

Abstract
We have identified a novel pro-apoptotic p53 target gene named CDIP (Cell Death Involved p53-target). Inhibition of CDIP abrogates p53-mediated apoptotic responses, demonstrating that CDIP is an important p53 apoptotic effector. CDIP itself potently induces apoptosis that is associated with caspase-8 cleavage, implicating the extrinsic cell death pathway in apoptosis mediated by CDIP. siRNA-directed knockdown of caspase-8 results in a severe impairment of CDIP-dependent cell death. In investigating the potential involvement of extrinsic cell death pathway in CDIP-mediated apoptosis, we found that TNF-α expression tightly correlates with CDIP expression, and that inhibition of TNF-α signaling attenuates CDIP-dependent apoptosis. We also demonstrate that TNF-α is upregulated in response to p53 and p53 inducing genotoxic stress, in a CDIP-dependent manner. Consistently, knockdown of TNF-α impairs p53-mediated stress-induced apoptosis. Together, these findings support a novel p53 → CDIP → TNF-α apoptotic pathway that directs apoptosis after exposure of cells to genotoxic stress. Thus, CDIP provides a new link between p53-mediated intrinsic and death receptor-mediated extrinsic apoptotic signaling, providing a novel target for cancer therapeutics aimed at maximizing the p53 apoptotic response of cancer cells to drug therapy.
Keywords: apoptosis, CDIP, p53, TNF-α
Introduction
Aberrant cellular proliferation and the evasion of apoptosis are recognized as key events in the progression of normal cells to a cancerous phenotype (Hanahan and Weinberg, 2000). The p53 tumor suppressor protein is activated in response to inappropriate growth signals and various forms of cellular stress (e.g., DNA damage), and functions as a transcription factor to promote antiproliferative responses, including cell cycle arrest and apoptosis (Oren, 2003). The apoptotic function of p53 has been suggested to be foremost with respect to its ability to function as a tumor suppressor (Symonds et al, 1994).
Although the mechanism by which p53 activates G1 arrest is well characterized and involves primarily the transcriptional activation of the cyclin-dependent kinase inhibitor p21Waf1 (el-Deiry et al, 1993; Brugarolas et al, 1995; Deng et al, 1995), apoptosis induced by p53 is proving to be a much more complex cellular process. Two general mechanisms of apoptosis have been described for mammalian cells: the intrinsic pathway, activated in response to cellular stresses (e.g., exposure to DNA damaging agents), and the extrinsic pathway initiated at the cell surface upon activation of death receptors by binding of their cognate ligands (Peter and Krammer, 2003; Lowe et al, 2004). p53-mediated apoptosis has been shown to be dependent upon the Apaf1/caspase-9 intrinsic cell death pathway, which involves the release of cytochrome c from the mitochondria into the cytosol and caspse-9 activation (Soengas et al, 1999; Schuler et al, 2000). To date, numerous p53-induced gene targets have been identified, a subset of which have an apparent proapoptotic role in the intrinsic cell death pathway; Bax, Noxa, PUMA and p53AIP proteins localize to the mitochondria and potentiate the release of cytochrome c (Benchimol, 2001).
The extrinsic death pathway is initiated by the TNF-α family of cytokines (e.g., TNF-α, FasL and TRAIL), all of which can act as extracellular activators of apoptosis upon binding to their cognate receptors (Baud and Karin, 2001; Chen and Goeddel, 2002). The resulting form of cell death involves caspase-8 activation, which directly activates downstream effector caspases, and/or amplifies the death signal through the mitochondria (Thorburn, 2004). Increasing evidence suggests that p53 may signal to the extrinsic death receptor pathway when triggering an apoptotic response. Two members of the TNFR superfamily, Fas/CD95/APO-1 and KILLER/DR5, have been shown to be regulated in a p53-dependent manner in response to genotoxic drugs (Owen-Schaub et al, 1995; Wu et al, 1997; Muller et al, 1998), and Fas/CD95/APO-1 has been shown to be required for an optimum apoptotic response upon exposure to DNA damage (Friesen et al, 1996; Muller et al, 1997; Embree-Ku et al, 2002). The precise nature of the p53 transcriptional program required for apoptosis has not yet been fully elucidated.
We identified CDIP as a novel p53 induced protein, and key downstream effector of p53-dependent apoptosis. CDIP itself is a potent inducer of apoptosis. CDIP-dependent apoptosis is associated with caspase-8 activation, implicating the extrinsic death receptor-signaling pathway as the mechanism by which CDIP mediates cell death. This line of investigation revealed that TNF-α is induced in a CDIP-dependent manner downstream of p53 expression or DNA damage. Importantly, apoptosis under similar conditions was compromised in cells depleted of TNF-α or inhibited of FADD, another mediator of the extrinsic cell death signal. Thus, CDIP is an important component of the p53-mediated apoptotic response, and represents a novel link between p53 and the extrinsic death receptor-mediated pathway of apoptosis.
Results
CDIP is upregulated in a p53-dependent manner
We identified a novel gene termed CDIP as p53-upregulated transcript by microarray analysis of cDNA expression following p53 induction in EJ-p53 tet-off cells. The CDIP cDNA encodes a 208-amino acid protein, with a predicted molecular mass of ∼24 kDa (Supplementary Figure 1). p53-dependent upregulation of the CDIP transcript was confirmed by Northern blotting in several cell lines (Figure 1A and B). Consistently, treatment of cells containing functional p53 with the DNA damaging agent etoposide (ETO) induced CDIP mRNA and protein expression; this effect was not observed in p53-null Saos2 cells (Figure 1B). Inhibition of p53 activation in response to ETO by siRNA targeting abrogated CDIP induction, demonstrating that CDIP induction in response to DNA damage is dependent upon p53 (Figure 1B, lower panel).
Figure 1.

Identification of CDIP as a direct target of p53 transcriptional activation. (A) p53-dependent induction of CDIP. The left panels show induction of CDIP mRNA after infection with recombinant adenovirus expressing p53 (Ad-p53) in Saos2 and EJ cells. Northern blots were performed sequentially using a 32P-labeled probe against CDIP, p53, p21Waf1 and 36B4 (loading control). (B) p53-dependent induction of CDIP in response to DNA damage. Upper panel shows MCF7, IMR90-E1A, U2OS and Saos2 (p53 null) cells that were either left untreated (−) or treated with the DNA damaging agent ETO (25 μM) for 1 day (+1) or 2 days (+2). Western blot analysis with a custom CDIP antibody also shows p53-associated CDIP upregulation in U2OS cells treated with ETO (upper right panel). The lower panel shows MCF7 cells transfected with either siRNA oligos targeting wild-type p53, or control siRNA oligos (GFP), followed by exposure to ETO for 24 h. Northern blots were performed sequentially using a 32P-labeled probe against p53, CDIP and 36B4 (loading control); Western blotting with an anti-p53 antibody confirmed that siRNA targeting of p53 resulted in the knockdown of p53 protein (lower panels). (C) A putative p53 recognition site located in the promoter region of the CDIP gene (p53-BS, set in bold, from −1618 to −1639) is shown; four such sites were identified within the CDIP promoter region. Three constructs containing these putative p53-BSs or a mutated copy of the 5′ p53-BS, as indicated, were cloned into a luciferase reporter vector (pGL3-promoter vector). Saos2 cells were cotransfected with either of the four CDIP promoter constructs (T1, T2, T3 or M1, plus pGL3 basic), and either wild-type p53, or mutant p53 (V143A) expression constructs, as well as a control pcDNA3.1 empty vector. Luciferase assay results for each construct are shown. Additionally, U2OS cells were transfected with the various CDIP promoter constructs described, treated with ETO to induce endogenous p53, and the relative luciferase activity was similarly determined. (D) ChIP assay of p53 DNA binding activity at the CDIP promoter in U2OS and Saos2 cells. U2OS and Saos2 cells were infected with Ad-p53 or Ad-GFP for 24 h (upper panel). ChIP assays were performed using an anti-p53 antibody or an anti-HA antibody as a negative control. U2OS cells were also treated with ETO for 24 h ChIP assays were performed with an anti-p53 antibody or a control anti-IgG antibody. A sequence map of the CDIP promoter region containing the ChIP primers (shown as arrows) used to amplify the p53-responsive element at −1618 to −1639 is shown (bold); the amplification product was 229 bp. Primers amplifying a 417-bp product of the p21Waf1 promoter were used as positive control.
