

Abstract
The c-Myc oncoprotein promotes cell growth by enhancing ribosomal biogenesis through upregulation of RNA polymerases I-, II-, and III-dependent transcription. Overexpression of c-Myc and aberrant ribosomal biogenesis leads to deregulated cell growth and tumorigenesis. Hence, c-Myc activity and ribosomal biogenesis must be regulated in cells. Here, we show that ribosomal protein L11, a component of the large subunit of the ribosome, controls c-Myc function through a negative feedback mechanism. L11 is transcriptionally induced by c-Myc, and overexpression of L11 inhibits c-Myc-induced transcription and cell proliferation. Conversely, reduction of endogenous L11 by siRNA increases these c-Myc activities. Mechanistically, L11 binds to the Myc box II (MB II), inhibits the recruitment of the coactivator TRRAP, and reduces histone H4 acetylation at c-Myc target gene promoters. In response to serum stimulation or serum starvation, L11 and TRRAP display inverse promoter-binding profiles. In addition, L11 regulates c-Myc levels. These results identify L11 as a feedback inhibitor of c-Myc and suggest a novel role for L11 in regulating c-Myc-enhanced ribosomal biogenesis.
Keywords: cell cycle, C-Myc, L11, ribosomal proteins, transcription, TRRAP
Introduction
c-Myc is a transcription factor that regulates the expression of numerous genes involved in proliferation, cell growth, differentiation, apoptosis, metabolism, and neoplastic transformation (reviewed by Grandori et al, 2000; Pelengaris et al, 2002a; Adhikary and Eilers, 2005). Maintaining the proper level of c-Myc expression is essential for normal cellular functions and animal development, as homozygous deletion of c-myc is embryonic lethal in mice (Davis et al, 1993) and cells lacking c-Myc exit the cell cycle and do not proliferate (de Alboran et al, 2001; Trumpp et al, 2001). However, deregulated expression of c-Myc contributes to tumorigenesis (Pelengaris et al, 2002b; Adhikary and Eilers, 2005). Overexpression of c-Myc is observed in various human tumors (reviewed by Nesbit et al, 1999; Pelengaris et al, 2002a). Constitutive or inducible expression of c-myc transgene leads to neoplastic pre-malignant and malignant phenotypes in mice (Adams et al, 1985; Felsher and Bishop, 1999; Pelengaris et al, 1999, 2002b). Thus, precise regulation of c-Myc expression and transcriptional activity is critical for normal cellular function.
Two critical domains are required for c-Myc biological activity. The C-terminal basic helix-loop-helix leucine zipper (bHLH/LZ) domain of c-Myc heterodimerizes with its partner protein Max (Amati et al, 1992; Luscher and Larsson, 1999; Pelengaris et al, 2002a; Adhikary and Eilers, 2005) and mediates sequence-specific DNA recognition of E-box elements. The N-terminal transcriptional activation domain (TAD) of c-Myc contains two conserved segments termed Myc box (MB) I and II. The MB I is implicated in regulation of c-Myc protein stability through a phosphorylation-dependent ubiquitylation-mediated proteasomal degradation pathway, which involves multiple protein kinases, protein phosphotase 2A, prolyl isomerase Pin 1, and Fbw7 containing E3 ubiquitin ligase (Moberg et al, 2004; Sears, 2004; Welcker et al, 2004; Yada et al, 2004; Yeh et al, 2004; Arnold and Sears, 2006, and reviewed by Dai et al, 2006a). The MB II is critical for all known c-Myc functions. Several critical coactivators of c-Myc that have been shown to be essential for c-Myc-mediated transcription bind to the MB II. First, TRRAP (McMahon et al, 1998), a core component of the TIP60 and GCN5 histone acetyltransferase (HAT) complexes, is recruited to c-Myc target gene promoters through binding to MB II. These complexes mediate histone acetylation and facilitate transcription of c-Myc target genes (McMahon et al, 2000; Park et al, 2001; Frank et al, 2003; Bernardi et al, 2004). Second, TIP48/TIP49 ATPases, components of chromatin remodeling complexes, also interact with c-Myc at MB II and their ATPase activity is required for c-Myc-induced transformation (Wood et al, 2000). Third, the F-box protein Skp2, a component of the SCFskp2 E3 ligase complex, binds to c-Myc at the MB II and mediates its ubiquitylation and degradation (Kim et al, 2003; von der Lehr et al, 2003). However, Skp2-mediated degradation is required for c-Myc transactivation, suggesting that Skp2 is an important coactivator for c-Myc-regulated transcription as well (Kim et al, 2003; von der Lehr et al, 2003). Recently, an HECT domain containing E3 ligase, named HectH9, has been shown to bind to the TAD of c-Myc and to ubiquitylate this protein through a Lysine-63 linked ubiquitin (Adhikary et al, 2005). This ubiquitylation also enhances c-Myc activity by facilitating recruitment of coactivator p300 acetyltransferase (Adhikary et al, 2005). Hence, the N-terminal TAD is crucial for regulating c-Myc stability and activity.
The importance of c-Myc activity for normal cell growth may be in part attributed to its recently identified role in ribosomal biogenesis (reviewed by Ruggero and Pandolfi, 2003; Oskarsson and Trumpp, 2005). Ribosomal biogenesis is a tightly regulated process requiring coordinated transcription mediated by all three RNA polymerases to ensure efficient and accurate production of ribosomes (reviewed by Ruggero and Pandolfi, 2003; White, 2005). Deregulation of ribosomal biogenesis also contributes to tumorigenesis (reviewed by Ruggero and Pandolfi, 2003). c-Myc appears to be a major player in controlling ribosomal biogenesis, as it has been shown to regulate transcription by these RNA polymerases (Adhikary and Eilers, 2005; Oskarsson and Trumpp, 2005). Specifically, c-Myc has recently been shown to enhance Pol I-catalyzed rRNA synthesis (Arabi et al, 2005; Grandori et al, 2005; Grewal et al, 2005) as well as Pol III-mediated 5S and tRNA transcription (Gomez-Roman et al, 2003). In addition, Pol II-mediated transcription of genes encoding ribosome assembly proteins, and translation initiation and elongation factors is stimulated by c-Myc as well (Coller et al, 2000; Guo et al, 2000; Boon et al, 2001; Menssen and Hermeking, 2002). In spite of the expanding list of c-Myc target genes, it remains unclear how c-Myc activity is regulated during ribosomal biogenesis to maintain homeostasis.
Here, we report that ribosomal protein L11 controls c-Myc function through a negative feedback mechanism. L11 is transcriptionally activated by c-Myc, whereas L11 in turn inhibits c-Myc transactivational activity by binding to the MB II region of c-Myc and inhibiting the recruitment of the TRRAP coactivator and subsequent histone H4 acetylation at the c-Myc target gene promoters. Consistently, reduction of endogenous L11 increases c-Myc activity. Our results demonstrate that L11 is a feedback inhibitor of c-Myc.
Results
Overexpression of L11 inhibits c-Myc transactivation activity
Previous work from our lab and others revealed that several ribosomal proteins including L11 negatively regulate cell cycle progression by inhibiting MDM2 and thus activating p53 (Lohrum et al, 2003; Zhang et al, 2003; Bhat et al, 2004; Dai and Lu, 2004; Dai et al, 2004, 2006b). L11 has also been identified as a potential c-Myc transcriptional target by several gene-expression profile-based studies (Coller et al, 2000; Guo et al, 2000; Boon et al, 2001; Menssen and Hermeking, 2002) and c-Myc enhances ribosomal biogenesis through upregulation of transcription of rRNA, tRNA, as well as genes encoding ribosome assembly proteins, translation initiation, and elongation factors. Altogether, these studies prompted us to examine whether L11's ability to negatively regulate cellular proliferation and growth also involves suppression of c-Myc activity in a negative feedback manner. To this end, we first examined the effect of L11 on c-Myc-dependent transcription of a luciferase reporter gene driven by a c-Myc-responsive E box-containing E2F2 promoter. As expected, c-Myc increased luciferase activity in H1299 cells (Figure 1A), and this effect was not observed when the E boxes were mutated to prevent c-Myc-binding (Figure 1A). By contrast, coexpression of L11 drastically reduced c-Myc-dependent luciferase activity in a dose-dependent manner. However, such effect was not observed when the mutant E box E2F2 promoter was used (Figure 1A). This inhibition was p53-independent, as H1299 cells are null for p53. This inhibition was also ARF-independent, as L11 also inhibited c-Myc transcriptional activity in ARF-null U2OS cells (Figure 1B). Interestingly, the ability of c-Myc to activate luciferase expression was more potent in ARF-deficient U2OS cells (Figure 1B) than in ARF-proficient H1299 cells (Figure 1A). This result possibly reflects the inhibitory effect of ARF on c-Myc activity (Datta et al, 2004; Qi et al, 2004). This inhibition was specific to wild-type L11, as the N-terminus-deleted mutant of L11 (L1166−178) that did not bind to c-Myc (Figure 3A) had no effect on c-Myc transcriptional activity (Figure 1B). These results suggest an inhibitory effect of L11 on c-Myc transactivation activity independently of p53 and ARF.
Figure 1.
