3/-7 kinase and NF-kappaB in response to double-stranded RNA and virus infection: common and unique pathways. Genes Cells 6: 375–388 [DOI] [PubMed] [Google Scholar]
- Janeway CA Jr, Medzhitov R (2002) Innate immune recognition. Annu Rev Immunol 20: 197–216 [DOI] [PubMed] [Google Scholar]
- Kawai T, Takahashi K, Sato S, Coban C, Kumar H, Kato H, Ishii KJ, Takeuchi O, Akira S (2005) IPS-1, an adaptor triggering RIG-I- and Mda5-mediated type I interferon induction. Nat Immunol 6: 981–988 [DOI] [PubMed] [Google Scholar]
- Kerscher O, Felberbaum R, Hochstrasser M (2006) Modification of proteins by ubiquitin and ubiquitin-like proteins. Annu Rev Cell Dev Biol 22: 159–180 [DOI] [PubMed] [Google Scholar]
- Kuranaga E, Kanuka H, Tonoki A, Takemoto K, Tomioka T, Kobayashi M, Hayashi S, Miura M (2006) Drosophila IKK-related kinase regulates nonapoptotic function of caspases via degradation of IAPs. Cell 126: 583–596 [DOI] [PubMed] [Google Scholar]
- Lemaitre B (2002) The road to Toll. Nat Rev Immunol 20: 521–527 [DOI] [PubMed] [Google Scholar]
- Lo RS, Massague J (1999) Ubiquitin-dependent degradation of TGF-beta-activated smad2. Nat Cell Biol 1: 472–478 [DOI] [PubMed] [Google Scholar]
- Massoumi R, Chmielarska K, Hennecke K, Pfeifer A, Fassler R (2006) Cyld inhibits tumor cell proliferation by blocking Bcl-3-dependent NF-kappaB signaling. Cell 125: 665–677 [DOI] [PubMed] [Google Scholar]
- May MJ, Larsen SE, Shim JH, Madge LA, Ghosh S (2004) A novel ubiquitin-like domain in IkappaB kinase beta is required for functional activity of the kinase. J Biol Chem 279: 45528–45539 [DOI] [PubMed] [Google Scholar]
- Panneerselvam S, Marx A, Mandelkow EM, Mandelkow E (2006) Structure of the catalytic and ubiquitin-associated domains of the protein kinase MARK/Par-1. Structure 14: 173–183 [DOI] [PubMed] [Google Scholar]
- Pomerantz JL, Baltimore D (1999) NF-kappaB activation by a signaling complex containing TRAF2, TANK and TBK1, a novel IKK-related kinase. EMBO J 18: 6694–6704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin BY, Liu C, Lam SS, Srinath H, Delston R, Correia JJ, Derynck R, Lin K (2003) Crystal structure of IRF-3 reveals mechanism of autoinhibition and virus-induced phosphoactivation. Nat Struct Biol 10: 913–921 [DOI] [PubMed] [Google Scholar]
- Sato M, Suemori H, Hata N, Asagiri M, Ogasawara K, Nakao K, Nakaya T, Katsuki M, Noguchi S, Tanaka N, Taniguchi T (2000) Distinct and essential roles of transcription factors IRF-3 and IRF-7 in response to viruses for IFN-alpha/beta gene induction. Immunity 13: 539–548 [DOI] [PubMed] [Google Scholar]
- Sato S, Sugiyama M, Yamamoto M, Watanabe Y, Kawai T, Takeda K, Akira S (2003) Toll/IL-1 receptor domain-containing adaptor inducing IFN-beta (TRIF) associates with TNF receptor-associated factor 6 and TANK-binding kinase 1, and activates two distinct transcription factors, NF-kappa B and IFN-regulatory factor-3, in the Toll-like receptor signaling. J Immunol 171: 4304–4310 [DOI] [PubMed] [Google Scholar]
- Schauber C, Chen L, Tongaonkar P, Vega I, Lambertson D, Potts W, Madura K (1998) Rad23 links DNA repair to the ubiquitin/proteasome pathway. Nature 391: 715–718 [DOI] [PubMed] [Google Scholar]
- Sessa F, Mapelli M, Ciferri C, Tarricone C, Areces LB, Schneider TR, Stukenberg PT, Musacchio A (2005) Mechanism of Aurora B activation by INCENP and inhibition by hesperadin. Mol Cell 18: 379–391 [DOI] [PubMed] [Google Scholar]
- Sharma S, tenOever BR, Grandvaux N, Zhou GP, Lin R, Hiscott J (2003) Triggering the interferon antiviral response through an IKK-related pathway. Science 300: 1148–1151 [DOI] [PubMed] [Google Scholar]
- Shimada T, Kawai T, Takeda K, Matsumoto M, Inoue J, Tatsumi Y, Kanamaru A, Akira S (1999) IKK-i, a novel lipopolysaccharide-inducible kinase that is related to IkappaB kinases. Int Immunol 11: 1357–1362 [DOI] [PubMed] [Google Scholar]
- Takahasi K, Suzuki NN, Horiuchi M, Mori M, Suhara W, Okabe Y, Fukuhara Y, Terasawa H, Akira S, Fujita T, Inagaki F (2003) X-ray crystal structure of IRF-3 and its functional implications. Nat Struct Biol 10: 922–927 [DOI] [PubMed] [Google Scholar]
- Takeda K, Akira S (2003) Toll-like receptors in innate immunity. Int Immunol 1010: 1–14 [DOI] [PubMed] [Google Scholar]
- Taniguchi T, Ogasawara K, Takaoka A, Tanaka N (2001) IRF family of transcription factors as regulators of host defense. Annu Rev Immunol 19: 623–655 [DOI] [PubMed] [Google Scholar]
- tenOever BR, Sharma S, Zou W, Sun Q, Grandvaux N, Julkunen I, Hemmi H, Yamamoto M, Akira S, Yeh WC, Lin R, Hiscott J (2004) Activation of TBK1 and IKKvarepsilon kinases by vesicular stomatitis virus infection and the role of viral ribonucleoprotein in the development of interferon antiviral immunity. J Virol 78: 10636–10649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walters KJ, Goh AM, Wang Q, Wagner G, Howley PM (2004) Ubiquitin family proteins and their relationship to the proteasome: a structural perspective. Biochim Biophys Acta 15: 73–87 [DOI] [PubMed] [Google Scholar]
- Wishart DS, Sykes BD (1994) The 13C chemical-shift index: a simple method for the identification of protein secondary structure using 13C chemical-shift data. J Biomol NMR 4: 171–180 [DOI] [PubMed] [Google Scholar]
- Yamamoto M, Sato S, Hemmi H, Hoshino K, Kaisho T, Sanjo H, Takeuchi O, Sugiyama M, Okabe M, Takeda K, Akira S (2003a) Role of adaptor TRIF in the MyD88-independent toll-like receptor signaling pathway. Science 301: 640–643 [DOI] [PubMed] [Google Scholar]
- Yamamoto M, Sato S, Hemmi H, Uematsu S, Hoshino K, Kaisho T, Takeuchi O, Takeda K, Akira S (2003b) TRAM is specifically involved in the Toll-like receptor 4-mediated MyD88-independent signaling pathway. Nat Immunol 198: 1144–1150 [DOI] [PubMed] [Google Scholar]
- Yoneyama M, Kikuchi M, Natsukawa T, Shinobu N, Imaizumi T, Miyagishi M, Taira K, Akira S, Fujita T (2004) The RNA helicase RIG-I has an essential function in double-stranded RNA-induced innate antiviral responses. Nat Immunol 5: 730–737 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figures
Supplementary Materials and Methods