We further investigated whether CDIP was involved in p53-dependent stress responses to γ-irradiation. Several cell lines (i.e., U2OS, HCT116 and IMR90-E1A) were investigated for CDIP induction after exposure to γ-irradiation, however, cells either did not upregulate CDIP or showed relatively weak induction compared to what is observed for ETO and camptothecin (Figures 1B and 6B; Supplementary Figure 2, and unpublished observations). Moreover, γ-irradiated cells appeared large and flattened, expressed high levels of p21Waf1 and had very little cleaved PARP, pointing to an arrest rather than apoptotic response (Supplementary Figure 2). Thus, it is likely that p53 activation of CDIP is specific to apoptosis. p53 and CDIP did show a similar pattern of upregulation in the mouse small intestine after exposure to γ-irradiation, suggesting that CDIP may play a role in in vivo responses to γ-irradiation where the cellular outcome is apoptosis (Supplementary Figure 3).
CDIP is a direct target of p53-dependent transcriptional regulation
CDIP contains four potential p53 binding sites (p53-BS; el-Deiry et al, 1992) within a ∼2 kb genomic region, 5′ of the CDIP transcriptional start site (Figure 1C). We evaluated this putative CDIP promoter region for p53-dependent transcriptional activity using a heterologous reporter assay. Several deletion mutants of this promoter sequence were generated and linked to the luciferase reporter gene of the pGL3-basic vector (Figure 1C). Co-expression of a wt-CDIP-promoter luciferase construct (T1) together with a wt-p53 expression plasmid in p53-null Saos2 cells increased luciferase activity significantly over basal levels, while co-expression of either mutant p53 (V143A) or pcDNA3.1 empty vector failed to do so (Figure 1C). Deletion of a 562-bp fragment encompassing the most 5′ potential p53-BS (T2) reduced CDIP promoter activity three-fold in response to wt-p53 (Figure 1C). In addition, the reporter construct containing a point mutation of this site (at the −1618 position, M1) showed a similar decrease of wt-p53-dependent transcriptional activity.
Consistently, transfection of these CDIP-promoter constructs into p53-intact U2OS cells, followed by treatment with ETO, showed a similar requirement of the −1618 to −1639 p53-BS for DNA damage-induced CDIP promoter activation (Figure 1C). To verify whether p53 could bind to this candidate p53-BS in vivo, we used chromatin immunoprecipitation (ChIP) assay to analyze cell lysates extracted from Ad-p53-infected Saos2 and U2OS cells (Figure 1D). A 229-bp genomic fragment spanning the candidate −1618 and −1639 p53-BS was specifically immunoprecipitated as a p53 protein–chromatin complex, with an anti-p53 antibody in cells infected with Ad-p53 (Figure 1D, middle panel). This amplification product was not detected in lysates precipitated with an anti-HA antibody or in control Ad-GFP-infected cells (Figure 1D, middle panel). To examine whether endogenous p53 could bind to the CDIP promoter after DNA damage, ChIP assays were performed in ETO-treated U2OS cells. The same 229-bp CDIP genomic fragment spanning the −1618 to −1639 p53-BS was amplified from complexes immunoprecipitated with anti-p53 specific antibody, but not with IgG, specifically under ETO-treated conditions (Figure 1D, lower panel). In both ChIP experiments, p53 binding to the consensus p53-BS of the p21Waf1 promoter was detected under conditions of p53 expression/activation, which served as a positive control. These results suggest that the CDIP promoter is likely a direct target for p53 transcriptional activation, and that the consensus p53 binding site located at position −1618 to −1639 of the CDIP promoter is responsible for p53-dependent CDIP transcriptional expression.
Inhibition of CDIP impairs p53-mediated apoptotic responses
To investigate the role of CDIP in mediating p53-dependent apoptosis, we used the pBabe-U6-shRNA retroviral vector system to create CDIP knockdown (shCDIP) and control (scrambled) constructs, and evaluated the effect of inhibiting endogenous CDIP expression on apoptosis induced by p53 and genotoxic stress. shCDIP-transfected U2OS cells did not upregulate CDIP mRNA in response to ETO treatment, validating our shCDIP construct (Figure 2A). Conversely, ETO-induced CDIP mRNA expression in control (scrambled-shCDIP)-transfected U2OS cells was similar to that of non-transfected U2OS cells also treated with ETO (Figure 2A).
Figure 2.

Effect of CDIP inhibition on genotoxic stress- and p53-mediated apoptosis. (A) CDIP repression compromises p53-mediated apoptosis in response to genotoxic stress. U2OS cells were transfected with an shRNA vector targeting CDIP or a control vector of identical scrambled sequence (scrambled CDIP-shRNA), and treated with ETO (30 μM, 12 and 24 h). Northern blotting was performed to determine the levels of CDIP and 36B4 (upper panel). U2OS or IMR90-E1A cells were transfected with either CDIP-shRNA, scrambled CDIP-shRNA or a control shRNA (luciferase) for 24 h, and treated with ETO (30 μM) for the indicated time points. Cells were then assessed for apoptosis by TUNEL, DNA fragmentation assay or PI staining to detect the sub-G1 cell population. Both TUNEL-positive and sub-G1 PI-stained cells were enumerated by flow cytometry. The middle left panel shows a representative experiment; apoptotic cells containing 3′-hydroxyl DNA strand breaks are detected in the FITC fluorescence range (FL1-H: 515–565 nm). In the right panel, the percentage of TUNEL-positive cells is shown as the mean±s.e.m. (n=3). U2OS cells and IMR90-E1A used for the DNA fragmentation (lower left panels) are represented as the amount of extranuclear or extracellular DNA fragments relative to untreated controls. Error bars indicate ±s.d. of three independent experiments with each sample in triplicate. (B) Combined effects of Puma and CDIP depletion, and Noxa and CDIP depletion on genotoxic stress-induced apoptosis. U2OS and U2OS/shCDIP cells were transfected with siRNAs directed against Puma or Noxa, and apoptosis was assessed by TUNEL assay 40 h after treatment with 30 μM ETO (right panel, mean percent of TUNEL-positive cells, ±s.e.m. n=3). Western blot analysis of Puma and Noxa was performed for cells similarly treated to establish effective knockdown of protein expression by siRNA (right panel). (C) Effect of CDIP depletion on p53-dependent apoptosis. Saos2 cells were transfected with CDIP-shRNA or control GFP-shRNA, followed by infection with adenovirus expressing GFP (Ad-GFP) or p53 (Ad-p53) at an MOI of 20. The apoptotic response of Ad-infected Saos2 was similarly assessed by DNA fragmentation assay and PI staining, as described (lower panels).
shCDIP-transfected U2OS cells showed a significant reduction in the percentage of TUNEL-positive cells compared to cells either transfected with scrambled-shCDIP, or left untransfected, 24 and 40 h after exposure to ETO (Figure 2A, middle panels). Moreover, ETO-treated U2OS cells and IMR90-E1A cells transfected with shCDIP contained fewer fragmented nuclei, in comparison to control scrambled-shCDIP transfectants (Figure 2A, lower left panel). Last, inhibition of CDIP expression by shCDIP resulted in a marked decrease in the sub-G1 population of ETO-treated U2OS cells, in comparison to control treated cells (Figure 2A, lower right panel). Although cell cycle profiles indicate that approximately 50% of cells are viable 2 days post-ETO (i.e., in G1, S and G2/M phases), we have found that the sub-G1 population is an underrepresentation of apoptosis, since not all TUNEL-positive cells have sub-G1 DNA content (Supplementary Figure 4). This is in agreement with previous reports indicating that TUNEL-positive apoptotic cells can be found in all phases of the cell cycle (Lin and Benchimol, 1995). Thus, by three independent apoptotic assays, depletion of CDIP expression by shCDIP was effective in inhibiting apoptosis induced by DNA damage after exposure to ETO in cell lines containing endogenous wild-type p53.