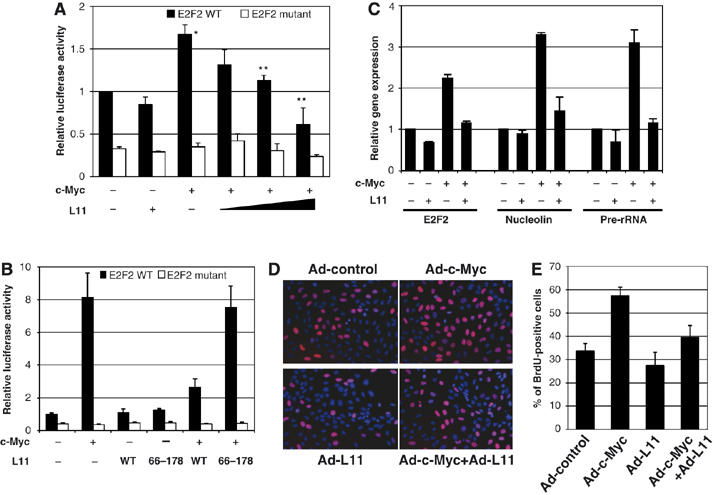
L11 inhibits c-Myc-dependent transactivation activity and reduces the percentage of cells in S phase. (A) L11 inhibits c-Myc-induced luciferase reporter expression driven by wild-type (WT), but not mutant, E box-containing E2F2 promoter. H1299 cells transfected with plasmids, as indicated, were subjected to luciferase assays to determine the reporter activity. *Indicates P<0.01 compared to control vector transfection; **indicates P<0.01 compared to transfection with c-Myc only. The protein expression of soluble c-Myc and L11 in this assay is shown in Supplementary Figure S1A. (B) Wild-type L11, but not its N-terminal deletion mutant (L1166−178), inhibited c-Myc-dependent luciferase reporter expression driven by WT, but not mutant, E box-containing E2F2 promoter. U2OS cells were transfected with plasmids as indicated and c-Myc driven luciferase activity were measured. (C) L11 inhibits c-Myc-dependent transcription of the endogenous c-Myc target genes. U2OS cells were infected with Ad-L11 and Ad-c-Myc individually or together and the expression of E2F2, nucleolin, and rRNA were measured by quantitative RT–PCR assays. The expression of total Ad-c-Myc and Ad-L11 proteins in this assay is shown in Figure 7H. (D, E) L11 reduces the percentage of cells in S phase determined by BrdU incorporation assay. U2OS cells infected with Ad-c-Myc and/or Ad-L11 were labeled with BrdU and subjected to anti-BrdU staining (red) and counter-stained with DAPI. Percentage of BrdU-positive cells is shown in (E).
To test whether L11 inhibits c-Myc-induced expression of endogenous target genes, we performed real-time PCR assays to analyze two of the c-Myc target genes, E2F2 and nucleolin, in cells. As shown in Figure 1C, overexpression of adenoviral-encoded c-Myc induced the expression of both E2F2 and nucleolin in U2OS cells. This effect was drastically reduced when L11 is also overexpressed. Similar effects were also observed in WI38 cells using adenovirus-encoding c-Myc and L11 (Supplementary Figure S1B). These results indicate that L11 inhibits c-Myc-activated transcription by Pol II.
c-Myc has also been shown to activate RNA Pol I-mediated transcription of precursor rRNA (Arabi et al, 2005; Grandori et al, 2005; Grewal et al, 2005). To test whether L11 can affect this c-Myc activity, we conducted transfection experiments followed by real-time reverse transcriptase–polymerase chain reaction (RT–PCR) assays. Indeed, L11 significantly reduced c-Myc-activated precursor rRNA synthesis as analyzed by detecting the expression of the 5′-external transcribed spacer (ETS) sequence (nt 852–972) of precursor rRNA (Figure 1C). We also performed a 3H-uridine-labeling rRNA assay, followed by quantitative analysis of all rRNA precursors. As expected (Arabi et al, 2005; Grandori et al, 2005; Grewal et al, 2005), c-Myc stimulated the synthesis of precursor rRNA, and again L11 suppressed this stimulation (Supplementary Figure S1C and D). Altogether, these results indicate that L11 also suppresses c-Myc-induced RNA Pol I-catalyzed transcription.
Overexpression of L11 inhibits c-Myc-driven cell proliferation
To determine whether L11 affects c-Myc-induced cell proliferation, BrdU incorporation was measured in serum-starved U2OS cells infected with recombinant adenoviruses encoding c-Myc (Ad-c-Myc) and L11 (Ad-L11) individually or together (Figure 1D). Quantification of BrdU incorporation showed that c-Myc significantly increased the number of cells undergoing DNA synthesis as expected (Datta et al, 2004) (Figure 1E). Consistent with the results in Figure 1A–C, coexpression of L11 reduced the number of c-Myc-induced S phase cells (Figure 1E). Furthermore, this inhibitory effect was observed in p53-null mouse embryonic fibroblast, 10.1, cells (Supplementary Figure S1E and F). These results demonstrate that L11 inhibits c-Myc-mediated cell proliferation independently of p53 and ARF.
L11 binds to c-Myc in cells
To elucidate possible mechanisms underlying the inhibition of c-Myc activity by L11, we tested whether L11 physically associates with c-Myc. To this end, we expressed V5-tagged c-Myc and Flag-tagged L11 individually or together in H1299 cells followed by co-immunoprecipitation (co-IP)-immunoblot analyses with either anti-V5 or anti-Flag antibodies. Indeed, c-Myc specifically co-immunoprecipitated with L11 by the anti-Flag antibody when coexpressed with Flag-L11 (Figure 2A). Conversely, L11 also specifically co-immunoprecipitated with c-Myc by the anti-V5 antibody (Figure 2B). We also observed that the untagged c-Myc specifically co-immunoprecipitated with Flag-L11 by the anti-Flag antibody (Figure 2C), suggesting that the N-terminal V5 tag would not account for the Myc interaction with L11. Moreover, endogenous c-Myc and L11 were specifically co-immunoprecipitated by either anti-L11 or anti-c-Myc antibodies (Figures 2D and E). This association was ARF, p53, and MDM2 independent, as L11 interacted with c-Myc in ARF-null U2OS cells (Supplementary Figure 2A) and in p53−/−mdm2−/− MEF cells (Supplementary Figure 2B). This interaction was also rRNA independent, as it was detected when cell lysates were pre-treated with RNase (Supplementary Figure 2C). Hence, L11 can specifically interact with c-Myc independently of p53, MDM2, ARF, and rRNA in cells.
Figure 2.
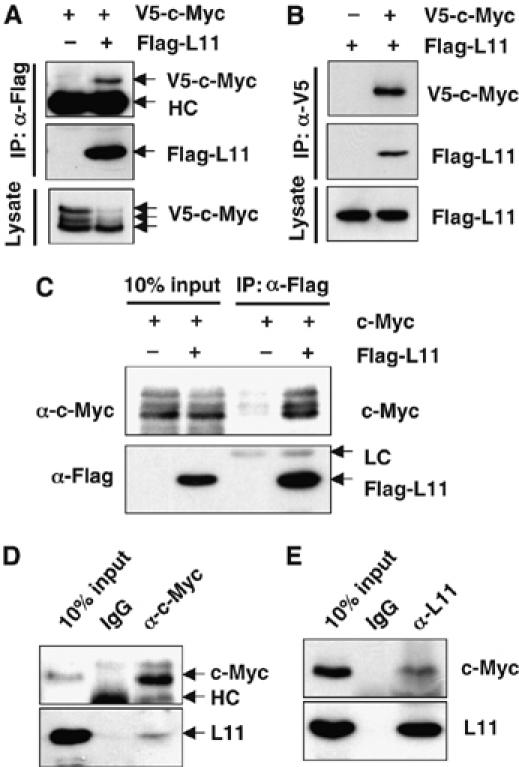
L11 interacts with c-Myc in cells. (A, B) Co-IP of ectopic L11 with ectopic c-Myc. H1299 cells were transfected with V5-c-Myc and Flag-L11 individually, or together and subjected to IP with anti-Flag (A) or anti-V5 (B), followed by IB with anti-Flag or anti-V5 antibody. (C) Overexpressed L11 interacts with untagged c-Myc. H1299 cells transfected with untagged c-Myc in the presence or absence of Flag-L11 were immunoprecipitated with anti-Flag antibody, followed by IB with indicated antibodies. (D, E) Endogenous L11 interacts with endogenous c-Myc. H1299 cell lysates were immunoprecipitated with monoclonal anti-c-Myc (C33) (D) or anti-L11 (E) antibody, followed by IB with anti-L11, polyclonal anti-c-Myc (N262) (D), or monoclonal anti-c-Myc (9E10) antibody (E).
N-terminal domain of L11 binds to MB II of c-Myc
To map c-Myc-binding domains of L11, we first constructed a panel of Flag-tagged L11 deletion mutants. We co-introduced wild-type L11 or its deletion mutants with V5-c-Myc into H1299 cells and performed co-IP assays. As shown in Figure 3A, c-Myc specifically co-immunoprecipitated with the mutants containing N-terminal 65 amino acids (Flag-L111−65 and Flag-L111−125), but not the mutant lacking these residues (Flag-L1166−178), indicating that c-Myc interacts with the N-terminus of L11 (Figure 3B), which was required for suppression of c-Myc-activated transcription by L11 (Figure 1B).
Figure 3.
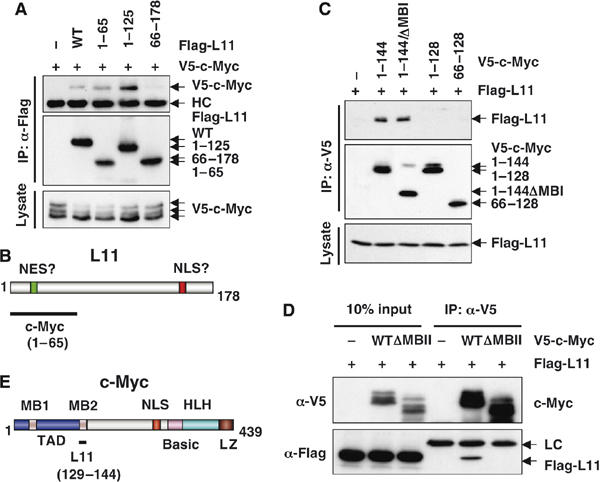
The N-terminal domain of L11 interacts with the MB II of c-Myc. (A) c-Myc interacts with the N-terminal domain of L11. H1299 cells transfected with plasmids encoding wild-type L11 or its deletion mutants together with the c-Myc plasmid were subjected to IP using anti-Flag antibodies, followed by IB with anti-V5 or anti-Flag antibodies. (B) Schematic diagram of L11 and its c-Myc-binding domains. (C) L11 binds to the MB II region of c-Myc. H1299 cells were transfected with the V5-tagged TAD of c-Myc (c-Myc1−144) and its deletion mutants and subjected to IP using anti-V5 antibodies, followed by IB using anti-Flag or anti-V5 antibodies. (D) The MB II region of c-Myc is required for c-Myc interaction with L11. H1299 cells were transfected with the V5-tagged WT c-Myc or MB II-deleted mutant of c-Myc (ΔMB II), together with Flag-L11, and subjected to IP using anti-Flag antibodies, followed by IB using antibodies as indicated. (E) Schematic diagram of c-Myc showing the L11 binding MB II of the TAD.
