

Abstract
We have used a chromatin immunoprecipitation (ChIP)-based cloning strategy to isolate and identify genes associated with estrogen receptor α (ERα) in MCF-7 human breast cancer cells. One of the gene regions isolated was a 288 bp fragment from the ninth intron of the breast cancer 1 associated ring domain (BARD1) gene. We demonstrated that ERα associated with this region of the endogenous BARD 1 gene in MCF-7 cells, that ERα bound to three of five ERE half sites located in the 288 bp BARD1 region, and that this 288 bp BARD1 region conferred estrogen responsiveness to a heterologous promoter. Importantly, treatment of MCF-7 cells with estrogen increased BARD1 mRNA and protein levels. These findings demonstrate that ChIP cloning strategies can be utilized to successfully isolate regulatory regions that are far removed from the transcription start site and assist in identifying cis elements involved in conferring estrogen responsiveness.
Keywords: MCF-7 cells, Estrogen receptor, BARD1, Chromatin immunoprecipitation, Transcription, BRCA1, Intron
1. Introduction
Estrogens are critical for maintenance and function of male and female mammalian reproductive systems, but also influence the function of cardiovascular, neural, and skeletal cells and proliferation of breast cancer cells (Katzenellenbogen et al., 1987; Hess et al., 1997; Couse and Korach, 1999; Dubal et al., 1999; Wise et al., 2001). The genomic effects of 17β-estradiol (E2) are initiated by binding of hormone to the estrogen receptor (ER). The E2-occupied ERα interacts with regulatory sequences referred to as estrogen response elements (EREs) in target genes to initiate changes in gene expression. Although some estrogen-responsive genes contain a consensus ERE sequence (GGTCAnnnTGACC, Klein-Hitpass et al., 1988), lower affinity, imperfect EREs, which deviate from the consensus ERE sequence by one or more basepairs, are far more common (Wood et al., 1998, 2001; Klinge, 2001; Loven et al., 2001; Bourdeau et al., 2004). In addition to binding directly to consensus and imperfect EREs to modulate gene expression, ERα also regulates transcription by interacting with DNA-bound transcription factors such as Sp1, NFκB, or AP1 proteins (Kushner et al., 2000; Nilsson et al., 2001; Safe, 2001). While both transcriptional induction and repression have been observed at AP-1 sites (Kushner et al., 2000; Petz et al., 2002, 2004a,b), transcription is generally increased when ERα interacts with DNA-bound Sp1 (Safe, 2001) and decreased when ERα interacts with NFκB (Nilsson et al., 2001).
Given the increased incidence of breast cancer in women with long term exposure to E2 and the role of E2 in proliferation of breast cancer cells (Harris et al., 1992; Colditz, 1998), there has been significant interest in identifying genes that mediate estrogen's effects. Computational screens have been used to search the genome in silico for EREs (Bourdeau et al., 2004). While these screens have been useful in identifying potential ERE binding sites, divergence in DNA sequence can make it difficult to identify imperfect EREs, ERE half sites, and other cis elements that associate with ERα through DNA-bound transcription factors. Even when a cis element is identified in silico, it does not necessarily mean that the site is functional (Vega et al., 2006). Microarray analysis has also been used to identify estrogen responsive genes and has provided a wealth of information (Frasor et al., 2003; Hewitt et al., 2003; Stossi et al., 2004; Weisz et al., 2004). However, these studies are limited in the number of gene regions that can be interrogated and in the ability to distinguish between the primary and secondary effects of hormone.
To identify genes in human breast cancer cells that are regulated by E2 and ERα, we utilized a chromatin immuno-precipitation (ChIP)-based cloning strategy to perform a genome-wide screen in MCF-7 cells in order to isolate and clone DNA regions associated with endogenously expressed ERα. Using this methodology we were assured that the gene regions isolated were associated with ERα and were biologically relevant. One of the gene regions isolated using this method was the breast cancer gene 1 (BRCA1) associated ring domain 1 (BARD1) gene. Herein, we have characterized the interaction of ERα with the BARD1 gene and demonstrate the efficacy of using this modified ChIP procedure to identify novel genes and functional cis elements involved in regulating gene expression.
2. Materials and methods
2.1. MCF-7 cell culture and chromatin immunoprecipitation (ChIP)
MCF-7 cells were maintained in phenol red containing minimal essential medium (MEM) with 5% calf serum. Three days prior to treatment, cells were transferred to phenol red-free MEM with 5% charcoal dextran treated calf serum. MCF-7 cells were exposed to ethanol vehicle or 10 nM E2 for 24 h and immunoprecipitation of chromatin was carried out essentially as described previously (Petz et al., 2002). The ERα-specific antibody SC-8002 (Santa Cruz Biotechnology, Inc., Santa Cruz, CA) or the monoclonal antibody BZ1 combined with the polyclonal antibody LP1 were used to immunoprecipitate ERα-associated chromatin. Both BZ1 and LP1 were produced at the Immunological Resource Center (University of Illinois, Urbana, IL) and are directed against the human ERα LBD. DNA from the immunoprecipitated chromatin was purified, treated with Klenow to create blunt ends, and ligated to oligos containing BamHI restriction sites as described (Weinmann and Farnham, 2002) with modifications. Following digestion with BamHI, the DNA fragments were subcloned into BamHI-cut pTZ18R (Pharmecia, Picataway, NJ). The ligated plasmid was used to transform DH5α E. coli. White colonies were screened for the presence of insert using restriction digestion with BamHI and subclones were sequenced. DNA sequences were used in BLAST searches and the location of each clone was determined using the Ensembl database (www.ensembl.org). One of these plasmids, pTZ18R BARD, contained DNA sequence from +73,683 to 73,971 of the BARD1 gene. Conventional ChIP assays (Petz et al., 2002, 2004a,b) were performed to confirm the association of ERα with the BARD1 gene using an ERα-specific antibody or a nonspecific antibody directed against fluorescein (Immunological Resource Center, University of Illinois, Urbana, IL). Primers P1 (5′-CCT TGG GTA TTA TCT TAG C-3′) and P2 (5′-GAT GGG TAA TCA TAG ACG-3′), which anneal to DNA sequence in the ninth intron of the BARD1 gene, were used for amplification and visualization of the DNA on ethidium stained gels.