CDIP was also studied in the context of other p53 pro-apoptotic targets. Puma and Noxa were depleted by siRNA targeting in U2OS and U2OS cells with stable expression of shCDIP. Cells depleted of Puma and Noxa are compromised in there apoptotic response to ETO (Figure 2B). This likely represents a CDIP-independent contribution of p53 to the overall drug-induced apoptotic response. Importantly, stable knockdown of CDIP in U2OS-shCDIP cells results in an even more pronounced impairment of ETO-induced apoptosis (Figure 2, compare panel B with A), which is minimally reduced further when combined with either Puma or Noxa knockdown (Figure 2B).
To test the effect of CDIP depletion on a purely p53-dependent apoptotic response, p53-null Saos2 cells were transfected with shCDIP, and the ability of exogenous p53 expression (by Ad-p53 infection) to induce apoptosis was assessed. shCDIP was effective in blocking Ad-p53-induced CDIP expression in Saos2 cells (Figure 2C, upper panel). This block of CDIP induction by Ad-p53 in shCDIP-transfected Saos2 cells corresponded to a decreased propensity for Ad-p53 infected Saos2 cells to undergo apoptosis (Figure 2C, lower panel). Together, these results demonstrate a requirement for CDIP in p53-mediated apoptosis, and suggest that it is an important component of the p53 death signal.
CDIP is a potent inducer of apoptosis
Next, we examined whether ectopic expression of CDIP had any effect on the viability of cultured U2OS and MCF7 cells. A tetracycline-regulated CDIP cell line (U2OS/CDIP-tet-on) was generated from U2OS cells, and the effect of CDIP expression on apoptosis was assessed. Cells cultured under conditions of tetracycline promoter-driven CDIP activation (+doxycyclin) upregulated HA-tagged CDIP in a time-dependent manner (Figure 3A). Crystal violet staining of U2OS/CDIP-tet-on cells revealed that cell growth was severely restricted 40 h after the introduction of doxycyclin (dox) to the culture media, and control U2OS/GFP-tet-on were not affected by the addition of dox (Figure 3B). This attenuation of cell growth corresponded with the ability of CDIP to induce significant apoptosis under tet-induced (+dox) conditions. For example, 48 h after the administration of dox the percentage of CDIP expressing cells containing double-stranded DNA breaks characteristic of apoptotic cells (as determined by TUNEL assay followed by enumeration of dUTP-FITC-positive cells by flow cytometry) averaged 78% (Figure 3A, lower left panel). Additional independent markers consistent with apoptosis were also apparent in U2OS/CDIP-tet-on cells, specifically under conditions of CDIP expression, including the appearance of cells with nucleosomal DNA fragments and sub-G1 DNA content (Figure 3A, right panel). By similar assays, the ectopic expression of CDIP in MCF7 cells, introduced by infection with adenovirus expressing CDIP (Ad-CDIP), resulted in significant apoptosis in comparison to control Ad-GFP-infected cells (Figure 3C). Thus, CDIP is a potent pro-apoptotic protein.
Figure 3.

Induction of apoptosis by CDIP expression. (A) Apoptosis analysis of U2OS/CDIP-tet-on cells grown in the absence (−) or presence (+) of the tetracycline analogue, doxycyclin (dox). Western blots were performed using an anti-HA antibody to detect CDIP; β-actin antibody was used as a loading control (upper panel). U2OS/CDIP-tet-on cells were grown ±dox for the indicated time periods, and apoptosis was assessed by three independent cell death assays: TUNEL, DNA fragmentation by cell death ELISA and PI staining to detect the sub-G1 population. (B) Viability of U2OS/CDIP- and U2OS/GFP-tet-on cells by crystal violet staining, 40 h after the administration of dox (2 μg/ml). (C) Western blotting and cell death analyses of MCF7 cells infected with a recombinant adenovirus expressing GFP (Ad-GFP) or CDIP (Ad-CDIP) for 12 and 24 h, as indicated. The upper panel shows anti-HA detection of CDIP, and the lower panel shows β-actin levels as a loading control. Apoptotic MCF-7 cells infected with Ad-CDIP or Ad-GFP were detected by DNA fragmentation assay (lower left panel) and flow cytometric analysis of PI-stained cells (lower right panel). Error bars indicate ±s.d. of three independent experiments.
Apoptosis induced by CDIP is dependent upon caspase-8 activation
As a first step in delineating the death pathway mediating CDIP-induced apoptosis, we looked for cleavage of caspase-9 and caspase-8 from their pro- to active forms in CDIP expressing U2OS/CDIP-tet-on cells (i.e., implicating either the intrinsic or extrinsic pathway, respectively). Surprisingly little caspase-9 cleavage was detected in cells after the induction of CDIP with dox, and by 30 h only a small amount of cytosolic cytochrome c was detected (Figure 4A). On the other hand, cleavage of caspase-8 from its pro- to active form was readily detected in cells 18 h after the administration of dox, and correlated with a 2.5-fold increase in caspase-8 activity, and the appearance of cleaved PARP, a pro-apoptotic cellular marker (Figure 4A).
Figure 4.

Caspase-8 is required for CDIP-dependent apoptosis. (A) U2OS/CDIP-tet-on cells were grown in the absence (−) or presence (+) of dox and harvested at the time intervals shown. Whole-cell lysates were either directly assessed for caspase-8, caspase-9, PARP and β-actin expression by Western blot analysis (left panel), or fractionated. Active caspase-8 species detected are consistent with three N-terminal Asp cleavage fragments. Fractionates were assessed for the presence of cytochrome c (cyt.-c), oxidative complex II (OxPhos II) and α-tubulin, by Western blotting; the latter were used as mitochondria- and cytoplasm-specific markers, respectively (upper right panel). Caspase-8 enzyme activity was measured by incubating cell lysates from similarly treated U2OS/CDIP-tet-on cells with the caspase-8 substrate, Ac-IETD-pNA. DNA fragmentation was also determined by cell death ELISA in dox-treated U2OS/CDIP-tet-on cells in the absence (+DMSO) and presence of the caspase-8 inhibitor II (lower right panel). (B) Inhibition of caspase-8 by siRNA directed against caspase-8 blocks CDIP-induced apoptosis. Caspase-8 siRNA and control siRNAs were transfected into U2OS/CDIP-tet-on cells. After 24 h, cells were grown in the absence or presence of dox and harvested at the time intervals shown. CDIP, caspase-8, PARP and β-actin (as a loading control) expression were assessed by Western blot analysis. Apoptosis was assessed in U2OS/CDIP-tet-on cells similarly transfected by TUNEL assay (40 h post-dox treatment) and trypan blue exclusion (24 and 48 h post-dox treatment). The percentages of TUNEL- and trypan blue-positive cells are shown as the mean±s.e.m. (n=3). (C) Inhibition of caspase-8 by siRNA attenuates apoptosis induced by genotoxic stress. ETO induces p53, caspase-8 and PARP cleavage 24 h post-ETO (30 μM) treatment (upper left panel). U2OS cells were transfected with siRNAs directed against caspase-8 and apoptosis was assessed by TUNEL assay 40 h after ETO treatment (middle panel is a representative experiment; lower panel is the mean percent of TUNEL-positive cells, ±s.e.m. n=3). Western blot analysis of p53 and caspase-8 cells similarly treated, establishes effective knockdown of caspase-8 protein expression by siRNA (right panel).