To define L11-binding domains of c-Myc, we first constructed a set of c-Myc deletion mutants and found by similar co-IP experiments that deletion of the N-terminal TAD of c-Myc abolished its interaction with L11, whereas the truncated N-terminal TAD-containing c-Myc, c-Myc1−144, efficiently bound to L11 (Supplementary Figure S2D), indicating that L11 specifically associates with the TAD of c-Myc. The c-Myc TAD contains conserved MB I and II motifs that are critical for regulation of c-Myc protein stability and transactivation activity. To test which motif interacts with L11, we further constructed a set of V5-tagged deletion mutants of TAD, and found by similar co-IP assays that deletion of MB I (c-Myc1−144/ΔMB I) retained its binding capacity to L11 similar to c-Myc1−144, whereas the c-Myc N-terminal fragment that retains MB I, but lacks MB II (c-Myc1−128), completely abolished its binding to L11 (Figure 3C). The requirement of MB II for c-Myc to bind to L11 was further verified using a V5-tagged c-Myc mutant that lacks MB II only (c-Myc/ΔMB II) (Figure 3D). Taken together, these results reveal that L11 binds to the c-Myc MB II (Figure 3E), which is critical for c-Myc function (Pelengaris et al, 2002a; Adhikary and Eilers, 2005).
L11 binds to c-Myc at c-Myc target gene promoters
As L11 binds to the c-Myc MB II (Figure 3D) and this MB II is known to bind to several critical c-Myc coactivators, such as TRRAP, GCN5, TIP60 (McMahon et al, 1998; McMahon et al, 2000; Frank et al, 2003), we wanted to test whether L11 suppresses c-Myc activity by impairing the recruitment of TRRAP to c-Myc target gene promoters. To this end, we first tested whether L11 associates with the E box-containing promoters of E2F2, nucleolin, and eIF-4E genes as well as rDNA genes by chromatin immunoprecipitation (ChIP)-PCR analysis. As shown in Figure 4A, endogenous L11 specifically associated with each of these c-Myc target gene promoters, but not the control intron 5 region of l11 in H1299 cells. These results suggest that L11 associates with c-Myc target gene promoters. To test whether the association of L11 with c-Myc target gene promoters requires c-Myc, we first determined whether manipulating the levels of c-Myc in cells would affect the binding of endogenous L11 to these promoters. As shown in Figure 4B, overexpression of c-Myc enhanced the binding of L11 to the nucleolin promoter. Conversely, knockdown of c-Myc markedly reduced the binding of L11 to the nucleolin gene promoter (Figure 4C). These results suggest that the L11 binding to a c-Myc target gene promoter may require the binding of c-Myc in the same promoter.
Figure 4.
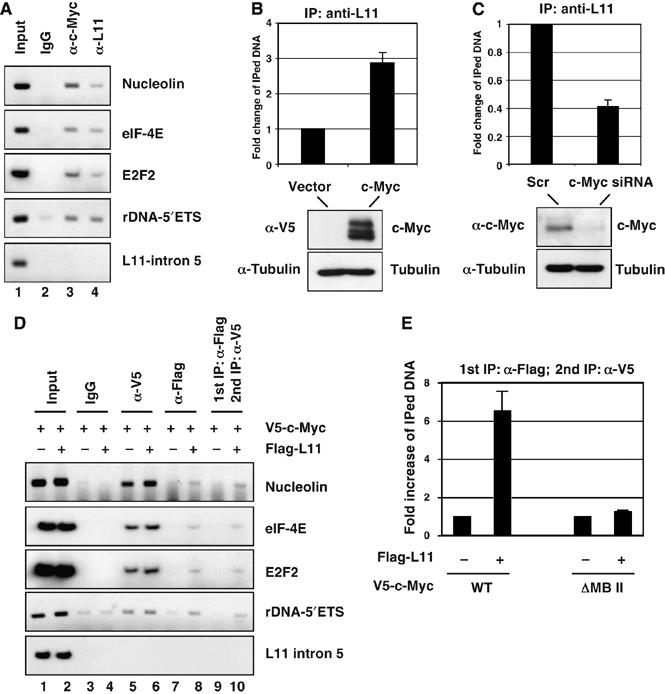
L11 co-resides with c-Myc in c-Myc target gene promoter. (A) Endogenous L11 binds to c-Myc target gene promoters. ChIP-PCR assays were conducted to detect the recruitment of endogenous c-Myc and L11 in c-Myc target gene promoters in H1299 cells, using anti-c-Myc (N262) or anti-L11 antibodies. (B) Overexpression of c-Myc enhances L11 binding to the nucleolin gene promoter. H1299 cells were transfected with or without V5-c-Myc. ChIP-PCR assays were conducted using anti-L11 antibodies to detect the recruitment of endogenous L11 at the nucleolin gene promoter. The expression of V5-c-Myc is shown in the bottom panels. (C) Knockdown of endogenous c-Myc decreases L11 binding to the nucleolin gene promoter. U2OS cells were transfected with scrambled or c-Myc siRNA. ChIP-PCR assays were conducted using anti-L11 antibodies to detect the recruitment of endogenous L11 at the nucleolin gene promoter. The expression of endogenous c-Myc is shown in the bottom panels. (D) L11 co-resides with c-Myc in its target gene promoters. H1299 cells were transfected with plasmids as indicated. ChIP-PCR assays were performed using anti-V5 or anti-Flag antibodies. Ten fold of initial lysates were used for anti-Flag IP compare to IP with anti-V5. The Flag-L11 protein–DNA complexes were eluted with Flag peptide competition and 10% of the elutes was used for DNA purification and PCR amplification (lane 7 and 8). The rest of elutes was subjected to a second IP using anti-V5 antibody, followed by PCR amplification (lanes 9 and 10). (E) Co-residence of L11 with c-Myc in the nucleolin gene promoter requires the MB II of c-Myc. H1299 cells were transfected with WT or ΔMB II mutant of c-Myc plasmid in the presence or absence of Flag-L11. Sequential ChIP-PCR assays were performed using anti-Flag antibody, followed by anti-V5 antibody as described in above (D). Real-time PCR was then performed to detect the immunoprecipitated nucleolin promoter DNA.
To further examine whether L11 associates with c-Myc at these promoters, we introduced V5-c-Myc or V5-c-Myc plus Flag-L11 plasmids into H1299 cells, followed by sequential ChIP assays. Flag-L11-associated protein–DNA complexes were first affinity purified by anti-Flag IP and eluted by Flag peptide competition (Dai et al, 2004). Eluted protein–DNA complexes were then immunoprecipitated with the anti-V5 antibody. As shown in Figure 4D, the anti-Flag antibody readily immunoprecipitated the promoter regions of E2F2, nucleolin, and eIF-4E genes and 5′-ETS sequence of rDNA (Figure 4D, lane 8). Importantly, subsequent anti-V5 IP, following anti-Flag IP showed the co-occupancy of L11 and c-Myc at the aforementioned gene promoters (Figure 4D, lane 10) as compared to control IP without Flag-L11 expression (Figure 4D, lane 9). Again, this association required the MB II, as deletion of the MB II abolished the co-occupancy of L11 and c-Myc at the nucleolin gene promoter (Figure 4E). Taken together, these results indicate that L11 binds to c-Myc at its target gene promoters.
L11 inhibits the recruitment of c-Myc coactivator TRRAP to and histone H4 acetylation at c-Myc target nucleolin gene promoter
To test whether L11 affects the recruitment of the c-Myc coactivator TRRAP to above c-Myc target gene promoters, we conducted ChIP followed by real-time PCR assays in H1299 cells transfected with control or Flag-L11 vector. Indeed, the binding of TRRAP to the promoters of nucleolin (Figure 5B), rDNA, eIF-4E, and E2F2 (Supplementary Figure S3A–C) was reduced by two- to threefold by overexpression of L11 in all cases. This reduction by L11 was dose-dependent, as increasing amounts of L11 as shown in Figure 5C did not significantly affect the level of ectopic TRRAP, but remarkably inhibited the association of TRRAP with the nucleolin promoter (Figure 5D). These results suggest that L11 inhibits the recruitment of TRRAP to c-Myc target gene promoters by competing with this c-Myc coactivator.
Figure 5.
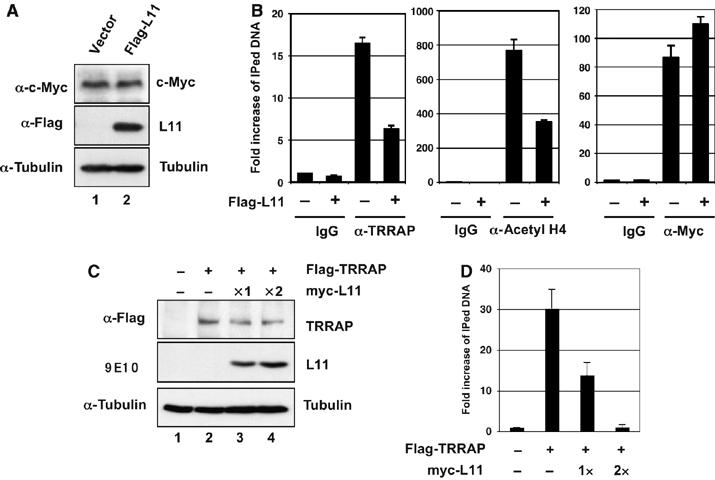
L11 reduces the recruitment of TRRAP to c-Myc target gene promoters and histone H4 acetylation at these promoters. (A) H1299 cell transfected with control or Flag-L11 plasmid were subjected to IB with anti-c-Myc or anti-Flag antibodies. (B) L11 reduces the recruitment of TRRAP to and histone H4 acetylation at c-Myc target gene promoters. The transfected cells in (A) were subjected to ChIP assays using goat anti-TRRAP, control goat IgG, rabbit polyclonal anti-acetyl histone H4, anti-c-Myc (N262), or control rabbit IgG followed by detection of the nucleolin gene promoter sequence using real-time PCR assays. (C, D) L11 competes the TRRAP for binding to the nucleolin gene promoter. H1299 cells transfected with Flag-TRRAP in the presence of increasing amounts of myc-tagged L11 were immunblotted with antibodies as indicated (C). The similar cells were also subjected to ChIP assays using anti-Flag antibody, followed by detection of the TRRAP binding to the nucleolin gene promoter using real-time PCR assays (D).