For real time quantitative PCR analysis of ChIP DNA, primers specific to BARD1 (5′-ACT CCA TCT CTC CAA AAG CAG-3′ and 5′-CGT CCT GAG TGT GAG TGT TG-3′), C14orf159 (5′-CCG AGG CAG GCA GAT CAC TTG-3′ and 5′-GCA CTC GCC ACC ACA CCT G-3′), C2orf30 (5′-TCC AGA CAG ACC GAC CAC AG-3′ and 5′-CTC AGC CTC GCC AGA CAT TC-3′), DOCK7 (Dedicator of cytokinesis 7, 5′-TGT GAG AAT CTT CTG AGG CTT GGG-3′ and 5′-AGT CCT GTC CTA AGG CAA ACC AAG-3′), LTBP1 (Latent transforming growth factor beta binding protein 1, 5′-AAA TCC TTG TCT CCG TTT ACG-3′ and 5′-AGG GAA TGG CAA AGA ATA AGC-3′), MLL/HRX (Myeloid/lymphoid or mixed-lineage leukemia/drosophila trithorax homolog, 5′-CAC TGT GCC TGG CGG AAG-3′ and 5′-CCT GAC TAG ATG AGA GGA GAT GG-3′), NPC1L1 (Nieman-Pick disease, type C1 gene-like 1, 5′-ATC TCT GAC CTT TGT CTT CTT G-3′ and 5′-GGT TAT GCT GAT GCT GCT G-3′), PABPC1 (poly(A) binding protein, cytoplasmic 1, 5′-AGA GAG AAA CAG AGA GTA TGT ATG-3′ and 5′-ACT TTG GGA GGC TGA GAC-3′), PPIH (peptidylprolyl isomerase H, 5′-TGA CAC CTC TCC CCA TTC TCT TG-3′and 5′-GCC TCA GGA AGC TCA CAA TCA TG-3′), PGR1 (progesterone receptor, 5′-GGG AAT TAG GGA AGG ATA AAG AGG-3′ and 5′-CCA ACC ACT ACA TTG TCA TCT ATC-3′), RALGPS1 (RAL GEF with PH domain and SH3 binding motif 1, 5′-TTG CCT GCT CTG AAG AAG-3′ and 5′-GCC AAA CAC TGA TTC CAA G-3′), RPS6KA5 (ribosome protein S6 kinase, 90 kDa peptide 5, 5′-AGA CCA CTG CTG CCT ATG C-3′ and 5′-TGT CTC CCT GCC CTT GTT G-3′), TANC2 (tetratricopeptide repeat, ankyrin repeat coiled coil containing 2, 5′-GGC AAG AAT ACC ACG TAA GCA ATG-3′ and 5′-TCT AGG GTA GCC AAG TGA CTG AG-3′), TLR5 (Toll-like receptor 5, 5′-TCA CTC TGA CTC TGT TCC TCA TG-3′ and 5′-TCT GTG CCC TGG GGA TGG-3′), or TNS3 (tensin 3, 5′-AGT GAA AGA AAG AGC GGT CTA TCC-3′and 5′-TCA AGC CAG CAA ACA AAG TTA GC-3′) were combined with iQ SYBR Green Supermix (Bio-Rad Laboratories, Inc., Richmond, CA) and used for quantitative PCR on the Bio-Rad iCycler (Bio-Rad Laboratories, Inc., Richmond, CA). One thousand, 5000, 10,000, and 50,000 genomic DNA copies were run in parallel with each primer set during each experiment to derive a standard curve. The relative copy number for each sample was determined from the standard curve. Each sample was run in triplicate and three independent experiments were performed. Data were normalized to the 36B4 gene, which was not affected by E2 treatment and analyzed using ezANOVA (C. Rorden, www.mricro.com, Columbia, SC).
2.2. Quantitative real time PCR
MCF-7 cells were cultured as described above and treated with ethanol vehicle or 10 nM E2 for 0.3, 2, 24, or 48 h. Cells were harvested with Trizol (Sigma, St. Louis, MO) and RNA was isolated according to the manufacturer's instructions. Isolated total RNA was treated with RQ1 RNase free DNase (Promega, Madison, WI) and cDNA was synthesized using the Reverse Transcription System (Promega, Madison, WI). The cDNA and iQ SYBR Green Supermix were combined with primers specific for 36B4 (5′-GCT GTT TCG ACG ACA CCG TT-3′ and 5′-TTC TGG AGG GAC GTC GAT G-3′), which is not affected by E2 treatment, or BARD1 (5′-TCA AAC ACC ATC CAA AGG ACA AC-3′ and 5′-GTC TGA GTC ACG TCA CTG TCT G-3′) and quantitative real time PCR was carried out on the Bio-Rad iCycler. The ΔΔCt method (Karsai et al., 2002) was used to calculate the fold change in BARD1 mRNA levels.
2.3. Western blot analysis
MCF-7 cells were cultured as described above, treated with ethanol vehicle or 10 nM E2 for 0.3, 2, 24, or 48 h or with 100 nM raloxifene, tamoxifen, or ICI 182,780 alone or in combination with 10 nM E2 for 24 h. Cells were harvested with TNE (40 mM Tris, pH 7.5, 140 mM NaCl, and 1.5 mM EDTA), pelleted, resuspended in 250 mM Tris–HCl, pH 7.8, lysed using three freeze/thaw cycles, and subjected to centrifugation. Protein concentrations of the whole cell extracts were determined using the Bio-Rad protein assay (Bio-Rad Laboratories, Inc., Richmond, CA) with BSA as a standard.
For Western analysis, 20–30 μg of whole cell extracts from MCF-7 cells that had been treated with E2 for various times were fractioned on a 10% SDS-PAGE gel and transferred to a nitrocellulose membrane. BARD1, Sp1, and β-actin were detected using sc-11438, sc-59 (Santa Cruz Biotechnology, Santa Cruz, CA) and A-5491 (Sigma, St. Louis, MO) antibodies, respectively. Horseradish peroxidase conjugated secondary antibodies and Super Signal West Femto chemiluminescent detection kit (Pierce Chemical Co., Rockford, IL) were used to visualize the proteins.
2.4. Preparation of purified ERα
ERα was expressed in Sf9 cells using ERα recombinant baculovirus stock generously provided by James Kadonaga (University of California, San Diego, CA) and Lee Kraus (Cornell University, Ithaca, NY). Seventy-two hours after infection, cells were harvested and ERα, which was fused at the N-terminus to a Flag tag, was immunopurified and eluted from M2 antibody resin (Sigma, St. Louis, MO) with elution buffer (20 mM Tris, pH 7.5, 400 mM KCl, 0.2 mM EDTA, 10% glycerol, 0.1% NP-40, 2 mM DTT, 0.5 mg/mL ovalbumin, and 0.2 mg/mL flag peptide). ERα concentrations were determined as previously described (Wood et al., 2001).
2.5. DNase I footprinting analysis
Primer P4 (5′-TCC TGA GTG TGA GTG TTG-3′) or P5 (5′-GTA AAA CGA CGG CCA GTG-3′, which was specific for vector sequence) was 32P-labeled and used with primer P3 (5′-GGT ATT ATC TTA GCA TGA TGT-3′) in 35 rounds of PCR amplification with the vector pTZ18R-BARD. The 204 bp (P3 and P4) or 304 bp (P3 and P5) singly end-labeled amplified fragments were purified using a Quick Spin Column (Roche, Indianapolis, IN). End-labeled, amplified DNA (100,000 cpm) was combined with increasing amounts of baculovirus-expressed, purified ERα in binding buffer (60 mM KCl, 15 mM Tris, pH 7.9, 0.2 mM EDTA, 10% glycerol, 4 mM DTT, 100 ng poly-deoxyinosine/deoxycytosine, and 10 nM E2) with 12.5 mM MgCl2 and 0.5 mM CaCl2 in a total volume of 20 μL. After 10 min at room temperature, 0.4 U of RQ1 RNase free DNase was added and the reaction was incubated for 1.5 min at room temperature. DNase digestion was terminated by adding stop solution (200 mM NaCl, 1% SDS, 30 mM EDTA, and 100 ng/μL tRNA). The DNA was extracted with phenol/chloroform, ethanol precipitated, and resuspended in formamide loading buffer. Fragments were resolved on 8% denaturing acrylamide gels and bands were visualized by autoradiography.