We disabled the extrinsic death pathway by treating cells with a caspase-8 inhibitor (z-IETD-fmk) as well as short interfering RNAs (Casp-8 siRNA), designed to specifically ablate caspase-8 expression, and tested the ability of CDIP to induce apoptosis. Treatment of U2OS/CDIP-tet-on cells with z-IETD-fmk substantially abrogated cell death in response to CDIP activation (Figure 4A). Consistently, transfection of casp-8 siRNA before the induction of CDIP resulted in a dramatic decrease in TUNEL-positive cells, and significantly decreased trypan blue-positive non-viable cells, in comparison to control siRNA-transfected cells (Figure 4B). The casp-8 siRNA was effective at abrogating CDIP-induced caspase-8 cleavage, and cleaved PARP was also reduced in casp-8 siRNA-transfected CDIP expressing cells (Figure 4B, upper left panel). These results demonstrate that CDIP-dependent apoptosis is dependent on caspase-8, and implicate the extrinsic cell death pathway as the principle means by which CDIP mediates cell death. A further prediction from these results is that inhibition of caspase-8 should attenuate apoptosis induced by genotoxic stress. Indeed, cells treated with casp-8 siRNA show a marked reduction in apoptosis in response to ETO (Figure 4C).
CDIP upregulates TNF-α expression downstream of p53
Having implicated the extrinsic cell death pathway downstream of CDIP, we then were interested in delineating how this pathway may be activated by CDIP. The extrinsic cell death pathway initiates at the cell surface when members of the TNF-α family of cytokines bind to their cognate receptors. We looked for changes in the expression of these proteins in response to CDIP induction and found that CDIP expression tightly correlated with the expression of TNF-α but not TRAIL ligand or Fas receptor (Figure 5A; Supplementary Figure 5). To determine if the increase in the TNF-α mRNA translated to an increase in TNF-α protein, we measured the amount of TNF-α in dox-treated U2OS/CDIP-tet-on cells using an immuno-absorbent assay. A marked increase in TNF-α protein (i.e., <5 pg/ml −dox to ∼110 pg/ml after 24 h +dox), was observed (Figure 5B).
Figure 5.
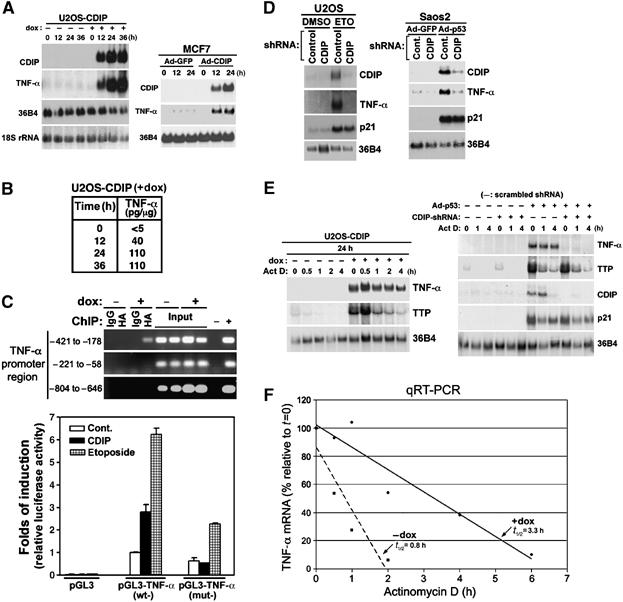
CDIP and p53 activate TNF-α, and p53 induction of TNF-α requires CDIP. (A) TNF-α mRNA increases upon induction of CDIP. U2OS/CDIP-tet-on cells grown in the absence (−) or presence (+) of dox, and MCF7 cells infected with a recombinant adenovirus expressing GFP (Ad-GFP) or CDIP (Ad-CDIP), were subjected to Northern blot analysis sequentially using 32P-labeled probes that detect CDIP, TNF-α and 36B4, or 18S rRNA (loading controls). (B) CDIP induces TNF-α protein. U2OS/CDIP-tet-on cells were grown in the presence or absence of dox for the indicated times, and TNF-α protein was quantified by ELISA. (C) CDIP binds and activates the TNF-α promoter. CDIP binding to the TNF-α promoter at the indicated regions was assessed by ChIP in U2OS/CDIP-tet-on cells in the absence (−) and presence (+) of dox, 24 h post-treatment. Bound chromatin was immunoprecipitated with anti-HA antibody to detect CDIP and anti-rabbit IgG as a negative control. Negative (−) and positive (+) PCR controls consisted of H2O and a pGL3-TNF-α promoter plasmid using the same amplification mixture, respectively. U2OS cells were transfected with a pcDNA3.1-CDIP expression vector (and empty vector control), together with either the pGL3-basic-promoter-luciferase construct or pGL3-TNF-α promoter-driven luciferase constructs with a wild-type promoter (wt) or a –421 to –178 deletion mutant (mut). Luciferase activity is shown relative to pGL3-luciferase basal activity. pGL3-basic-, -TNF-α (wt)- and -TNF-α (mut)-transfected cells were additionally stimulated with ETO to activate endogenous CDIP (lower panel). (D) p53- and genotoxic stress-mediated TNF-α induction is inhibited by shRNA-directed knockdown of CDIP. CDIP-shRNA vector and control shRNA vectors (scrambled CDIP-shRNA) were transfected into U2OS cells, followed by ETO treatment for 24 h. p53-null Saos2 cells were transfected with CDIP-shRNA or control scrambled CDIP-shRNA, and infected with adenovirus expressing wt-p53. The expression of CDIP, TNF-α, p21Waf1 and 36B4 (loading control) was assessed by sequential Northern blotting with 32P-labeled probes specific for these transcripts. (E) CDIP stabilizes TNF-α mRNA. U2OS/CDIP-tet-on cells grown ±dox were treated with actinomycin D (Act D, 8 μg/ml), and harvested at the indicated time intervals. TNF-α, TTP and 36B4 (loading control) mRNA expression was assessed by Northern blot analysis. Saos2 cells were also transfected with CDIP-shRNA or control scrambled CDIP-shRNA, and infected with adenovirus expressing wt-p53 (Ad-p53) or Ad-GFP (−). After 24 h, the actinomycin D chase and Northern blot experiments were performed as described above. (F) CDIP expression leads to an increase in TNF-α half-life. Cells were cultured ±dox, and TNF-α transcript levels were assessed by quantitative real-time RT-PCR (qRT-PCR), after treatment with actinomycin D. The amount of TNF-α transcript relative to that present after 24 h ±dox (i.e., t=0 for actinomycin treatment), was quantitated, and the half-life was determined by regression analysis.
Cellular fractionation and localization experiments revealed that CDIP is found predominantly in the nucleus, where it displays a punctate staining pattern, particularly after DNA damage (Supplementary Figure 6). This suggested that CDIP might function as a transcriptional regulator. Therefore, we evaluated whether CDIP could activate TNF-α gene transcription using an ∼2 kb genomic fragment of TNF-α promoter region, in a heterologous luciferase reporter gene assay. Co-expression of CDIP (by exogenous pcDNA3.1-CDIP expression or by induction with dox) in U2OS cells together with the TNF-α promoter-luciferase fusion construct resulted in TNF-α promoter activation (Figure 5C). LPS, a known activator of TNF-α transcription, did not induce CDIP expression in macrophages or U2OS cells, supporting that the CDIP → TNF-α signal is distinct from that of TNF-α induction by LPS (Supplementary Figure 7). ChIP analysis shows that CDIP binds to a 200-bp region (−421 to –221) of the TNF-α promoter, and when this region is deleted, TNF-α promoter activation in response to CDIP expression and ETO is severely attenuated (Figure 5C).