As TRRAP is associated with TIP60- or GCN5-containing HAT complexes (McMahon et al, 2000; Frank et al, 2003), we also examined histone H4 acetylation in these gene promoters. Indeed, overexpression of L11 reduced the histone H4 acetylation at all of these promoters (Figure 5B and Supplementary Figure S3A–C), but not in the control intron 5 region of l11 (Supplementary Figure S3D). It has been shown that binding of c-Myc induces histone acetylation at a single nucleosome in a Myc MB II/TRRAP-dependent manner (Bouchard et al, 2001). We also tested whether the reduction of histone H4 acetylation by L11 also occurs at the nucleosome containing the E-box elements in the nucleolin gene promoter. Using several pairs of primers encompassing the E-box elements (P0) and the regions approximately two nucleosomes away (P1) from the 5′ and three nucleosomes away (P2) from the 3′ of the E-box elements (Supplementary Figure S3E), we observed that the inhibition of histone H4 acetylation by L11 indeed occurred only at the nucleosome that contains E-box elements (P0) (Supplementary Figure S3F). These results suggest that L11 inhibits c-Myc transactivation activity by inhibiting the recruitment of its coactivator TRRAP and subsequently reducing histone H4 acetylation at c-Myc target gene promoters.
L11 and TRRAP inversely bind to a c-Myc target gene promoter in response to serum stimulation or serum starvation
To further analyze the physiological regulation of c-Myc by L11 through inhibition of TRRAP binding to a c-Myc target gene promoter, we performed ChIP-PCR assays to test the dynamic occupancy of L11 at the nucleolin promoter and compared to that of TRRAP following serum stimulation. As shown in Figure 6A, endogenous c-Myc levels were extremely low when U2OS cells were cultured in 0.2% fetal calf serum for 72 h. Upon serum stimulation, c-Myc levels displayed a ‘bell'-shaped curve with a peak at 3–6 h and a drastic decline at 12 h (Sears, 2004) (Figure 6A). Consistently, the expression of c-Myc target gene nucleolin displayed a similar curve to that of c-Myc levels as determined by real-time PCR assays (Figure 6B). Interestingly, TRRAP binding to the nucleolin promoter exhibited a curve (Figure 6C) similar to that for c-Myc levels (Figure 6A) after serum stimulation. The same was true for histone H4 acetylation at the nucleolin promoter (data not shown). By contrast, the association of L11 with the nucleolin promoter showed an inverse ‘bell'-shaped curve to that of TRRAP (Figure 6C). Conversely, when exponentially growing cells were starved in 0.2 % serum, L11 binding to the nucleolin promoter was significantly increased in contrast to the decrease of TRRAP binding to the same promoter (Figure 6D). Taken together, these results suggest that L11 may compete with TRRAP for binding to the c-Myc target gene promoter, in response to growth signals, further supporting the idea that L11 binds to the MB II at c-Myc target gene promoters (Figures 3C and 4) and inhibits the recruitment of TRRAP to the promoters (Figure 5)
Figure 6.
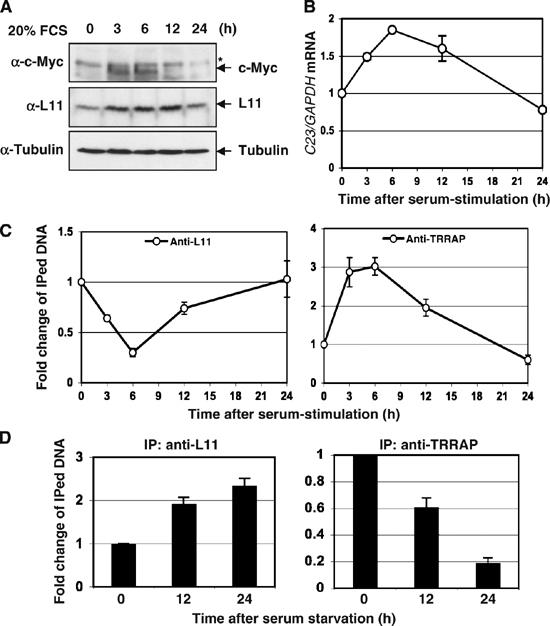
The dynamic binding of L11 to c-Myc target gene promoter inversely correlates with that of TRRAP in response to serum stimulation or serum starvation. (A) Dynamic expression of c-Myc and L11 in cells following serum-stimulation. U2OS cells cultured in 0.2% FCS containing medium for 48 h were stimulated with 20% FCS, and harvested at the indicated time points for IB assays using indicated antibodies. *Indicates nonspecific band. (B) The expression of the nucleolin in response to serum stimulation. Starved U2OS cells were stimulated with 20% FCS and harvested at the indicated time point as in (A) were assayed for expression of the nucleolin gene by real-time RT–PCR assays. (C) Dynamic association of L11 and TRRAP at the nucleolin gene promoter in response to serum stimulation. U2OS cells were serum starved and re-stimulated as in (A) and subjected to ChIP assays using anti-L11 or anti-TRRAP antibodies, followed by real-time PCR detection of the nucleolin promoter. (D) Dynamic association of L11 and TRRAP at the nucleolin gene promoter in response to serum starvation. Exponentially growing U2OS cells were starved in 0.2% FCS-containing medium and harvested at indicated time points. The cells were subjected to ChIP assays using anti-L11 or anti-TRRAP antibodies, followed by real-time PCR detection of the nucleolin promoter.
Reduction of endogenous L11 by siRNA enhances c-Myc activity
To further test whether c-Myc activity is regulated by endogenous L11, we determined whether transient depletion of endogenous L11 affects c-Myc activity using RNA interference technique. Consistent with results in Figure 1, knockdown of L11 by siRNA enhanced c-Myc-dependent luciferase reporter activity driven by wild-type, but not mutant, E Box containing E2F2 promoter (Figure 7A). Also, knockdown of endogenous L11 enhanced the expression of endogenous nucleolin, E2F2, and precursor rRNA determined by the real-time RT–PCR assays (Figure 7B). This enhancement was c-Myc-dependent, as further ablation of endogenous c-Myc by siRNA drastically reduced the L11 siRNA-induced expression of the above genes (Figure 7B). Correspondingly, knockdown of endogenous L11 significantly increased the percentage of cells with BrdU incorporation (Figure 7C and D). This effect was also c-Myc-dependent, as concomitant knockdown of c-Myc inhibited the L11 siRNA-induced BrdU incorporation (Figure 7C and D). These results suggest that endogenous L11 also inhibits c-Myc transactivation activity and cell cycle progression.
Figure 7.
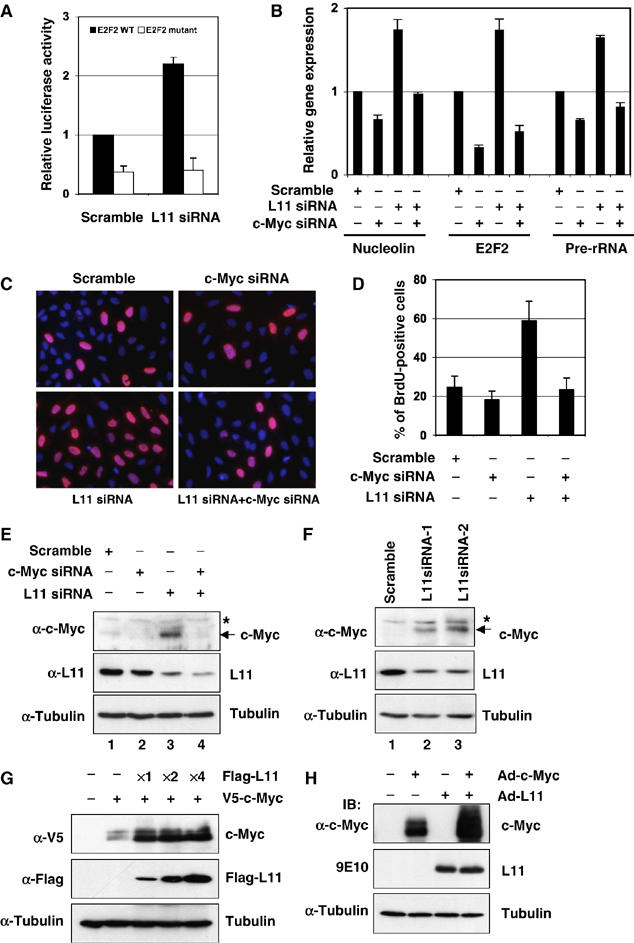
Endogenous L11 regulates c-Myc activity and levels. (A) Reduction of L11 by siRNA enhances c-Myc-induced luciferase reporter expression driven by WT, but not mutant, E box-containing E2F2 promoters in U2OS cells. (B) Knockdown of endogenous L11 enhances c-Myc-dependent transcription of the c-Myc target genes. U2OS cells were transfected with siRNAs as indicated and real-time RT–PCR assays were performed to determine the expression of E2F2, nucleolin, and rRNA genes. (C, D) Knockdown of endogenous L11 induces the percentage of cells in S phase. BrdU incorporation assay were performed in serum-starved U2OS cells transfected with siRNAs as indicated. Percentage of BrdU-positive cells is shown in (D). (E) The representative expression of endogenous c-Myc and L11 in above experiments (B), (C), and (D) as determined by IB with antibodies as indicated. (F) Knockdown of endogenous L11 by L11 siRNAs against two different sequences increases the protein levels of endogenous c-Myc in U2OS cells. *Indicates a nonspecific band. (G) Overexpression of L11 stabilizes ectopically expressed c-Myc. H1299 cells were transfected with plasmids as indicated. Whole-cell lysates were subjected to IB using antibodies as indicated. (H) Adenoviral-mediated overexpression of L11 stabilizes adenoviral encoded c-Myc. U2OS cells were infected with adenoviruses as indicated. Whole-cell lysates were subjected to IB using antibodies as indicated.