2.6. Gel mobility shift assays
Gel mobility shift assays were carried out essentially as previously described (Loven et al., 2001) using three 50 bp oligos with sequences corresponding to three regions of the BARD1 intron nine region: 5′-TAT CTT AGC ATG ATG TGA ATA TGT CAA ATG CTT AAA AGC TGC TTA AAT TG-3′ and its complementary strand, which contain the first ERE half site (HS1), 5′-GAT GTT GTT TAT TAT AAA AAG GTC AGA AGA CTA ACA ACT GAA TAC TCC AT-3′ and its complementary strand, which contain the second ERE half site (HS2), and 5′-GCA GAG GTT TGT CAA GAC GCT GAG TCT TTC CTG AAT TGG GTC ATT AAT GC-3′ and its complementary strand, which contain ERE half sites three and four (HS3 and HS4). 32P labeled oligos (10,000 cpm) were combined with 50 fmol of purified ERα in binding buffer in a final volume of 20 μL and incubated at room temperature for 10 min. BSA was included to maintain a constant total protein concentration of 20 μg. The ERα-specific monoclonal antibody BZ1, 100-fold excess of unlabeled A2 ERE-containing competitor, or 100-fold excess of nonspecific competitor DNA was added to the binding reactions as indicated before addition of labeled oligos. Complexes were fractioned on non-denaturing acrylamide gels using low ionic strength buffer and protein–DNA complexes were visualized with audioradiography.
2.7. Reporter vectors for transient transfections
The Sst/Nco1 fragment from pTZ18R BARD containing the BARD1 gene region was isolated and inserted into Sst1/Nco1-cut TransLucent Luciferase Reporter Vector (Panomics, Redwood City, CA), which contains a TATA sequence. Mutations of BARD 1 ERE half sites were generated using the Quick Change II Site-Directed Mutagensis Kit (Stratagene, La Jolla, CA) with primer sets specific for ERE1 (5′-CAT GAT GTG AAT AAA TCA AAT GCT TAA AAG CTG C-3′ and 5′-GCA GCT TTT AAG CAT TTG ATT TAT TCA CAT CAT GC-3′), ERE2 (5′-GTT TAT TAT AAA AAA ATC AGA AGA CTA ACA ACT GA-3′ and 5′-TCA GTT GTT AGT CTT CTG ATT TTT TTA TAA TAA AC-3′), ERE4 (5′-GAG TCT TTC CTG AAT TGA ATC ATT AAT GCA AAT TC-3′ and 5′-GAA TTT GCA TTA ATG ATT CAA TTC AGG AAA GAC TC-3′), or in combination to produce the reporter plasmids mHS1, mHS2, mHS4, or mHS 1,2,4, respectively. The luciferase coding sequence from TransLucent Luciferase Reporter Vector was removed and replaced with Renilla coding sequence to produce pTLC Renilla, which was used as an internal control.
2.8. Transient transfection assays
MDA-MB231 breast cancer cells were maintained in Leibovitz's L-15 medium (Invitrogen, Carlsbad, CA) supplemented with 10% fetal calf serum. Forty-eight hours before transfection cells were transferred to phenol red-free improved MEM supplemented with 5% charcoal dextran-treated calf serum. Cells were transfected with the human ERα expression vector CMV-hERα, 1 μg of a BARD1 reporter vector, and 5 ng of the renilla reporter plasmid TK Renilla (Fig. 5A) or pTLC Renilla (Fig. 5B) in serum-free, phenol red free improved MEM using Lipofectin (Invitrogen, Carlsbad, CA) as described previously (Loven et al., 2001). After 6 h, DNA and medium was removed and cells were transferred to phenol red-free improved MEM supplemented with 5% charcoal dextran-treated calf serum and ethanol vehicle or 10 nM E2 for 24 h. Cells were harvested after 24 h and luciferase assays were carried out using the Dual-Luciferase Reporter Assay System (Promega, Madison, WI) according to the manufacturer's instructions. Renilla activity was used to normalize for transfection efficiency. Four independent experiments were carried out in duplicate. Data were normalized to compensate for variation between experiments and the Student's t-test was utilized to determine whether there was a statistical difference between the E2- and ethanol-treated cells.
Fig. 5.
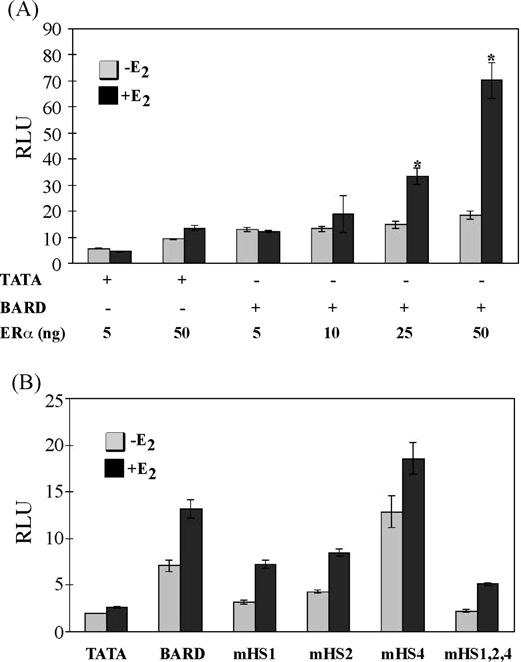
Intron nine of the BARD1 gene confers estrogen responsiveness. MDAMB231 breast cancer cells were transfected with an ERα expression vector (ERα), a renilla control vector, and a reporter vector with a TATA sequence alone (TATA) or a TATA sequence combined with the 288 bp region of the BARD1 gene containing wild type DNA sequence (BARD) or one (mHS1, mHS2, mHS4) or three (mHS1,2,4) mutant ERE half sites as indicated in Panels A and B. Cells were treated with ethanol vehicle (−E2) or 10nM E2 (+E2) for 24 h. Data from four independent experiments were combined and are expressed as the mean in relative luciferase units (RLUs)±S.E.M. for all samples. Note that some error bars are too small to visualize. Statistical differences were determined using the Student's t-test as indicated by an asterisk in Panel A. Significant differences were determined by ANOVA in Panel B.