Thus far we have shown that CDIP is a transcriptional target of p53, and that CDIP induction by p53 is associated with an increase in TNF-α expression. This raises the possibility that TNF-α is also a component of the p53-signaling pathway. To address this possibility, we asked if induction of p53 expression correlated with an increase in TNF-α expression, and further tested whether knockdown of CDIP under these conditions inhibited TNF-α induction. U2OS and Saos2 cells were transfected with CDIP shRNA (shCDIP) or control scrambled CDIP shRNA, and TNF-α mRNA levels were assessed by Northern blotting after treatment with ETO or the expression of exogenous p53, respectively. As shown in Figure 5D, ETO treatment (of U2OS cells containing wt-p53) and exogenous p53 expression (in p53-null Saos2 cells) resulted in the upregulation of both TNF-α and CDIP. Notably, in cells similarly treated, knockdown of CDIP with shRNA completely inhibited TNF-α mRNA induction (Figure 5D). p53 was effective in upregulating an unrelated p53 target gene (i.e., p21Waf1) under these conditions, indicating that the absence of TNF-α induction upon p53 activation/expression was in fact due to the inhibition of CDIP (Figure 5D).
Since increased mRNA levels can be a result of an increase in gene transcription and/or an increase in mRNA half-life, we examined the effect of the RNA synthesis inhibitor, actinomycin D, on CDIP-induced TNF-α mRNA expression. Northern blot analysis revealed that the level of the TNF-α transcript was sustained in U2OS/CDIP-tet-on cells after the administration of actinomycin D (Figure 5E). This effect was quantified by real-time PCR, and the half-life of the TNF-α transcript in CDIP expressing cells was found to be four-fold increased over that of non-expressing cells (Figure 5F). In contrast, transcript levels of tristetraproline (TTP), a known regulator of TNF-α mRNA stability (Carballo et al, 1998), rapidly decreased upon treatment with actinomycin D, irrespective of CDIP expression (Figure 5E, left panel). To further verify that CDIP acts downstream of p53 in the regulation of TNF-α mRNA, and confirm that this regulation can occur at the post-transcriptional level, we investigated whether CDIP, induced by Ad-p53, resulted in an increase in TNF-α mRNA stability. Consistently, the expression of Ad-p53 led to an increase in TNF-α mRNA expression, together with CDIP and p21Waf1, and TNF-α mRNA levels were stable for over 4 h in actinomycin D-treated cells; shCDIP-transfected cells showed no such effect (Figure 5E, right panel). Taken together, these results suggest that CDIP upregulates TNF-α downstream of p53, and that CDIP-mediated TNF-α upregulation occurs through transcriptional and post-transcriptional mechanisms.
Inhibition of TNF-α expression attenuates DNA damage-induced apoptosis
Our findings that CDIP is a transcriptional target of p53, capable of upregulating TNF-α downstream of p53, indicate a putative role for TNF-α in p53-mediated apoptotic signaling. To investigate this possibility, we used TNF-α shRNA to inhibit endogenous TNF-α expression and assessed the effect of this on DNA damage-induced apoptosis in U2OS and IMR90-E1A cells, which contain intact wild-type p53. U2OS cells were transfected with two TNF-α shRNAs (shTNF-α A and shTNF-α B), treated with ETO, and cell viability and apoptosis were assessed by trypan blue exclusion and TUNEL assay, respectively. Importantly, ETO treatment did not induce TNF-α mRNA in cells transfected with shTNF-α (Figure 6A, upper left panel). TNF-α shRNA-transfected cells showed a marked reduction in apoptosis in response to ETO treatment compared with control shRNA-transfected cells (Figure 6A, lower panel). This was consistent with a decrease in the percentage of non-viable shTNF-α-transfected cells (i.e., an increase in the viable cell population), after similar treatment with ETO (Figure 6A, right panel). IMR90-E1A cells upregulate CDIP in response to camptothecin (CPT; Figure 6B, upper left panel). Again, shTNF-α was effective at blocking TNF-α induction in response to CPT (Figure 6B, upper middle panel). In comparison to control-transfected cells, both shCDIP- and shTNF-α-transfected cells were comparably decreased in their apoptotic response to CPT (Figure 6B, lower panels), and displayed an increase in long-term viability, despite the presence of CPT in the culture media (Figure 6B, upper right panel). Thus, TNF-α is important for p53-induced apoptosis in response to ETO and CPT, and taken together with our findings that TNF-α is upregulated by p53 in a CDIP-dependent manner, suggests that a p53 → CDIP → TNF-α death signal directs apoptosis after exposure of cells to genotoxic stress.
Figure 6.

Inhibition of TNF-α attenuates apoptosis induced by DNA damage. (A) U2OS cells were transfected with shTNF-α constructs (A and B, where indicated, otherwise an equal amount of A combined with B) and treated with ETO (30 μM, 36 h). Northern blots were performed using 32P-labeled probes (p21Waf1, TNF-α and 36B4). Apoptosis was assessed by TUNEL assay. A representative experiment is shown in the lower left panel; apoptotic cells are positive for dUTP-FITC (FL1-H: 515–565 nm). In the lower right panel, the percentage of TUNEL-positive cells is shown as the mean±s.e.m. (n=3). Cell viability was also determined by trypan blue exclusion (upper right panel); average values±s.e.m. (n=3). (B) IMR90-E1A cells were transfected with either control (Cont.-) or CDIP-shRNA and treated with CPT (1 μM, 24 h). Western blot analysis confirmed the activation of p53 (s15p) and upregulation of CDIP by CPT. In contrast, CDIP-shRNA-transfected cells did not upregulate CDIP. TNF-α expression was assessed by qRT-PCR in cells similarly treated (upper middle panel). CPT-induced apoptosis was assessed by TUNEL assay in control (Cont.-), CDIP- and TNF-α-shRNA-transfected cells; apoptotic cells are positive for dUTP-TMR red (lower left, FL2-H: 570–620 nm). The percentage of TUNEL-positive cells is shown as the mean±s.e.m. (n=3; lower right panel). Long-term cell viability was determined by crystal violet staining of IMR90-E1A cells similarly transfected and treated with DMSO or CPT (1 μM) for 48 h (upper right panel).
CDIP induces apoptosis through the TNF-α death receptor-signaling cascade
To further substantiate this p53 → CDIP → TNF-α apoptotic signal, U2OS/CDIP-tet-on cells were cultured in the presence of a TNF-α neutralizing antibody, and CDIP-induced apoptosis was assessed. The TNF-α neutralizing antibody was effective in abrogating apoptosis in dox-treated U2OS/CDIP-tet-on cells, as measured by a decrease in TUNEL-positive apoptotic cells, compared to control rabbit-IgG-treated cells (Figure 7A, left panel). Additionally, U2OS/CDIP-tet-on cells treated with TNF-α neutralizing antibody exhibited a significant decrease in DNA fragmentation and histone release induced by CDIP (Figure 7A, right panel). Further, CDIP expressing TNF-α antibody-treated cultures remained viable up to 48 h; whereas control-treated cells were ∼50% trypan blue positive (i.e., non-viable) at this time point (Figure 7A, right panel). These results demonstrate that CDIP-induced cell death is largely dependent on TNF-α, which is consistent with our findings that CDIP and TNF-α are components of the p53-mediated apoptotic signal.
Figure 7.