L11 regulates c-Myc levels
Whereas examining the level of endogenous c-Myc in the above experimental setting using IB analyses, we found that c-Myc levels were increased when L11 was transiently knocked down by siRNA (Figure 7E and F). This increase was, in part, due to the induction of c-myc mRNA levels (data not shown). These results suggest that in addition to directly regulating c-Myc transcriptional activity, endogenous L11 may be also involved in regulating c-Myc levels. Seemingly, in line with this possibility was that overexpression of L11 led to the decrease of exogenous c-Myc as shown in Figures 2A and 3A and Supplementary Figures S1A. However, it is important to note that these assays only detected soluble c-Myc extracted in the NP-40 lysis buffer. When whole-cell lysates were used for IB assays, the total steady-state level of c-Myc was markedly increased by overexpression of L11 in both transfection experiments (Figure 7G) and adenoviral infection assays (Figure 7H). These results suggest that ectopic L11 may increase ectopic c-Myc levels by moving it into NP-40-insoluble fractions, which contain chromatin-bound and nucleolar proteins. Indeed, overexpression of L11 relocalized ectopic c-Myc into the nucleolus (data not shown), which might account, in part, for the induction of exogenous c-Myc by L11. Of note, the possible role of endogenous L11 in downregulating native c-myc mRNA and protein levels as implied in the L11 knockdown assays (Figure 7E and F) might not apply to the expression of the exogenous c-myc gene, as the recombinant c-myc gene and its mRNA do not contain intrinsic regulatory elements, such as native promoters, introns, 5′- or 3-′UTRs, and thus would not be subjected to the regulation at the mRNA levels, as would endogenous c-Myc. This is probably why we could observe only the increase of ectopic c-Myc levels upon overexpression of L11 (Figure 7G and H). In accordance with this assumption, overexpression of L11 did not cause any significant change of endogenous c-Myc levels in H1299 cells (Figure 5A). The latter might result from the combined outcome of the dual, but inverse, effects of L11 on c-Myc protein levels through a direct interaction and on c-myc mRNA levels by an unknown mechanism(s). Taken together, these results (Figures 1, 2, 3, 4, 5, 6 and 7) suggest that L11 may affect c-Myc function through at least two mechanisms: direct binding to the MB II of c-Myc as demonstrated above, and regulating c-Myc protein and mRNA levels through yet unknown mechanisms.
c-Myc activity is not regulated by ribosomal protein L29
Next, we wanted to test whether the inhibitory effect of L11 on c-Myc activity is a general effect of all individual ribosomal proteins. To this end, we coexpressed V5-c-Myc with each of three other Flag-tagged ribosomal proteins, L29, L30, and S12, in H1299 cells followed by co-IP assays using the anti-Flag antibody. As shown in Figure 8A, L11, but not L29, L30, or S12, specifically co-immunoprecipitated with c-Myc, indicating that none of L29, L30, and S12 binds to c-Myc in cells. To further test whether the inability to bind to c-Myc would be translated into the defect in regulating its activity, we chose L29 for this test. Unlike L11, which increases total levels, but decreases soluble levels of ectopic c-Myc, L29 did not significantly affect the soluble or total levels of ectopic c-Myc protein (Figure 8C). Consistently, overexpression of L29 did not inhibit the c-Myc-induced expression of the endogenous nucleolin gene (Figure 8B). Also, unlike the case of L11 knockdown, knocking down endogenous L29 did not increase the c-Myc level (Figure 8E) and the expression of the endogenous nucleolin gene (Figure 8D). Altogether, these results suggest that the inhibitory effect of L11 on c-Myc activity is specific to L11 rather than a general effect of all individual ribosomal proteins.
Figure 8.
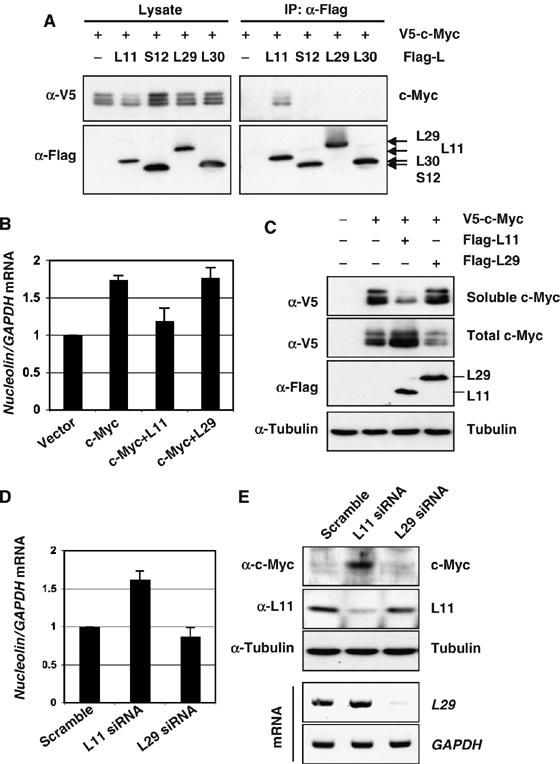
The inhibition of c-Myc activity by L11 is not a general effect of all individual ribosomal proteins. (A) L11, but not L29, L30, or S12, binds to c-Myc. H1299 cells were transfected with Flag-tagged L11, L29, L30, or S12 together with V5-c-Myc plasmids as indicated. Co-IPs were preformed using anti-Flag antibodies followed by IB with anti-V5 or anti-Flag antibodies. (B, C) Overexpression of L11, but not L29, inhibits the expression of the endogenous nucleolin gene. U2OS cells were transfected with plasmids as indicated. Half of the cells were used for IB assays to detect the protein expression of transfected c-Myc, L11, and L29 (C). The soluble c-Myc was determined from cleared lysates using NP40 lysis buffer, whereas the total level of c-Myc was determined using whole-cell lysates. The other half of the transfected cells were subjected to RT– real-time PCR to determine the expression of the nucleolin mRNA (B). (D, E) Knockdown of L11, but not L29, enhances the expression of the endogenous nucleolin gene. U2OS cells were transfected with siRNA as indicated. Half of the cells were used for IB assays to detect the protein expression of endogenous c-Myc and L11 (top panels) (E). The other half of the transfected cells were subjected to RNA extraction and RT, followed by real-time PCR to determine the relative expression of the nucleolin mRNA (D). The knockdown efficiency of L29 mRNA is shown in bottom panels of (E), as determined by semiquantitative RT–PCR.
c-Myc induces transcription of L11
Results from above observations suggest that L11 inhibits c-Myc activity. Interestingly, ribosomal proteins including L11 have been identified as potential c-Myc transcriptional targets (Coller et al, 2000; Guo et al, 2000; Boon et al, 2001; Menssen and Hermeking, 2002). We next wanted to validate that c-Myc indeed regulates L11 expression as this would suggest a feedback-regulatory loop between c-Myc and L11. By analyzing the promoter and exon–intron sequences of the human l11 gene, we found two canonical consensus c-Myc-binding (E box) elements in introns 1 and 2, respectively, and one noncanonical E box in exon 2 (Figure 9A). After introducing V5-tagged c-Myc into human lung nonsmall cell carcinoma H1299 or human osteocarcinoma U2OS cells, we conducted ChIP-PCR assays using the anti-V5 antibody and the primers encompassing intron 1 and 2 E box elements, as well as the intron 5 as a control (Figure 9A). As shown in Figure 9B, c-Myc specifically bound to the intron 1 and 2 E box elements in both H1299 (lane 4) and U2OS cells (lane 8), but not to the intron 5 region (bottom panels). To compare the c-Myc-binding capacity to L11 promoter with other known c-Myc target gene promoters, ChIP-real-time PCR assays were performed to quantitate the immunoprecipitated DNAs. As shown in Figure 9D, a significant amount of L11 promoter sequence was immunoprecipitated by the anti-c-Myc antibody (30-fold change compare to IgG control), which was more than one-third of the amount of the nucleolin promoter (Figure 9D). Furthermore, serum stimulation for 6 h enhanced c-Myc binding to the L11 promoter by 3.5-fold, which is also almost the half of that for the nucleolin gene promoter (Figure 9E). These results indicate that L11 is indeed a physiological relevant target gene of c-Myc. Consistent with above ChIP results, ectopic expression of c-Myc induced the expression of endogenous l11 mRNA in H1299 (Figure 9C, lane 2), U2OS (Figure 9C, lane 4), and human fibroblast WI38 cells (Figure 9C, lane 7), as well as L11 protein levels in both H1299 and U2OS cells (Figure 9F). Furthermore, knockdown of endogenous c-Myc by siRNA markedly reduced L11 mRNA and protein levels in U2OS cells (Figure 9G). Taken together, these results demonstrate that c-Myc activates transcription of l11, possibly through binding to its intron 1 and 2 E box elements. Thus, L11 inhibits c-Myc activity through a potential feedback mechanism.
Figure 9.