3. Results
In an effort to identify novel estrogen-responsive genes regulated by ERα in human breast cancer cells, we developed a ChIP-based cloning strategy. An ERα-specific antibody was used to immunoprecipitate formaldehyde-crosslinked, sheared chromatin from MCF-7 cells and the immunoprecipitated DNA was purified, subcloned, and sequenced. From this screening process, 36 sublones with an average length of ∼250 bp were identified. All 36 of the DNA regions were used in BLAST searches and 32 aligned to unique regions of the human genome. Using the Ensembl database (www.ensembl.org), we found 21 subclones that mapped to previously identified genes.
To verify that ERα was associated with the gene regions we had identified, ChIP analysis was carried out using an ERα-specific antibody and MCF-7 cells that had been treated with vehicle, E2 for 45 min or E2 for 24 h. Quantitative real time PCR analysis was utilized to amplify 15 of the 21 mapped regions. These assays revealed that although more ERα was associated with the BARD1, C14orf159, LTBP1, PABPC1, PPIH, and PRG1 genes in the presence of E2 at 45 min and 24 h than in the absence of E2 (Table 1, +++), increased association of ERα with the TLR5 and TNS3 genes was observed only at 45 min (++). Three genes, C3orf30, RALGPS1, and TANC2, were associated with ERα in both the absence and presence of E2 (+). ERα was not associated with the DOCK7, MLL/HRX, NPC1L1 or RPS6KA5 genes (−). Interestingly, intron 13 of TNS, intron 3 of LTBP1, and regions upstream of PGR1 were recently identified as ERα-associated gene regions in MCF-7 cells by other investigators who had used different times of hormone exposure (Carroll et al., 2005, 2006; Vega et al., 2006).
Table 1.
Gene regions identified by ChIP cloning
| Gene | Accession no. | Chromosome location | Gene location | ERα binding |
|---|---|---|---|---|
| BARD1 | NM_000465 | Chr. 2: 215,425,902–215,426,189 | Intron 9 | +++ |
| C14orf159 | NM_024952 | Chr. 14: 90,724,568–90,724,769 | Intron 10 | +++ |
| C3orf30 | NM_152539 | Chr. 3: 120,348,117–120,348,506 | Exon 1 | + |
| DOCK7 | NM_033407 | Chr. 1: 62,749,994–62,750,102 | Intron 23 | − |
| LTBP1 | NM_000627 | Chr. 2: 33,182,204–33,182,331 | Intron 3 | +++ |
| MLL/HRX | NM_005933 | Chr. 11: 117,828,720–117,829,230 | Intron 1 | − |
| NPC1L1 | NM_013389 | Chr. 7: 44,525,391–44,525,466 | Intron 14 | − |
| PABPC1 | NM_002568 | Chr. 8: 101,784,320–101,804,115 | Upstream | +++ |
| PPIH | NM_006347 | Chr. 1: 42,727,227–42,727,476 | Upstream | +++ |
| PRG1 | NM_000926 | Chr. 11: 100,718,397–100,718,656 | Upstream | +++ |
| RALGPS1 | NM_014636 | Chr. 9: 128,702,059–128,702,358 | Intron 4 | + |
| RPS6KA5 | NM_004755 | Chr. 14: 90,445,212–90,445,591 | Intron 7 | − |
| TANC2 | NM_371071 | Chr. 17: 58,750,076–58,853,443 | Intron 7 | + |
| TLR5 | NM_003268 | Chr. 1: 221,350,878–221,351,087 | Exon 4 | ++ |
| TNS3 | NM_022748 | Chr. 7: 47,216,239–47,216,539 | Intron 13 | ++ |
Symbols indicate a significant increase in the association of ERα in the presence of E2 at 45 min and at 24 h; (+++) compared to the corresponding ethanol control, a significant increase in the association of ERα in the presence of E2 at 45 min only (++) compared to the corresponding ethanol control, similar association of ERα in the absence and in the presence of E2 (+), or no association of ERα with the gene region in the absence or in the presence of E2 (−).
3.1. ERα associates with intron nine of the BARD1 gene
One of the gene regions isolated from our genome-wide screen was a 288 bp region from the ninth intron of the BARD1 gene from +73,683 to +73,971 relative to the transcription start site (Fig. 1A and B). Given the association of BARD1 with BRCA1 and the involvement of the BARD1/BRCA1 heterodimer in DNA repair (Wu et al., 1996; Westermark et al., 2003), this was a gene of substantial interest and was characterized in more detail using a variety of molecular approaches. Our quantitative ChIP analysis, which included 3 independent experiments, revealed a two-fold increase in the association of ERα with the ninth intron of the BARD1 gene in the presence of E2 (Table 1 and data not shown). Comparison with the pS2 and 36B4 genes suggested that ERα associated with the ninth intron of the BARD 1 gene in the absence and in the presence of E2 (data not shown).
Fig. 1.
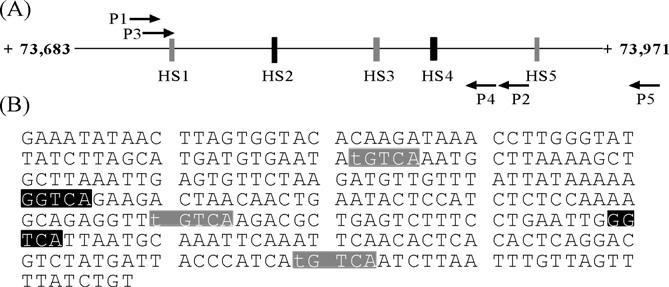
ERα associates with a 288 bp region from the BARD1 gene within intron 9. Schematic drawing (A) and DNA sequence (B) of the 288 bp region of the BARD1 ninth intron isolated from ERα-immunoprecipitated chromatin. The numbering (+73,683 to +73,971) indicates the distance of the BARD1 gene region from its transcription start site. Black boxes indicate consensus ERE half sites (HS2 and HS4) and gray boxes indicate imperfect ERE half sites (HS1, HS3, and HS5). Arrows show the positions of primers used to produce DNA fragments for ChIP analysis (P1 and P2) and DNAse I footprinting (P3–P5) Bases differing from the consensus ERE half site are in lower case.
3.2. BARD1 mRNA and protein levels increase in E2-treated MCF-7 cells
To determine whether E2 influenced BARD1 gene expression, MCF-7 cells were treated with E2, RNA was isolated, cDNA was synthesized, and quantitative real time PCR was utilized to determine the levels of BARD1 and 36B4 mRNA present. Although 36B4 mRNA levels were unaffected when MCF-7 cells were treated with E2 (data not shown), BARD1 mRNA levels were significantly increased 1.3-, 1.15-, 2.05-, and 2.17-fold (p ≤ 0.05) after MCF-7 cells had been treated with E2 for 0.3, 2, 24, and 48 h, respectively (Fig. 2A).
Fig. 2.