Induction of apoptosis by CDIP involves the TNF-α death receptor signaling pathway. (A) TNF-α neutralizing antibodies block CDIP-mediated apoptosis. U2OS/CDIP-tet-on cells (±dox) were incubated with either a neutralizing monoclonal antibody to human TNF-α (8 μg/ml) or Rb IgG (8 μg/ml), and assessed for apoptosis using DNA fragmentation (cell death ELISA) and TUNEL assays; cell viability was additionally measured by trypan blue exclusion. The percentage of TUNEL-positive cells is shown as the mean±s.e.m. (n=3). (B) The effect of FADD inhibition on CDIP-mediated cell death. U2OS/CDIP-tet-on cells were transfected either pcDNA3.1-DN-FADD or empty vector, and grown ±dox for 24 h, and then analyzed for cell death using trypan blue exclusion and DNA fragmentation/cell death ELISA. Error bars indicate ±s.d. of three independent experiments.
If CDIP-dependent apoptosis occurs primarily through the extrinsic death receptor-signaling pathway, we expect that other components known to be essential for this death-signaling pathway (i.e., FADD and caspase-8) are requisites of the CDIP-mediated death signal. Indeed, we have here shown that intact caspase-8 is essential for apoptosis induced by CDIP (Figure 4). To verify the importance of this pathway in mediating apoptosis induced by CDIP, we investigated the role of FADD, an intermediary effector of this pathway (acting upstream of caspase-8 and downstream of TNF-α), in CDIP-dependent apoptosis. The transfection of cells with a dominant-negative form of FADD (DN-FADD) resulted in reproducible decreases in apoptotic and non-viable cells after the induction of CDIP expression compared to control vector-transfected cells (Figure 7B). Taken together, these results indicate that the TNF-α pro-apoptotic signaling pathway is a downstream effector of CDIP-induced cell death.
Discussion
In this study, we identify CDIP as a novel pro-apoptotic p53 target gene. CDIP was identified as a p53-responsive gene through a cDNA expression array that compared gene expression under p53-induced and non-induced conditions. We demonstrate that CDIP is upregulated in a p53-dependent manner in several cell lines. p53-dependent CDIP upregulation likely occurs by direct transcriptional activation; CDIP contains four p53 consensus binding sites within its promoter region, the most 5′ of which is bound by p53 and required for p53-dependent transcriptional activation. CDIP is highly expressed in cells undergoing apoptosis in response to p53 expression and p53 activation by genotoxic stress. Significantly, shRNA-directed inhibition of CDIP abrogates p53-mediated apoptosis, placing CDIP among other important pro-apoptotic p53 targets that attenuate p53-dependent apoptosis when disrupted by RNA inhibition and gene depletion strategies. However, it is not yet possible to attribute the full apoptotic effects of p53 to any one of its pro-apoptotic gene targets, and further studies are required to elucidate how these genes interact (e.g., playing redundant or cooperative roles in p53-mediated apoptosis), and whether they act in a cell- and/or stimulus-specific manner. Indeed, we observed some attenuation of ETO-induced apoptosis upon siRNA targeting of Puma and Noxa that was likely a contribution of p53 acting independently of CDIP.
Interestingly, the expression of CDIP alone is sufficient to induce apoptosis. CDIP-dependent apoptosis is accompanied by caspase-8 cleavage, implicating the extrinsic cell death pathway as a mediator of the CDIP-induced death signal. Consistent with this, we demonstrate that CDIP-dependent apoptosis is almost entirely dependent on caspase-8 and also, to a large extent, on TNF-α and FADD. Inhibiting TNF-α (by shRNA and neutralizing antibody) and FADD (by the expression of a dominant-interfering mutant) produced a less complete block of CDIP-induced in comparison to the effects of caspase-8 inhibition; thus, we cannot rule out the possibility that CDIP utilizes other effectors of apoptosis (i.e., that also require caspase-8) to transduce its death signal.
CDIP expression tightly correlates with TNF-α expression, and we found that CDIP regulates TNF-α expression through mechanisms involving both transcriptional upregulation of the TNF-α promoter and increased TNF-α mRNA stability. As CDIP is here shown to be an important effector of p53-dependent apoptosis, these results suggest that TNF-α also plays a role in apoptosis induced by p53. In fact, exposure of cells to genotoxic stress, and the expression of exogenous p53 in Saos2 cells, results in TNF-α upregulation that is dependent upon CDIP. Moreover, depletion of TNF-α attenuates both drug- and CDIP-induced apoptosis, which taken together support a novel p53 → CDIP → TNF-α death-signaling pathway. This contrasts previous studies that show that primary lymphocyte cell populations, harboring defects in the extrinsic death-receptor pathway, display no impairment of p53-mediated apoptosis (Newton and Strasser, 2000; Salmena et al, 2003). It is plausible that in transformed cells, with an inherently high proliferative rate, thus, increased sensitivity to death stimuli, (Lowe et al, 2004), the p53 apoptotic program is very different in comparison to primary cells.
TNF-α orchestrates pleiotropic functions not only in the host immune response but also in control of cell proliferation, differentiation and apoptosis (Wajant et al, 2003; Muppidi et al, 2004). Physiologically, the secretion of TNF-α is highly localized and transient, and the regulatory mechanisms that control TNF-α expression allowing for such diverse cellular processes as proliferation and apoptosis have not been fully elucidated. The TNF-α promoter contains binding sites for a number of transcription factors (e.g., NF-κB , Ap-1, STAT1, CREBP), and the recently identified factor, LITAF (LPS-Induced TNF-α Factor) (Myokai et al, 1999; Tang et al, 2003), also known as PIG7 (Polyak et al, 1997). A search for conserved protein domains within the predicted CDIP amino-acid sequence identified a LITAF domain in the C-terminal region (amino acids 124–183; Supplementary Figure 1), originally described for the LITAF/PIG7 protein. Like CDIP, LITAF/PIG7 is a known target of p53 that is induced under conditions of cellular stress that cause apoptosis (Polyak et al, 1997). LITAF/PIG7 has also been shown to upregulate TNF-α expression in response to LPS (Myokai et al, 1999; Tang et al, 2003), however, it is unlikely that LPS can upregulate TNF-α through CDIP, since CDIP is not induced by LPS. The pro-apoptotic potential of LITAF/PIG7 has not been comprehensively tested, and it will be interesting to determine whether it promotes cell death through a mechanism similar to that of CDIP. It also is possible that p53 acts through LITAF/PIG7 to transcriptionally regulate TNF-α expression.
Although members of the TNF death receptor family signal apoptosis independently of p53, interactions between p53-dependent intrinsic apoptotic pathway and the death receptor-mediated extrinsic apoptotic pathway are well documented. For example, p53-dependent upregulation is believed to be responsible for the induction of Fas/CD95/APO-1 in response to DNA damaging agents in several different cell types (Muller et al, 1997, 1998; Miyake et al, 1998). Also, the TRAIL receptor, KILLER/DR5, is upregulated in cells undergoing apoptosis in response chemotherapeutic agents and γ-irradiation in a p53-dependent manner (Wu et al, 1997, 1999). Both Fas/CD95/APO1 and KIILER/DR5 contain genomic p53 consensus binding sites that likely mediate p53-dependent transcriptional activation of these death receptor genes (Muller et al, 1998; Takimoto and El-Deiry, 2000). Our demonstration that inhibiting TNF-α signaling attenuates apoptosis in response to genotoxic stress further supports a role for the extrinsic pathway in p53-mediated apoptosis.
Overall, our demonstration that CDIP is a p53 target provides additional insights into the p53-mediated apoptotic response, suggesting a novel network in which CDIP is important for regulating TNF-α expression downstream of p53. Thus, it is plausible that impaired CDIP induction in response to p53 activation by genotoxic stress, and the subsequent failure of consequent downstream signaling events leading to cell death, confers a survival advantage to tumor-prone cells, allowing them to escape apoptosis. In support of this view, the basal level of CDIP expression is higher in normal cell strains, such as non-diploid human fibroblasts, compared to transformed cells (unpublished observations), suggesting that loss of CDIP function may be a genetic event that promotes tumor progression. In summary, identifying the components of this unrecognized cell death pathway, from p53 → CDIP → TNF-α to death receptor-mediated cell death, provides an opportunity for therapeutic intervention, as it reveals a novel target for drug design that could potentially maximize the p53 response and sensitize tumor cells to cancer therapy.