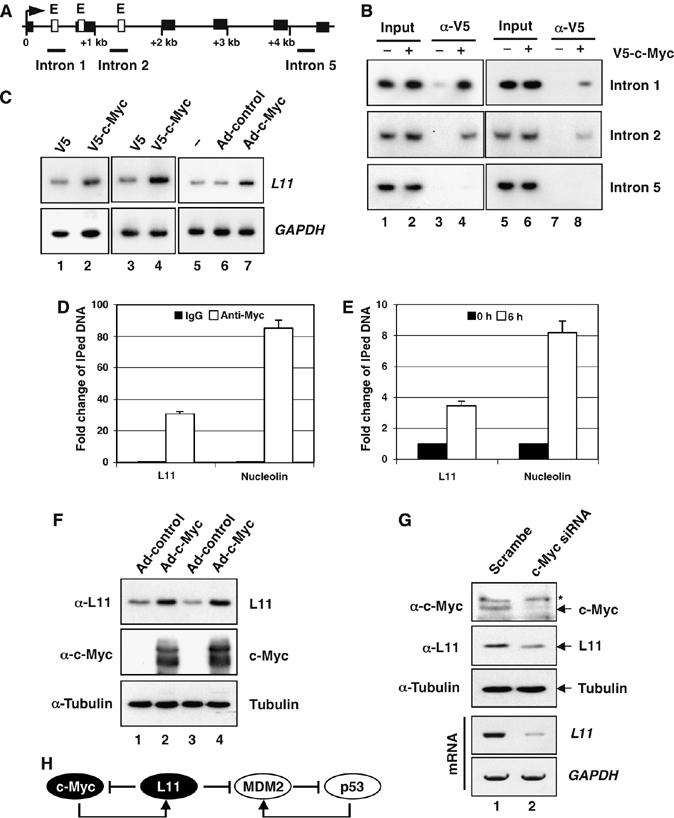
L11 is a target gene of c-Myc. (A) Diagram of the l11 gene exon–intron regions. Bars indicate the three regions amplified by ChIP-PCR assays. Empty boxes indicate E boxes. Filled boxes indicate exons. Arrow indicates the transcription start. (B) c-Myc specifically binds to the E box-containing introns 1 and 2, but not intron 5, regions of the l11 gene in H1299 (lanes 1 to 4) and U2OS (lanes 5 to 8) cells determined by ChIP-PCR assays. (C) c-Myc-induced l11 mRNA expression in cells. H1299 (lanes 1 to 2), U2OS (lanes 3 to 4), and WI38 cells (lanes 5 to 7) were transfected or infected with c-Myc-expressing plasmid or adenovirus and the expression of l11 was determined within 48 h by a semiquantitative RT–PCR assay. (D, E) Comparison of c-Myc binding to the L11 and the nucleolin gene promoters. (D) U2OS cells were subjected to ChIP assays using anti-c-Myc (N262) antibodies or control rabbit IgG followed by real-time PCR assays to detect the L11 and the nucleolin promoter DNAs. (E) U2OS cells were serum starved for 48 h, followed by stimulation with 20% FCS for 6 h as in Figure 6A. The serum-starved and re-stimulated cells were subjected to ChIP assays using anti-c-Myc (N262) antibodies, followed by real-time PCR to detect the L11 and the nucleolin promoter DNAs. (F) c-Myc induces L11 protein expression in cells. U2OS (lanes 1 and 2) and H1299 (lanes 3 and 4) cells were infected with c-Myc expressing adenovirus or control virus. Immunoblot (IB) was performed using antibodies as indicated. (G) Knockdown of endogenous c-Myc reduces L11 mRNA and protein levels. U2OS cells were transfected with c-Myc or scrambled siRNA. IB was performed using antibody as indicated and l11 mRNA was examined by a semiquantitative RT–PCR assay. *Indicates nonspecific band recognized by anti-c-Myc (N262). (H) A schematic for the L11-c-Myc feedback regulation versus the L11-MDM2-p53 regulatory loop.
Discussion
One of the most elegant natural mechanisms for cellular homeostasis is feedback regulation that allows autoregulation of a cellular process. Our study as described here unveils, for the first time, that L11, a component of the ribosome, controls c-Myc activity through a feedback-inhibitory mechanism. Overexpression of L11 inhibits the transactivation activity of c-Myc (Figure 1), whereas reduction of L11 by siRNA induces this activity (Figure 7). Interestingly, the expression of l11 gene itself is induced by c-Myc (Figure 9), and thus forming a negative autoregulatory feedback loop (Figure 9H). Mechanistically, we show that L11 binds to c-Myc at the MB II motif (Figure 3C and D), co-resides with c-Myc at its target gene promoters (Figure 4), prevents the recruitment of the coactivator TRRAP by c-Myc, and consequently inhibits histone H4 acetylation at these promoters (Figure 5). Thus, a key mechanism underlying the inhibition of c-Myc activity by L11 appears to involve L11 association with c-Myc at its target gene promoters, where it conceals c-Myc MB II, preventing the loading of TRRAP-containing HATs and remodeling of histones to a more transcriptionally active state.
The inhibition of c-Myc activity by overexpression of L11 is not due to a reduction of c-Myc protein levels, as L11 increased the total levels of ectopic c-Myc in cells (Figures 7G, H and 8C), although the soluble c-Myc was reduced by overexpression of L11 (Supplementary Figures S1A and Figures 2A, 3A and 8C). One explanation for these results is that L11 may block c-Myc degradation similar to the case of inhibition of MDM2 degradation by L11 (Dai et al, 2006b). It has been shown that c-Myc degradation occurs at the nucleolus (Welcker et al, 2004). We observed that overexpression of L11 relocalizes c-Myc into the nucleolus (data not shown). Thus, it is possible that L11 may block the degradation of c-Myc at the nucleolus and lead to the accumulation of c-Myc in this compartment. Another possibility is that binding of L11 to c-Myc may block Skp2 binding to c-Myc, as Spk2 also binds to the MB II of c-Myc (Kim et al, 2003; von der Lehr et al, 2003). Skp2 mediates c-Myc ubiquitination and degradation. This degradation is required for c-Myc transactivation activity (Kim et al, 2003; von der Lehr et al, 2003). Therefore, it is possible that, by blocking Skp2-mediated c-Myc ubiquitination and degradation pathway, L11 impairs the dynamic turnover of c-Myc, most possibly at the promoters of c-Myc target genes. Consequently, L11 inhibits c-Myc activity. Although this hypothesis would be interesting for future research, our results as presented here demonstrate that binding to the MB II represents a crucial mechanism for L11 to inhibit c-Myc activity, as this binding would displace the binding of the c-Myc coactivator TRRAP to the same region.
Surprisingly, knocking down endogenous L11 also increased the level of c-Myc (Figure 7E and F). This increase is probably, in part, due to the increase of c-myc mRNA level when knocking down L11 (data not shown). It remains unclear how L11 regulates c-myc mRNA levels. One possibility is that L11 may regulate c-Myc transcription. It has been recently shown that several Drosophila ribosomal proteins are associated with linker histone H1 and suppress transcription of a set of genes (Ni et al, 2006). Interestingly, we recently purified an L11-associated complex that also contains the linker histone H1 using an affinity purification method (data not shown). Thus, one of our future studies would be to test whether L11 could associate with histone H1 in chromatin and repress c-Myc transcription. Another testable possibility would be that L11 might regulate c-myc mRNA turnover. However, the regulation of c-myc mRNA and protein by knockdown of L11, although worthwhile for further investigation, would not discount or conflict with our findings that L11 represses c-Myc activity by directly binding to this protein as discussed above. Also, this regulation may be through an indirect and more complex process, as overexpression of L11 did not simply reduce endogenous c-Myc protein levels (Figure 5A), as discussed in the result section. Taken together, at least two mechanisms may account for the inhibitory effect of L11 on c-Myc: inhibiting the recruitment of TRRAP to c-Myc target gene promoters by directly binding to the MB II domain of c-Myc, and regulating c-Myc levels.
The regulation of c-Myc by L11 is specific to L11, because other tested ribosomal proteins, such as L29, L30, and S12, did not bind to c-Myc (Figure 8A). Further analyses show that overexpression of L29 neither changes the c-Myc protein levels nor reduces its activity in regulating the expression of the target gene, nucleolin (Figure 8B and C). Also, unlike the case of L11 knockdown, L29 knockdown does not increase c-Myc levels and its activity (Figure 8D and E). Thus, not all individual ribosomal proteins would regulate c-Myc. Although our study does not absolutely rule out the possibility that other untested ribosomal proteins may also play a role in regulating c-Myc activity similar to that of L11, our results as presented strongly demonstrate the role of L11 in repressing c-Myc activity as a feedback regulator.
The inhibition of c-Myc activity by L11 is also suggested in cells in response to growth signals. In early response to serum stimulation, c-Myc levels rapidly increased, whereas L11 binding to c-Myc target gene promoter inversely decreased (Figure 6C), indicating that the repression of L11 on c-Myc activity is de-repressed at a stage when c-Myc activity is required for cells to proliferate. The elevated L11 molecules by this enhanced c-Myc activity might be used for limiting c-Myc activity at the later stage of serum stimulation to prevent aberrant cell growth. Consistent with this notion, when growth signals are removed, as in the case of serum starvation (Figure 6D), L11 increasingly associates with c-Myc in a target gene promoter to block TRRAP binding to the same promoter, thus leading to inhibition of c-Myc-dependent transcription. It has been shown that serum starvation led to release of L11 from ribosome (Bhat et al, 2004). Thus, growth inhibition would reduce ribosomal biogenesis and result in enhanced free form of L11 to target c-Myc. Another possibility is that post-transcriptional modification may play a role in regulating the L11-c-Myc interaction at c-Myc target gene promoter in response to growth signals. Nevertheless, these results suggest that L11-c-Myc regulatory loop is highly regulated in cells.
In summary, our findings extend the role of L11 in cell proliferation regulation beyond regulating the MDM2-p53 feedback loop (Figure 9H) (Lohrum et al, 2003; Zhang et al, 2003; Bhat et al, 2004; Dai et al, 2006b). In this respect, L11 resembles the function of ARF, which regulates both the MDM2-p53 pathway (Sherr and Weber, 2000) and c-Myc function (Datta et al, 2004; Qi et al, 2004); but L11 works independently of ARF and p53. It is likely that L11 may primarily act as a ‘sensor' of aberrant ribosomal biogenesis, whereas ARF primarily acts as a ‘sensor' of oncogenic stress (Sherr and Weber, 2000). Because L11 activates p53 (Lohrum et al, 2003; Zhang et al, 2003) and represses c-Myc activity, one important question would be if L11 could prevent cell transformation. More studies, such as genetic studies in animals and identification of L11 mutations in human cancers, are necessary to verify this prediction.