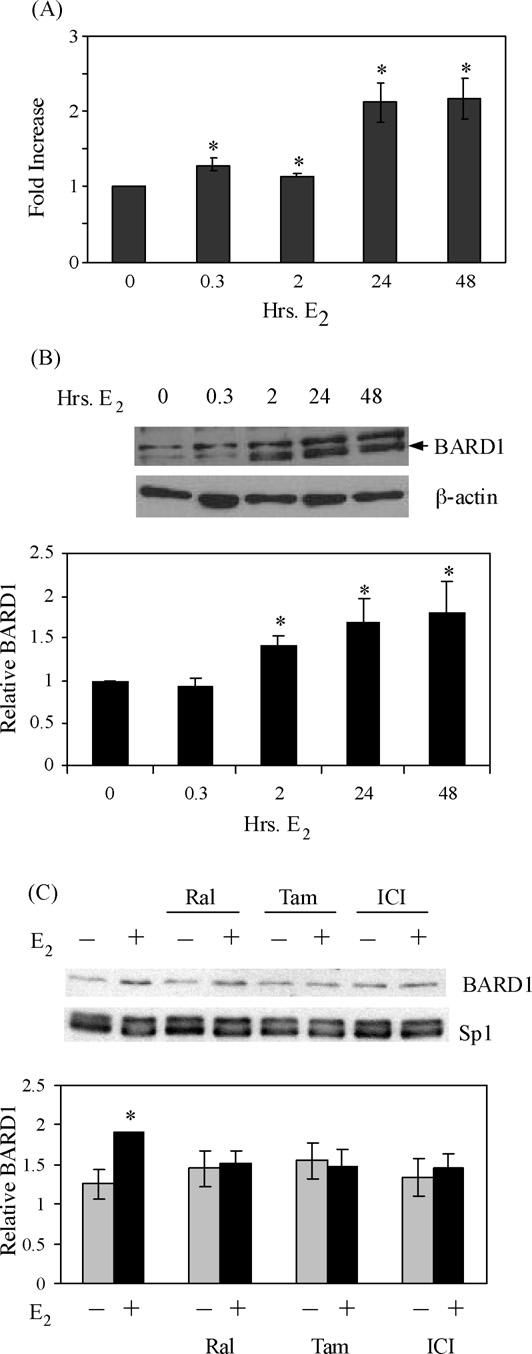
BARD1 mRNA and protein levels increase when MCF-7 cells are exposed to E2. MCF-7 cells were treated with ethanol vehicle, 10 nM E2 or 100 nM raloxifene (Ral), tamoxifen (Tam), or ICI 182,780 (ICI) as indicated. (A) RNA was isolated and utilized in quantitative real time PCR experiments to determine the levels of BARD1 and 36B4 mRNA. Values were calculated using the ΔΔCt method, which was normalized to 36B4 mRNA levels. Data from three independent experiments were combined and are expressed as the mean-fold increase in BARD1 mRNA levels ± S.E.M. Student's t-tests revealed that samples from MCF-7 cells that had been treated with E2 were statistically different (indicated by an asterisk) from cells that had been treated with ethanol (p ≤ 0.05). (B and C) Whole cell extracts from MCF-7cells that had been treated as indicated were subjected to Western analysis with BARD1 antibody or control β-actin- or Sp1-specific antibody. Each blot shown is representative of three independent experiments. Combined, normalized data is shown graphically as the relative BARD1 protein level (mean ± S.E.M.). Asterisks indicate significant increases in BARD1 protein levels compared to ethanol control (p ≤ 0.05). Error bars are sometimes too small to be visualized.
Our quantitative real time PCR assays provided evidence that BARD1 mRNA levels increased in MCF-7 cells upon E2 treatment. However, increased mRNA levels do not always reflect the level of protein present. Therefore, MCF-7 cells were treated with E2 for 0, 0.3, 2, 24, and 48 h, whole cell extracts were prepared, and Western blot analysis was performed. These experiments revealed that the expression of BARD1, but not β-actin, increased upon treatment of MCF-7 cells with E2 (Fig. 2B). Furthermore, quantitative analysis demonstrated that while neither tamoxifen, raloxifene, nor ICI 182,780 alone altered BARD1 expression, each of these compounds significantly reduced the E2-mediated increase in BARD1 levels (Fig. 2C). These findings demonstrate that E2 regulates expression of the endogenous BARD1 gene in MCF-7 cells.
3.3. ERα binds to three ERE half sites in intron nine of the BARD1 gene
The E2-induced expression of BARD1 mRNA and protein levels in MCF-7 cells combined with our ChIP analysis, which demonstrated that ERα interacts with intron nine of the BARD1 gene, suggested that there must be one or more cis elements responsible for recruiting the receptor to this region of the BARD1 gene. Inspection of the 288 bp BARD1 gene region revealed that it contained three imperfect ERE half sites (HS1, HS3, and HS5, Fig. 1A and B, gray boxes) with identical nucleotide sequence (TGTCA) and two consensus ERE half sites (HS2 and HS4, GGTCA, Fig. 1A and B, black boxes). Thus, it seemed possible that ERα might interact with one or more of these five ERE half sites to regulate BARD1 gene expression.
To identify the cis element(s) in the BARD1 intron nine gene region that interact with ERα, DNase I footprinting was performed. 32P-labeled DNA fragments containing a 204 bp portion of the BARD1 intron nine gene region were incubated with increasing amounts of purified ERα and exposed to DNase I. In the presence of ERα, HS1 and HS2 were protected from DNase I cleavage (Fig. 3A) and were flanked by a hypersensitive site at their 3′-ends. In contrast, the imperfect ERE half site HS3 was not protected. A larger 304 bp region of the BARD1 intron nine gene region was also radiolabeled and utilized in DNase I footprinting experiments so that HS4 and HS5 could be visualized. We again observed protection of the consensus ERE half site HS2 (HS2, Fig. 3B) and an adjacent DNase I hypersensitive site. The other consensus ERE half site, HS4, was also protected from DNase I cleavage and was flanked by a hypersensitive site, but neither of the imperfect ERE half sites (HS3 and HS5) was protected. 32P-labeled DNA, which had been chemically cleaved at each G residue, was also included for reference (Fig. 3A and B, lanes 1). From these experiments we learned that only one of the three imperfect ERE half sites (HS1), but both of the consensus ERE half sites (HS2 and HS4) were protected from DNase I cleavage and that each protected ERE half site was flanked by a hypersensitive site.
Fig. 3.