Materials and methods
Cell lines and culture conditions
MCF7, IMR90-E1A, U2OS, Saos2 and EJ cells were cultured in DMEM containing 10% fetal bovine serum (FBS) (Invitrogen, Carlsbad, CA), 100 U/ml penicillin and 100 μg/ml streptomycin at 37°C. U2OS/CDIP-tet-on cells were cultured in the presence or absence of dox (2 μg/ml). Adenoviruses expressing p53, HA-CDIP, GFP-CDIP and GFP were generated, amplified and titrated as previously described (Ohtsuka et al, 2004). Cells were grown to 50–70% confluency and infected with recombinant adenovirus at a multiplicity of infection (MOI) of ∼20 for the indicated times.
ChIP
ChIP assays were performed according to the manufacturer's instruction, using the Chromatin Immuno-precipitation Assay kit (Upstate Biotechnology, NY). ChIP DNA (1 μl) extracted from the cell lysates was next used as a template for PCR. CDIP primers were as follows: 5′ ATGACCTACAAATCTCAACATGACTTGGAT 3′ and 5′ AGAGGAAGGGACGCCCTAACAGAGTTAC 3′, designed to yield a 229-bp product, including the putative p53-responsive element (position –1618 to −1639) in the CDIP promoter. p21Waf1 primers 5′-CTGAAAGCTGACTGCCCCTA-3′ and 5′-TCTCCTACCATCCCCTTCCTC-3′, designed to yield a 417-bp product spanning the p53-responsive element, were used as a positive control. TNF-α promoter primers were as follows: 5′-GCCCCTCCCAGTTCTAGTTC-3′ and 5′-AAAGTTGGGGACACACAAGC –3′ at position –421 to –178; 5′-GGGAGTGTGAGGGGTATCCT-3′ and 5′-CATTCAACCAGCGGAAAACT-3′ at position –221 to –58; and 5′-CAGGACCTCCAGGTATGGAA-3′ and 5′-TTTCATTCTGACCCGGAGAC-3′ at position –804 to –646. The PCR protocol was as follows: 35 cycles of a 30 s denaturation step at 94°C, a 1-min annealing step at 55°C and a 1 min extension step at 72°C.
Propidium iodide staining and flow cytometry
Cells were harvested and washed with PBS. Ice-cold 80% ethanol was then added dropwise over the pellets with periodic vortexing to mix cells. After fixation, propidium iodide (PI) was added to 50 μg/ml in phosphate-buffered saline (PBS) and samples were analyzed by flow cytometry. Sub-G1 populations were analyzed using the CellQuest program.
Apoptosis assays
Cell death, as determined by the fragmentation of DNA, was measured by photometric enzyme immunoassay using the Cell Death Detection ELISA kit (Roche, IN), following the manufacturer's protocol. For cell death as determined by TUNEL assay, cells were trypsinized, recovered by centrifugation at 300 g and fixed in 2% paraformaldehyde containing PBS for 16 h. Permeabilization and enzymatic labeling with either fluorescein- or TMR red-conjugated-dUTP were performed according to the manufacturer's protocol (Roche, IN). The percentage of cells that incorporated the fluorescence-conjugated dUTP was determined by flow cytometry. Trypan blue exclusion assay was also performed to analyze cell death. For caspase-8 assay, 5 × 106 cells were seeded in a 156-cm2 dish and cultured in the absence and presence of dox (2 μg/ml), for the time periods indicated. Active caspase-8 was assessed by direct incubation of lysates with the substrate Ac-IETD-pNA, according to assay kit instructions (Sigma-Aldrich, MO).
Subcellular fractionation
Cells (1 × 107) were washed with PBS and harvested by centrifugation at 350 g for 5 min. Cells were resuspended in 0.5 ml of hypotonic buffer (5 mM Tris (pH 7.4), 5 mM KCl, 1.5 mM MgCl2, 0.1 mM EGTA, 1 mM DTT) containing 1 mM PMSF, 10 mg/ml aprotinin and 10 mg/ml leupeptin, and incubated for 30 min on ice. Cells were homogenized by 30 strokes in a 20-gauge needle. Homogenates were centrifuged at 500 g for 5 min at 4°C to collect the nuclear fraction. Supernatants were centrifuged at 10 000 g for 30 min at 4°C to remove the heavier membrane fraction containing the mitochondria. Pellets were washed again with hypotonic buffer and recovered by centrifugation at 10 000 × g for 30 min to increase the purity of the subcellular fraction.
shRNA and siRNA experiments
Vectors expressing shRNAs against CDIP (5′-CGAGAUUGGCUUGAUGAAU-3′), scrambled CDIP (5′-GUUGAGGUGGUGAUCAUAAGUU-3′), GFP (5'-AAGACAAUCGGC UGCUCUGAU-3') and luciferase (5′-GGUGUUGGGCGCGUUAUUU-3′) were generated using shSTRIKE™ U6 Hairpin cloning systems (Promega, WI). Constructs expressing shTNF-α in pSIREN-DNR vectors were purchased from BD Biosciences. Caspase-8-, Noxa- and Puma-specific siRNAs were purchased from Santa Cruz Biotechnology. All shRNA and siRNA constructs were introduced into cells by transfection with Lipofectamine 2000 (Invitrogen, CA), according to the manufacturer's protocol.
TNF-α assay
The amount of TNF-α present in the cell culture supernatants and the total cell lysates was measured by a quantitative sandwich enzyme immunoassay, using the Quantikine human TNF-α immunoassay kit (R&D Systems, MN), according to the manufacturer's protocol.
Antibodies and other reagents
Several commercially available antibodies and other reagents were used in this study. They were the following: anti-HA (Santa Cruz, CA, Covance, CA), anti-p53 (Cell Signaling Technologies, MA), anti-p21Waf1 (Calbiochem, CA), anti-β-actin (Sigma-Aldrich, MO), anti--α-tubulin (Sigma-Aldrich, MO), anti-caspase-8 and 9 (BD Biosciences Pharmingen, CA), anti-β-calnexin (Sigma-Aldrich, MO), anti-Histone (Upstate, NY) and anti-TNF-α (Axxora, CA), anti-oxidative complex II (Molecular Probes, CA). Caspase-8 inhibitor II (Calbiochem, CA), ER tracker (Molecular Probes, CA), cell death detection ELISA kit (Roche, IN) and Quantikine TNF-α ELISA kit (R&D Systems, MN). A custom rabbit polyclonal antibody to CDIP was raised against a GST-CDIP fusion recombinant protein and affinity purified (Animal Pharm Services Inc.).
Accession number. We have submitted the CDIP sequence to GenBank, and the EMBL/GenBank accession number is DQ167023.
Supplementary Material
Supplementary Information
Acknowledgments
We thank S Boswell and S Das for helpful discussion, and YR Novella for the TNF-α promoter construct. This work was supported by NIH grants CA85681, CA078356, CA097216 and CA80058, and by Shiseido Research Funding.