Materials and methods
Cell lines, adenoviruses, and plasmids
Human lung nonsmall cell adenocarcinoma H1299 cells, human oesteosarcoma U2OS cells, mouse p53−/−/mdm2−/− MEFs, mouse embryonic fibroblast 10–1 cells, human fibroblast WI38 cells, and IMR90 cells were cultured as described previously (Dai et al, 2004). Adenoviruses encoding full-length c-Myc and myc-tagged L11 were described previously (Bhat et al, 2004; Yeh et al, 2004). CMV-empty, CMV-β-gal, pD40-His/V5-c-Myc, E2F2-Luc, E2F2(-E-box)-Luc, and CMV-c-Myc plasmids were described previously (Arnold and Sears, 2006; Yeh et al, 2004). Flag-tagged L11 (Flag-L11) plasmid has also been described previously (Lohrum et al, 2003). Deletion mutants of L11 with N-terminal Flag tag were generated by PCR and cloned into pcDNA3-2Flag vector (Dai et al, 2006b). Deletion mutants of c-Myc with N-terminal V5 tag were generated by PCR and cloned into pcDNA3-V5 vector. Flag-tagged L29, L30, and S12 expression plasmids were generated by inserting the full-length cDNAs amplified by RT–PCR from HeLa cells into the pcDNA3-2Flag vectors (Dai et al, 2004). The primers used were: 5′-CGCGGATCCGCCAAGTCCAAGAACCACAC-3′ and 5′-CCGGAATTCCTACTCTGAAGCCTTTGTAGG-3′ for L29, 5′-CGCGGATCCGTGGCCGCAAAGAAGAC-3′ and 5′-CCGGAATTCTTACTTTTCACCAGTCTGTTCTGG-3′ for L30, and 5′-CGCGGATCCGCCGAGGAAGGCATTGC-3′ and 5′-CGCTCTAGATCATTTCTTGCATTTGAAATACTCTTC-3′ for S12. All inserts were confirmed by automatic sequencing. Flag-TRRAP expression plasmid (CβSBS-Flag-TRRAP) was kindly provided by Dr Michael D. Cole (Princeton University, New Jersey) (McMahon et al, 1998).
Immunoblot and immunoprecipitation analyses
Cell lysate preparation, immunoprecipitation, and immunoblot analysis were performed as described previously (Dai et al, 2004), except that 300 mM NaCl was used in the lysis buffer in co-IPs between endogenous c-Myc and L11. Anti-Flag M2 (Sigma), polyclonal anti-V5 (Sigma), monoclonal anti-V5 (Invitrogen), anti-c-Myc (N262, C33, and 9E10, Santa Crutz) antibodies were purchased. Rabbit polyclonal anti-L11 was generated using purified His-tagged full-length L11 protein (Dai and Lu, 2004) expressed in Escherichia Coli as an antigen.
Luciferase reporter and transcription assays
See Supplementary data for details.
BrdU incorporation assays
BrdU incorporation assays were conducted as described previously (Datta et al, 2004). Cells were incubated in the presence of 10 μM of BrdU for 5 h. Cells were then fixed with 95% of ethanol and 5% of acetic acid, treated with 2 M HCl containing 1% Triton X-100, and stained with the monoclonal anti-BrdU (Roche) antibody, followed by staining with Alexa Fluor 546 (red) goat anti-mouse antibodies and DAPI. Stained cells were analyzed under a Zeiss Axiovert 25 fluorescent microscope.
Reverse transcriptase-polymerase chain reaction and quantitative real-time PCR analysis
Total RNA extraction and 32P-dCTP-labeled semiquantitative RT–PCR reactions were performed as described previously (Dai and Lu, 2004). The primers for amplifying GAPDH were used as described previously (Dai and Lu, 2004). The primers for L11 were 5′-TACAGTCAGAGCCAACCTCAG-3′ and 5′-AGATGAAGCTCCCAGAATGCC-3′. The primers for L29 were 5′-GCCAAGTCCAAGAACCACAC-3′ and 5′-CTACTCTGAAGCCTTTGTAGG-3′. Quantitative real-time PCR was performed on an ABI 7300 real-time PCR system (Applied Biosystems) using SYBR Green Mix (Applied Biosystems). Relative gene expression was calculated using the ΔCτ method, following the manufacturer's instruction. All reactions were carried out in triplicate. The primer sequences used are listed in the Supplementary Table S1.
RNA interference
The 21-nt siRNA duplexes with a 3′ dTdT overhang were synthesized by Dharmacon (Lafayette, CO). The target sequences for L11 were 5′-AAGGTGCGGGAGTATGAGTTA-3′ (siRNA-1, used for all experiments, except where indicated) (Bhat et al, 2004) and 5′-AAGCATTGGTATCTACGGCCT-3′ (siRNA-2) (Bernardi et al, 2004). The target sequence for c-Myc was 5′-AACAGAAATGTCCTGAGCAAT-3′ (Williams et al, 2003). The target sequence for L29 was 5′-AAGTTCCTGAGGAACATGCGC-3′. The scrambled II RNA duplex was used as a control (Dai et al, 2004). Transfection of siRNA was performed as previously described using siLentFectTM Lipid (Bio-Rad), following the manufacturer's protocol (Dai et al, 2004).
Chromatin immunoprecipitation (ChIP)-PCR
ChIP analysis was performed as described previously (Zeng et al, 2002) using anti-c-Myc (N262), anti-L11, anti-TRRAP (sc-5405, Santa Cruz), or anti-acetyl histone H4 (06-866, Upstate Biotechnologies) antibodies. Immunoprecipitated DNA fragments were analyzed by semiquantitative and/or real-time PCR amplification using primers for E2F2, nucleolin, eIF4E, rDNA, or l11 genes. The primers are listed in Supplementary Table S1. For detection of co-occupancy of both c-Myc and L11 in promoters, H1299 cells were transfected with V5-c-Myc alone or together with Flag-L11. After crosslinking and sonication, the lysates were immunoprecipitated with anti-Flag antibodies. The immunoprecipitated Flag-L11 protein–DNA complexes were then eluted with 0.1 mg/ml of Flag peptide (Sigma) using methods as described previously (Dai et al, 2004). The elutes were then immunoprecipitated with anti-V5 antibody, followed by DNA purification and PCR amplification as described above.
Supplementary Material
Supplementary Data
Supplementary Figures
Acknowledgments
We thank Dr Yanping Zhang, Dr Michael D. Cole, Dr George Thomas, and Dr Michael Kastan for providing us with cDNA and antibody reagents and Jayme Gallegos for critically reading the manuscript. HA was supported by the NIH training grant 5-T32-GM08617, and RS by NIH/NCI grants R01-CA100855 and K01-CA086957. This work was supported, in part, by NCI grants CA93614, CA095441, and CA 079721 to HL
References
- Adams JM, Harris AW, Pinkert CA, Corcoran LM, Alexander WS, Cory S, Palmiter RD, Brinster RL (1985) The c-myc oncogene driven by immunoglobulin enhancers induces lymphoid malignancy in transgenic mice. Nature 318: 533–538 [DOI] [PubMed] [Google Scholar]
- Adhikary S, Eilers M (2005) Transcriptional regulation and transformation by Myc proteins. Nat Rev Mol Cell Biol 6: 635–645 [DOI] [PubMed] [Google Scholar]
- Adhikary S, Marinoni F, Hock A, Hulleman E, Popov N, Beier R, Bernard S, Quarto M, Capra M, Goettig S, Kogel U, Scheffner M, Helin K, Eilers M (2005) The ubiquitin ligase HectH9 regulates transcriptional activation by Myc and is essential for tumor cell proliferation. Cell 123: 409–421 [DOI] [PubMed] [Google Scholar]
- Amati B, Dalton S, Brooks MW, Littlewood TD, Evan GI, Land H (1992) Transcriptional activation by the human c-Myc oncoprotein in yeast requires interaction with Max. Nature 359: 423–426 [DOI] [PubMed] [Google Scholar]
- Arabi A, Wu S, Ridderstrale K, Bierhoff H, Shiue C, Fatyol K, Fahlen S, Hydbring P, Soderberg O, Grummt I, Larsson LG, Wright AP (2005) c-Myc associates with ribosomal DNA and activates RNA polymerase I transcription. Nat Cell Biol 7: 303–310 [DOI] [PubMed] [Google Scholar]
- Arnold HK, Sears RC (2006) Protein phosphatase 2A regulatory subunit B56alpha associates with c-myc and negatively regulates c-myc accumulation. Mol Cell Biol 26: 2832–2844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernardi R, Scaglioni PP, Bergmann S, Horn HF, Vousden KH, Pandolfi PP (2004) PML regulates p53 stability by sequestering Mdm2 to the nucleolus. Nat Cell Biol 6: 665–672 [DOI] [PubMed] [Google Scholar]
- Bhat KP, Itahana K, Jin A, Zhang Y (2004) Essential role of ribosomal protein L11 in mediating growth inhibition-induced p53 activation. EMBO J 23: 2402–2412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boon K, Caron HN, van Asperen R, Valentijn L, Hermus MC, van Sluis P, Roobeek I, Weis I, Voute PA, Schwab M, Versteeg R (2001) N-myc enhances the expression of a large set of genes functioning in ribosome biogenesis and protein synthesis. EMBO J 20: 1383–1393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouchard C, Dittrich O, Kiermaier A, Dohmann K, Menkel A, Eilers M, Luscher B (2001) Regulation of cyclin D2 gene expression by the Myc/Max/Mad network: Myc-dependent TRRAP recruitment and histone acetylation at the cyclin D2 promoter. Genes Dev 15: 2042–2047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coller HA, Grandori C, Tamayo P, Colbert T, Lander ES, Eisenman RN, Golub TR (2000) Expression analysis with oligonucleotide microarrays reveals that MYC regulates genes involved in growth, cell cycle, signaling, and adhesion. Proc Natl Acad Sci USA 97: 3260–3265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai MS, Jin Y, Gallegos JR, Lu H (2006a) Balance of Yin and Yang: ubiquitylation-mediated regulation of p53 and c-Myc. Neoplasia 8: 630–644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai MS, Lu H (2004) Inhibition of MDM2-mediated p53 ubiquitination and degradation by ribosomal protein L5. J Biol Chem 279: 44475–44482 [DOI] [PubMed] [Google Scholar]