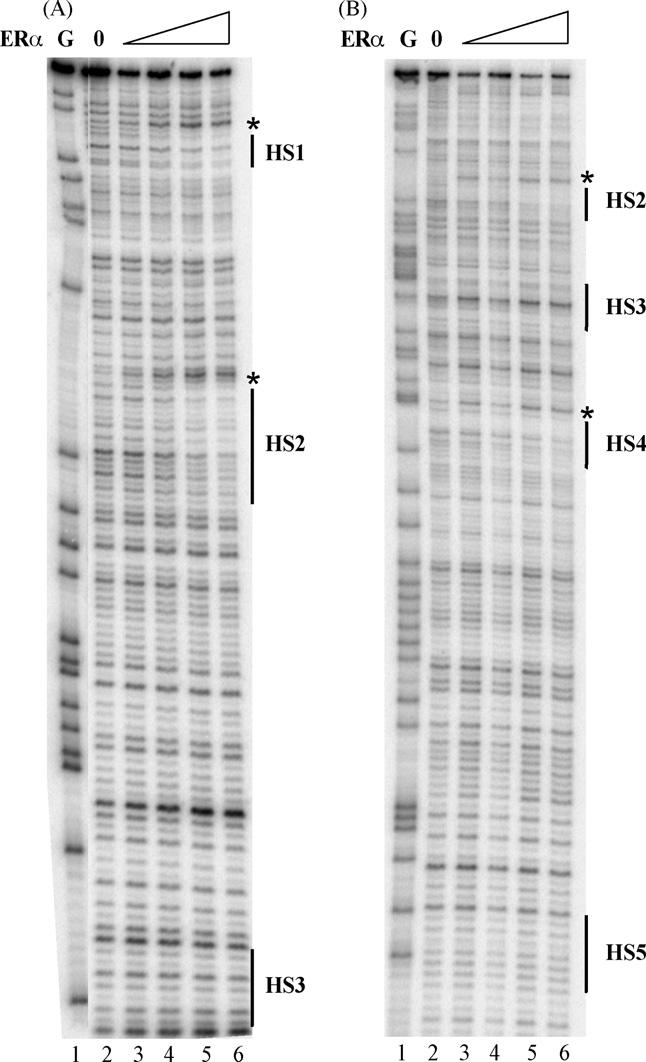
ERα limits DNase I cleavage at three ERE half sites in the ninth intron of the BARD1 gene. Noncoding strand primers were 32P-labeled and used to PCR amplify a 204 bp (A) or 304 bp (B) region of DNA. Labeled DNA was incubated with 0, 25, 100, 250, or 500 fmol of purified, baculovirus-expressed ERα (lanes 2–6) and exposed to DNase I. DNA that had been cleaved at each G residue was included for reference (lanes 1). Hypersensitive sites are indicated by an asterisk.
To confirm that HS1, HS2, and HS4 interacted with ERα, gel mobility shift assays were carried out with ERα and 32P-labeled DNA containing three different 50 bp portions of the BARD1 ninth intron region. Each 50 bp oligo contained one (HS1 or HS2) or two (HS3 and HS4) of the ERE half sites (Fig. 4). When each of the radiolabeled 50 bp probes was combined with purified ERα, one major ERα–ERE complex was observed (lanes 2, 7, and 12, →). The specificity of the receptor–DNA interactions was demonstrated by the altered migration of the receptor–DNA complexes in the presence of an ERα-specific antibody (lanes 3, 8, and 13) and the ability of oligos containing a consensus ERE (lanes 4, 9, and 14), but not a nonspecific DNA sequence (lanes 5, 10, and 15), to compete for ERα binding. A faster migrating, nonspecific band, which was unaffected by the presence of ERα-specific antibody, was sometimes observed. Interestingly, the migration of the receptor–DNA complexes was identical in spite of the fact that one of the probes contained two ERE half sites (HS3 and HS4). These findings are consistent with our DNase I footprinting experiments, which demonstrated that ERα binds to HS4, but not to HS3. Additional gel mobility shift experiments demonstrated that oligos containing HS5 failed to bind to ERα (data not shown).
Fig. 4.
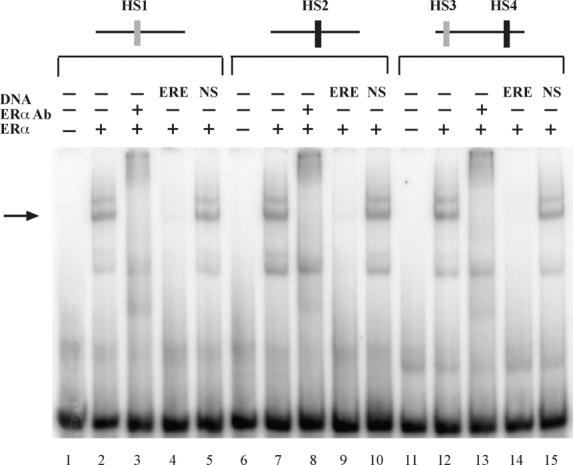
ERα binds specifically to ERE half sites in the ninth intron of the BARD1 gene. Gel mobility shift assays were carried out with 32P-labeled oligos containing 50 bp regions of intron 9 of the BARD1 gene. BARD1 regions containing one or two ERE half sites are represented schematically above the gel. 32P-labeled oligos were incubated with 50 fmol of purified, baculovirus-expressed ERα (lanes 2, 7, and 12). Antibody to ERα (lanes 3, 8, and 13), unlabeled oligos containing the A2 ERE (ERE, lanes 4, 9, and 14), or unlabeled oligos containing nonspecific (NS) DNA sequence (lanes 5, 10, and 15) were included as indicated. Reactions were fractionated on a nondenaturing gel and visualized by audioradiography.
3.4. The 288 bp region of the BARD1 gene ninth intron confers estrogen responsiveness
The association of ERα with intron nine of the endogenous BARD1 gene, the increase in BARD1 mRNA and protein levels in E2-treated MCF-7 cells, and the protection of ERE half sites in the ninth intron of the BARD1 gene suggested that the interaction of ERα with this 288 bp region of the BARD1 gene might modulate estrogen-responsive gene expression. Thus, transient transfections were performed in MDA-MB231 breast cancer cells, which do not express ERα. Transcription of a reporter plasmid containing only a TATA box was not significantly affected by expression of ERα or E2 (Fig. 5A and data not shown). However, when the 288 bp region of the BARD1 ninth intron was present in the reporter plasmid, transcription was significantly enhanced in the presence of E2 when 25 or 50 ng of the ERα expression vector was utilized (Fig. 5A). No changes in transcription were observed when the ERα expression vector was omitted (data not shown).