References
- Baud V, Karin M (2001) Signal transduction by tumor necrosis factor and its relatives. Trends Cell Biol 11: 372–377 [DOI] [PubMed] [Google Scholar]
- Benchimol S (2001) p53-dependent pathways of apoptosis. Cell Death Differ 8: 1049–1051 [DOI] [PubMed] [Google Scholar]
- Brugarolas J, Chandrasekaran C, Gordon JI, Beach D, Jacks T, Hannon GJ (1995) Radiation-induced cell cycle arrest compromised by p21 deficiency. Nature 377: 552–557 [DOI] [PubMed] [Google Scholar]
- Carballo E, Lai WS, Blackshear PJ (1998) Feedback inhibition of macrophage tumor necrosis factor-alpha production by tristetraprolin. Science 281: 1001–1005 [DOI] [PubMed] [Google Scholar]
- Chen G, Goeddel DV (2002) TNF-R1 signaling: a beautiful pathway. Science 296: 1634–1635 [DOI] [PubMed] [Google Scholar]
- Deng C, Zhang P, Harper JW, Elledge SJ, Leder P (1995) Mice lacking p21CIP1/WAF1 undergo normal development, but are defective in G1 checkpoint control. Cell 82: 675–684 [DOI] [PubMed] [Google Scholar]
- el-Deiry WS, Kern SE, Pietenpol JA, Kinzler KW, Vogelstein B (1992) Definition of a consensus binding site for p53. Nat Genet 1: 45–49 [DOI] [PubMed] [Google Scholar]
- el-Deiry WS, Tokino T, Velculescu VE, Levy DB, Parsons R, Trent JM, Lin D, Mercer WE, Kinzler KW, Vogelstein B (1993) WAF1, a potential mediator of p53 tumor suppression. Cell 75: 817–825 [DOI] [PubMed] [Google Scholar]
- Embree-Ku M, Venturini D, Boekelheide K (2002) Fas is involved in the p53-dependent apoptotic response to ionizing radiation in mouse testis. Biol Reprod 66: 1456–1461 [DOI] [PubMed] [Google Scholar]
- Friesen C, Herr I, Krammer PH, Debatin KM (1996) Involvement of the CD95 (APO-1/FAS) receptor/ligand system in drug-induced apoptosis in leukemia cells. Nat Med 2: 574–577 [DOI] [PubMed] [Google Scholar]
- Hanahan D, Weinberg RA (2000) The hallmarks of cancer. Cell 100: 57–70 [DOI] [PubMed] [Google Scholar]
- Lin Y, Benchimol S (1995) Cytokines inhibit p53-mediated apoptosis but not p53-mediated G1 arrest. Mol Cell Biol 15: 6045–6054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowe SW, Cepero E, Evan G (2004) Intrinsic tumour suppression. Nature 432: 307–315 [DOI] [PubMed] [Google Scholar]
- Miyake H, Hara I, Gohji K, Arakawa S, Kamidono S (1998) p53 modulation of Fas/Apo-1 mediated apoptosis in a human renal cell carcinoma cell line. Int J Oncol 12: 469–473 [DOI] [PubMed] [Google Scholar]
- Muller M, Strand S, Hug H, Heinemann EM, Walczak H, Hofmann WJ, Stremmel W, Krammer PH, Galle PR (1997) Drug-induced apoptosis in hepatoma cells is mediated by the CD95 (APO-1/Fas) receptor/ligand system and involves activation of wild-type p53. J Clin Invest 99: 403–413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller M, Wilder S, Bannasch D, Israeli D, Lehlbach K, Li-Weber M, Friedman SL, Galle PR, Stremmel W, Oren M, Krammer PH (1998) p53 activates the CD95 (APO-1/Fas) gene in response to DNA damage by anticancer drugs. J Exp Med 188: 2033–2045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muppidi JR, Tschopp J, Siegel RM (2004) Life and death decisions: secondary complexes and lipid rafts in TNF receptor family signal transduction. Immunity 21: 461–465 [DOI] [PubMed] [Google Scholar]
- Myokai F, Takashiba S, Lebo R, Amar S (1999) A novel lipopolysaccharide-induced transcription factor regulating tumor necrosis factor alpha gene expression: molecular cloning, sequencing, characterization, and chromosomal assignment. Proc Natl Acad Sci USA 96: 4518–4523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newton K, Strasser A (2000) Ionizing radiation and chemotherapeutic drugs induce apoptosis in lymphocytes in the absence of Fas or FADD/MORT1 signaling. Implications for cancer therapy. J Exp Med 191: 195–200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohtsuka T, Ryu H, Minamishima YA, Macip S, Sagara J, Nakayama KI, Aaronson SA, Lee SW (2004) ASC is a Bax adaptor and regulates the p53-Bax mitochondrial apoptosis pathway. Nat Cell Biol 6: 121–128 [DOI] [PubMed] [Google Scholar]
- Oren M (2003) Decision making by p53: life, death and cancer. Cell Death Differ 10: 431–442 [DOI] [PubMed] [Google Scholar]
- Owen-Schaub LB, Zhang W, Cusack JC, Angelo LS, Santee SM, Fujiwara T, Roth JA, Deisseroth AB, Zhang WW, Kruzel E, Radinsky R (1995) Wild-type human p53 and a temperature-sensitive mutant induce Fas/APO-1 expression. Mol Cell Biol 15: 3032–3040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peter ME, Krammer PH (2003) The CD95(APO-1/Fas) DISC and beyond. Cell Death Differ 10: 26–35 [DOI] [PubMed] [Google Scholar]
- Polyak K, Xia Y, Zweier JL, Kinzler KW, Vogelstein B (1997) A model for p53-induced apoptosis. Nature 389: 300–305 [DOI] [PubMed] [Google Scholar]
- Salmena L, Lemmers B, Hakem A, Matysiak-Zablocki E, Murakami K, Au PY, Berry DM, Tamblyn L, Shehabeldin A, Migon E, Wakeham A, Bouchard D, Yeh WC, McGlade JC, Ohashi PS, Hakem R (2003) Essential role for caspase 8 in T-cell homeostasis and T-cell-mediated immunity. Genes Dev 17: 883–895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuler M, Bossy-Wetzel E, Goldstein JC, Fitzgerald P, Green DR (2000) p53 induces apoptosis by caspase activation through mitochondrial cytochrome c release. J Biol Chem 275: 7337–7342 [DOI] [PubMed] [Google Scholar]
- Soengas MS, Alarcon RM, Yoshida H, Giaccia AJ, Hakem R, Mak TW, Lowe SW (1999) Apaf-1 and caspase-9 in p53-dependent apoptosis and tumor inhibition. Science 284: 156–159 [DOI] [PubMed] [Google Scholar]
- Symonds H, Krall L, Remington L, Saenz-Robles M, Lowe S, Jacks T, Van Dyke T (1994) p53-dependent apoptosis suppresses tumor growth and progression in vivo. Cell 78: 703–711 [DOI] [PubMed] [Google Scholar]
- Takimoto R, El-Deiry WS (2000) Wild-type p53 transactivates the KILLER/DR5 gene through an intronic sequence-specific DNA-binding site. Oncogene 19: 1735–1743 [DOI] [PubMed] [Google Scholar]
- Tang X, Fenton MJ, Amar S (2003) Identification and functional characterization of a novel binding site on TNF-alpha promoter. Proc Natl Acad Sci USA 100: 4096–4101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorburn A (2004) Death receptor-induced cell killing. Cell Signal 16: 139–144 [DOI] [PubMed] [Google Scholar]
- Wajant H, Pfizenmaier K, Scheurich P (2003) Tumor necrosis factor signaling. Cell Death Differ 10: 45–65 [DOI] [PubMed] [Google Scholar]
- Wu GS, Burns TF, McDonald ER III, Jiang W, Meng R, Krantz ID, Kao G, Gan DD, Zhou JY, Muschel R, Hamilton SR, Spinner NB, Markowitz S, Wu G, el-Deiry WS (1997) KILLER/DR5 is a DNA damage-inducible p53-regulated death receptor gene. Nat Genet 17: 141–143 [DOI] [PubMed] [Google Scholar]
- Wu GS, Burns TF, McDonald ER III, Meng RD, Kao G, Muschel R, Yen T, el-Deiry WS (1999) Induction of the TRAIL receptor KILLER/DR5 in p53-dependent apoptosis but not growth arrest. Oncogene 18: 6411–6418 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Information