- Dai MS, Shi D, Jin Y, Sun XX, Zhang Y, Grossman SR, Lu H (2006b) Regulation of the MDM2-p53 pathway by ribosomal protein L11 involves a post-ubiquitination mechanism. J Biol Chem 281: 24304–24313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai MS, Zeng SX, Jin Y, Sun XX, David L, Lu H (2004) Ribosomal protein L23 activates p53 by inhibiting MDM2 function in response to ribosomal perturbation but not to translation inhibition. Mol Cell Biol 24: 7654–7668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Datta A, Nag A, Pan W, Hay N, Gartel AL, Colamonici O, Mori Y, Raychaudhuri P (2004) Myc-ARF (alternate reading frame) interaction inhibits the functions of Myc. J Biol Chem 279: 36698–36707 [DOI] [PubMed] [Google Scholar]
- Davis AC, Wims M, Spotts GD, Hann SR, Bradley A (1993) A null c-myc mutation causes lethality before 10.5 days of gestation in homozygotes and reduced fertility in heterozygous female mice. Genes Dev 7: 671–682 [DOI] [PubMed] [Google Scholar]
- de Alboran IM, O'Hagan RC, Gartner F, Malynn B, Davidson L, Rickert R, Rajewsky K, DePinho RA, Alt FW (2001) Analysis of C-MYC function in normal cells via conditional gene-targeted mutation. Immunity 14: 45–55 [DOI] [PubMed] [Google Scholar]
- Felsher DW, Bishop JM (1999) Reversible tumorigenesis by MYC in hematopoietic lineages. Mol Cell 4: 199–207 [DOI] [PubMed] [Google Scholar]
- Frank SR, Parisi T, Taubert S, Fernandez P, Fuchs M, Chan HM, Livingston DM, Amati B (2003) MYC recruits the TIP60 histone acetyltransferase complex to chromatin. EMBO Rep 4: 575–580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez-Roman N, Grandori C, Eisenman RN, White RJ (2003) Direct activation of RNA polymerase III transcription by c-Myc. Nature 421: 290–294 [DOI] [PubMed] [Google Scholar]
- Grandori C, Cowley SM, James LP, Eisenman RN (2000) The Myc/Max/Mad network and the transcriptional control of cell behavior. Annu Rev Cell Dev Biol 16: 653–699 [DOI] [PubMed] [Google Scholar]
- Grandori C, Gomez-Roman N, Felton-Edkins ZA, Ngouenet C, Galloway DA, Eisenman RN, White RJ (2005) c-Myc binds to human ribosomal DNA and stimulates transcription of rRNA genes by RNA polymerase I. Nat Cell Biol 7: 311–318 [DOI] [PubMed] [Google Scholar]
- Grewal SS, Li L, Orian A, Eisenman RN, Edgar BA (2005) Myc-dependent regulation of ribosomal RNA synthesis during Drosophila development. Nat Cell Biol 7: 295–302 [DOI] [PubMed] [Google Scholar]
- Guo QM, Malek RL, Kim S, Chiao C, He M, Ruffy M, Sanka K, Lee NH, Dang CV, Liu ET (2000) Identification of c-myc responsive genes using rat cDNA microarray. Cancer Res 60: 5922–5928 [PubMed] [Google Scholar]
- Kim SY, Herbst A, Tworkowski KA, Salghetti SE, Tansey WP (2003) Skp2 regulates Myc protein stability and activity. Mol Cell 11: 1177–1188 [DOI] [PubMed] [Google Scholar]
- Lohrum MA, Ludwig RL, Kubbutat MH, Hanlon M, Vousden KH (2003) Regulation of HDM2 activity by the ribosomal protein L11. Cancer Cell 3: 577–587 [DOI] [PubMed] [Google Scholar]
- Luscher B, Larsson LG (1999) The basic region/helix-loop-helix/leucine zipper domain of Myc proto-oncoproteins: function and regulation. Oncogene 18: 2955–2966 [DOI] [PubMed] [Google Scholar]
- McMahon SB, Van Buskirk HA, Dugan KA, Copeland TD, Cole MD (1998) The novel ATM-related protein TRRAP is an essential cofactor for the c-Myc and E2F oncoproteins. Cell 94: 363–374 [DOI] [PubMed] [Google Scholar]
- McMahon SB, Wood MA, Cole MD (2000) The essential cofactor TRRAP recruits the histone acetyltransferase hGCN5 to c-Myc. Mol Cell Biol 20: 556–562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menssen A, Hermeking H (2002) Characterization of the c-MYC-regulated transcriptome by SAGE: identification and analysis of c-MYC target genes. Proc Natl Acad Sci USA 99: 6274–6279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moberg KH, Mukherjee A, Veraksa A, Artavanis-Tsakonas S, Hariharan IK (2004) The Drosophila F box protein archipelago regulates dMyc protein levels in vivo. Curr Biol 14: 965–974 [DOI] [PubMed] [Google Scholar]
- Nesbit CE, Tersak JM, Prochownik EV (1999) MYC oncogenes and human neoplastic disease. Oncogene 18: 3004–3016 [DOI] [PubMed] [Google Scholar]
- Ni JQ, Liu LP, Hess D, Rietdorf J, Sun FL (2006) Drosophila ribosomal proteins are associated with linker histone H1 and suppress gene transcription. Genes Dev 20: 1959–1973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oskarsson T, Trumpp A (2005) The Myc trilogy: lord of RNA polymerases. Nat Cell Biol 7: 215–217 [DOI] [PubMed] [Google Scholar]
- Park J, Kunjibettu S, McMahon SB, Cole MD (2001) The ATM-related domain of TRRAP is required for histone acetyltransferase recruitment and Myc-dependent oncogenesis. Genes Dev 15: 1619–1624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelengaris S, Khan M, Evan G (2002a) c-MYC: more than just a matter of life and death. Nat Rev Cancer 2: 764–776 [DOI] [PubMed] [Google Scholar]
- Pelengaris S, Khan M, Evan GI (2002b) Suppression of Myc-induced apoptosis in beta cells exposes multiple oncogenic properties of Myc and triggers carcinogenic progression. Cell 109: 321–334 [DOI] [PubMed] [Google Scholar]
- Pelengaris S, Littlewood T, Khan M, Elia G, Evan G (1999) Reversible activation of c-Myc in skin: induction of a complex neoplastic phenotype by a single oncogenic lesion. Mol Cell 3: 565–577 [DOI] [PubMed] [Google Scholar]
- Qi Y, Gregory MA, Li Z, Brousal JP, West K, Hann SR (2004) p19ARF directly and differentially controls the functions of c-Myc independently of p53. Nature 431: 712–717 [DOI] [PubMed] [Google Scholar]
- Ruggero D, Pandolfi PP (2003) Does the ribosome translate cancer? Nat Rev Cancer 3: 179–192 [DOI] [PubMed] [Google Scholar]
- Sears RC (2004) The life cycle of C-myc: from synthesis to degradation. Cell Cycle 3: 1133–1137 [PubMed] [Google Scholar]
- Sherr CJ, Weber JD (2000) The ARF/p53 pathway. Curr Opin Genet Dev 10: 94–99 [DOI] [PubMed] [Google Scholar]
- Trumpp A, Refaeli Y, Oskarsson T, Gasser S, Murphy M, Martin GR, Bishop JM (2001) c-Myc regulates mammalian body size by controlling cell number but not cell size. Nature 414: 768–773 [DOI] [PubMed] [Google Scholar]
- von der Lehr N, Johansson S, Wu S, Bahram F, Castell A, Cetinkaya C, Hydbring P, Weidung I, Nakayama K, Nakayama KI, Soderberg O, Kerppola TK, Larsson LG (2003) The F-box protein Skp2 participates in c-Myc proteosomal degradation and acts as a cofactor for c-Myc-regulated transcription. Mol Cell 11: 1189–1200 [DOI] [PubMed] [Google Scholar]
- Welcker M, Orian A, Grim JE, Eisenman RN, Clurman BE (2004) A nucleolar isoform of the Fbw7 ubiquitin ligase regulates c-Myc and cell size. Curr Biol 14: 1852–1857 [DOI] [PubMed] [Google Scholar]
- White RJ (2005) RNA polymerases I and III, growth control and cancer. Nat Rev Mol Cell Biol 6: 69–78 [DOI] [PubMed] [Google Scholar]
- Williams NS, Gaynor RB, Scoggin S, Verma U, Gokaslan T, Simmang C, Fleming J, Tavana D, Frenkel E, Becerra C (2003) Identification and validation of genes involved in the pathogenesis of colorectal cancer using cDNA microarrays and RNA interference. Clin Cancer Res 9: 931–946 [PubMed] [Google Scholar]
- Wood MA, McMahon SB, Cole MD (2000) An ATPase/helicase complex is an essential cofactor for oncogenic transformation by c-Myc. Mol Cell 5: 321–330 [DOI] [PubMed] [Google Scholar]
- Yada M, Hatakeyama S, Kamura T, Nishiyama M, Tsunematsu R, Imaki H, Ishida N, Okumura F, Nakayama K, Nakayama KI (2004) Phosphorylation-dependent degradation of c-Myc is mediated by the F-box protein Fbw7. Embo J 23: 2116–2125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeh E, Cunningham M, Arnold H, Chasse D, Monteith T, Ivaldi G, Hahn WC, Stukenberg PT, Shenolikar S, Uchida T, Counter CM, Nevins JR, Means AR, Sears R (2004) A signalling pathway controlling c-Myc degradation that impacts oncogenic transformation of human cells. Nat Cell Biol 6: 308–318 [DOI] [PubMed] [Google Scholar]
- Zeng SX, Dai MS, Keller DM, Lu H (2002) SSRP1 functions as a co-activator of the transcriptional activator p63. EMBO J 21: 5487–5497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Wolf GW, Bhat K, Jin A, Allio T, Burkhart WA, Xiong Y (2003) Ribosomal protein L11 negatively regulates oncoprotein MDM2 and mediates a p53-dependent ribosomal-stress checkpoint pathway. Mol Cell Biol 23: 8902–8912 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Data
Supplementary Figures