Since ERα bound to HS1, HS2, and HS4 in gel mobility shift and DNase I footprinting assays (Figs. 3 and 4), each of these ERE half sites was mutated to AATCA individually and in combination in the context of the 288 bp BARD1 gene region. Transient transfections were carried out with reporter plasmids containing the BARD1 gene with wild type or mutant ERE half sites. Inclusion of the BARD1 fragment significantly increased transcription in the absence and in the presence of E2 compared to the parent vector (Fig. 5B, p ≤ 0.05), suggesting that even in the absence of hormone ERα interacted with the BARD1 DNA sequence and enhanced transcription. Mutation of HS1 or HS2 significantly reduced the magnitude of transcription in the absence and in the presence of E2 (p ≤ 0.01) when compared to the wild type BARD1 sequence. Furthermore, in the absence of hormone, mutation of HS1, but not HS2, reduced transcription to the level observed with the parent vector thereby providing evidence that HS1 is involved in sustaining basal BARD1 expression. Taken together, these findings support the notion that ERα is required for basal as well as E2-induced transcription and are in agreement with our ChIP results, which demonstrated that the receptor was associated with the BARD1 gene in the absence and in the presence of E2. Paradoxically, when HS4 was mutated, transcription was significantly increased (p ≤ 0.05) in the absence and in the presence of E2 indicating that, unlike HS1 and HS2, the role of HS4 is to limit estrogen responsiveness. Combined mutations in HS1, HS2, and HS4 more effectively reduced transactivation than mutation of HS1 or HS2 alone. These experiments suggest that although HS1, HS2, and HS4 play a role in estrogen responsiveness of the BARD1 gene, these individual ERE half sites can have opposing effects on transcription. The dual effects of these cis elements on transcription are consistent with previous studies of the progesterone receptor gene, where mutation of the ERα-associated +90 AP-1 site dramatically decreased transcription in the context of a 1.5 kb region of the progesterone receptor gene, but mutation of the ERα-associated +745 AP-1 site greatly enhanced transcription (Petz et al., 2002, 2004a,b).
4. Discussion
Using a ChIP-based cloning strategy, we isolated a 288 bp region of the human BARD1 gene that was associated with ERα in E2-treated MCF-7 breast cancer cells. Conventional ChIP assays demonstrated that ERα was associated with the ninth intron of the endogenous BARD1 gene in MCF-7 cells, gel mobility shift and DNase I footprinting assays showed that ERα bound to three ERE half sites in this region, and transient transfection assays revealed that the 288 bp region of the BARD1 gene isolated in our modified ChIP assay conferred estrogen responsiveness to a heterologous promoter in human breast cancer cells. Overall these findings suggest that this 288 bp region of the BARD1 gene ninth intron is involved in regulating BARD1 levels in MCF-7 cells and confirm the usefulness of our ChIP-based cloning strategy in isolating estrogen-responsive genes, identifying cis elements involved in mediating hormonal control, and providing information about the location of these regulatory elements within the genome. Although we have examined the ability of a discrete 288 bp region of the ninth intron of the BARD1 gene to regulate BARD1 gene expression, it is possible that other gene regions may also be involved in responding to E2.
4.1. Regulation of estrogen-responsive genes in MCF-7 cells
One of our goals in these investigations was to determine whether it would be possible to utilize our ChIP-based cloning strategy to identify novel gene regions associated with ERα. We felt that there were distinct advantages to this procedure over other established methods. First, we were able to perform a truly unbiased, genome-wide screen that included large regions of previously unexplored DNA sequence. Second, since the native chromatin environment had been preserved, we had confidence that the gene regions we isolated were associated with ERα, functional, and biologically relevant. Third, there was no preconceived notion about where a cis element would be located or what its DNA sequence might be.
Using our ChIP-based cloning strategy, we identified gene regions that were 100–200 kb upstream and 80–140 kb downstream of the genes' transcription start sites. Although this was somewhat unexpected, two groups recently reported that the majority of gene regions associated with ERα were at locations far removed from the transcription start site (Carroll et al., 2005, 2006; Vega et al., 2006). Thus, although the overwhelming majority of studies aimed at identifying DNA sequences regulating gene expression to date have focused on 1–2 kb proximal promoter regions, our studies and others suggest that simply screening these proximal promoters is not sufficient for identification of gene regions involved in regulating expression of endogenous estrogen-responsive genes. This also appears to be the case with a number of other nuclear receptors including the thyroid hormone receptor (Plateroti et al., 2006), androgen receptor (Heemers et al., 2004), peroxisome proliferative activated receptor γ (Neess et al., 2006), and retinoic acid receptor (Donato and Noy, 2005).
4.2. Regulation of the BARD1 gene in MCF-7 cells
Inspection of the ninth intron of the BARD1 gene revealed the presence of five ERE half sites, but ERα bound to only three of these sites. While the association of ERα with the two consensus ERE half sites (GGTCA) might have been anticipated from earlier work (Edinger et al., 1997; Martini et al., 2000; Petz and Nardulli, 2000; Petz et al., 2004a,b), the association of the receptor with only one of the three imperfect EREs, each of which had identical DNA sequence (TGTCA), was surprising. Upon closer inspection, we detected a DNA sequence 3 bp upstream of HS1 that differs from a consensus ERE half site (GGTCA) by 2 bp (TGTGA). It seems possible that this DNA sequence could provide an additional imperfect ERE half site for ERα monomer docking, facilitate ERα dimer formation, and help to explain why ERα bound to HS1, but not HS3 or HS5. It should be noted, however, that this DNA sequence upstream of HS1 was not protected in our DNase I footprinting assays.
The association of ERα with the BARD1 gene in the absence and in the presence of E2 was somewhat unexpected. It seems possible that ERα association with intron nine of the BARD1 gene may be needed to sustain lower level, basal expression of BARD1 in MCF-7 cells in the absence of hormone. Then when MCF-7 cells have been exposed to hormone, increased binding of ERα to this region of the BARD1 gene could help to enhance BARD1 mRNA and protein expression. It is important to note that the increased association of ERα with the ninth intron of the BARD1 gene in the presence of E2 increased BARD1 mRNA levels, BARD1 protein levels, and transcription of a reporter plasmid containing the intron nine BARD1 gene region.
4.3. Role of BARD1 in determining cell fate
BARD1 was originally isolated as a protein that interacted with the ring finger domain of BRCA1 to form a BARD1/BRCA1 heterodimer (Wu et al., 1996). The BARD1/BRCA1 heterodimer functions as an ubiquitin ligase and is essential, but not sufficient, for homology-directed DNA repair (Westermark et al., 2003) and also plays a role in double-strand DNA break repair (Liu and West, 2002). Upon DNA damage, the BARD1/BRCA1 heterodimer slows transcription and RNA processing and promotes cell cycle arrest so that DNA repair can take place (Fabbro et al., 2004; Kleiman et al., 2005; Schuchner et al., 2005; Starita et al., 2005). When BRCA1 levels are low, BARD1 accumulates in the cytoplasm where it plays a role in initiating programmed cell death (Irminger-Finger et al., 2001; Jefford et al., 2004; Rodriguez et al., 2004). Thus the relative expression of BRCA1 and BARD1 is critical in determining cell fate. The ability of the BARD1/BRCA1 heterodimer to set a cell on a course toward DNA repair and the capacity of BARD1 to initiate apoptosis has led to the idea that BARD1 serves as a guardian of the genome (Irminger-Finger et al., 2001).
By regulating the level of BARD1, ERα must also play a role in helping to determine cell fate. Entering into this equation, however, is the fact that BRCA1 levels are also increased by E2 (Gudas et al., 1995). Thus, the simultaneous induction of BARD1 and BRCA1 by E2 would favor DNA repair over apoptosis. Other regulatory mechanisms would be needed to decrease BRCA1 or increase BARD1 levels so that BARD1-mediated apoptosis could occur. Interestingly, it has also been suggested that the fluctuating levels of BARD1 and BRCA1 in the mouse uterus during the estrus cycle play a role in uterine remodeling (Irminger-Finger et al., 2001).
We chose to examine the role of ERα in regulating BARD1 gene expression because of the importance of BARD1 in mammary cell biology. Previous studies have shown that knocking down BARD1 gene expression in mammary cells results in altered cell morphology, multinucleated cells, and aberrant cell cycle progression (Irminger-Finger et al., 1998). Furthermore, mutations in the BARD1 gene have been associated with an increased risk of breast cancer (Thai et al., 1998; Ghimenti et al., 2002).
It has been clear for some time that E2 is required for mammary cell development. However, the E2-induced regulation of BARD1 gene expression provides a mechanism through which the cell fate of mammary cells might be influenced. While overproduction of BARD1 would lead to apoptosis of mammary cells, the combined effects of BARD1 and BRCA1 would promote DNA repair and an extension of mammary cell lineage.
Acknowledgement
These studies were supported by NIH Grant DK 0601463 (to AMN).
References
- Bourdeau V, Deschenes J, Metivier R, Nagai Y, Nguyen D, Bretschneider N, Gannon F, White JH, Mader S. Genome-wide identification of high-affinity estrogen response elements in human and mouse. Mol. Endocrinol. 2004;18:1411–1427. doi: 10.1210/me.2003-0441. [DOI] [PubMed] [Google Scholar]
- Carroll J, Meyer C, Song J, Li W, Geistlinger T, Eeckhoute J, Brodshy A, Keeton E, Fertuck K, Hall G, Wang Q, Bekiranov S, Sementchenko V, Fox E, Silver P, Gingeras T, Liu X, Brown M. Genome-wide analysis of estrogen receptor binding sites. Nat. Genet. 2006;38:1289–1297. doi: 10.1038/ng1901. [DOI] [PubMed] [Google Scholar]
- Carroll JS, Liu XS, Brodsky AS, Li W, Meyer CA, Szary AJ, Eeckhoute J, Shao W, Hestermann EV, Geistlinger TR, Fox EA, Silver PA, Brown M. Chromosome-wide mapping of estrogen receptor binding reveals long-range regulation requiring the forkhead protein FoxA1. Cell. 2005;122:33–43. doi: 10.1016/j.cell.2005.05.008. [DOI] [PubMed] [Google Scholar]
- Colditz GA. Relationship between estrogen levels, use of hormone replacement therapy, and breast cancer. J. Natl. Cancer Inst. 1998;90:814–823. doi: 10.1093/jnci/90.11.814. [DOI] [PubMed] [Google Scholar]
- Couse JF, Korach KS. Estrogen receptor null mice: what have we learned and where will they lead us? Endocr. Rev. 1999;20:358–417. doi: 10.1210/edrv.20.3.0370. [DOI] [PubMed] [Google Scholar]
- Donato LJ, Noy N. Suppression of mammary carcinoma growth by retinoic acid: proapoptotic genes are targets for retinoic acid receptor and cellular retinoic acid-binding protein II signaling. Cancer Res. 2005;65:8193–8199. doi: 10.1158/0008-5472.CAN-05-1177. [DOI] [PubMed] [Google Scholar]
- Dubal DB, Wilson ME, Wise PM. Estradiol: a protective and trophic factor in the brain. J. Alzheimers Dis. 1999;1:265–274. doi: 10.3233/jad-1999-14-507. [DOI] [PubMed] [Google Scholar]
- Edinger RS, Mambo E, Evans MI. Estrogen-dependent transcriptional activation and vitellogenin gene memory. Mol. Endocrinol. 1997;11:1985–1993. doi: 10.1210/mend.11.13.0037. [DOI] [PubMed] [Google Scholar]
- Fabbro M, Schuechner S, Au WW, Henderson BR. BARD1 regulates BRCA1 apoptotic function by a mechanism involving nuclear retention. Exp. Cell. Res. 2004;298:661–673. doi: 10.1016/j.yexcr.2004.05.004. [DOI] [PubMed] [Google Scholar]
- Frasor J, Danes JM, Komm B, Chang KC, Lyttle CR, Katzenellenbogen BS. Profiling of estrogen up- and down-regulated gene expression in human breast cancer cells: insights into gene networks and pathways underlying estrogenic control of proliferation and cell phenotype. Endocrinology. 2003;144:4562–4574. doi: 10.1210/en.2003-0567. [DOI] [PubMed] [Google Scholar]
- Ghimenti C, Sensi E, Presciuttini S, Brunetti IM, Conte P, Bevilacqua G, Caligo MA. Germline mutations of the BRCA1-associated ring domain (BARD1) gene in breast and breast/ovarian families negative for BRCA1 and BRCA2 alterations. Genes Chromos. Cancer. 2002;33:235–242. doi: 10.1002/gcc.1223. [DOI] [PubMed] [Google Scholar]
- Gudas JM, Nguyen H, Li T, Cowan KH. Hormone-dependent regulation of BRCA1 in human breast cancer cells. Cancer Res. 1995;55:4561–4565. [PubMed] [Google Scholar]
- Harris JR, Lippman ME, Veronesi U, Willett W. Breast cancer (1) N. Engl. J. Med. 1992;327:319–328. doi: 10.1056/NEJM199207303270505. [DOI] [PubMed] [Google Scholar]
- Heemers H, Verrijdt G, Organe S, Claessens F, Heyns W, Verhoeven G, Swinnen JV. Identification of an androgen response element in intron 8 of the sterol regulatory element-binding protein cleavage-activating protein gene allowing direct regulation by the androgen receptor. J. Biol. Chem. 2004;279:30880–30887. doi: 10.1074/jbc.M401615200. [DOI] [PubMed] [Google Scholar]
- Hess RA, Bunick D, Lee K-H, Bahr J, Taylor JA, Korach KS, Lubahn DB. A role for oestrogens in the male reproductive system. Nature. 1997;390:509–512. doi: 10.1038/37352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hewitt SC, Deroo BJ, Hansen K, Collins J, Grissom S, Afshari CA, Korach KS. Estrogen receptor-dependent genomic responses in the uterus mirror the biphasic physiological response to estrogen. Mol. Endocrinol. 2003;17:2070–2083. doi: 10.1210/me.2003-0146. [DOI] [PubMed] [Google Scholar]
- Irminger-Finger I, Leung WC, Li J, Dubois-Dauphin M, Harb J, Feki A, Jefford CE, Soriano JV, Jaconi M, Montesano R, Krause KH. Identification of BARD1 as mediator between proapoptotic stress and p53-dependent apoptosis. Mol. Cell. 2001;8:1255–1266. doi: 10.1016/s1097-2765(01)00406-3. [DOI] [PubMed] [Google Scholar]
- Irminger-Finger I, Soriano JV, Vaudan G, Montesano R, Sappino AP. In vitro repression of Brca1-associated RING domain gene, BARD1, induces phenotypic changes in mammary epithelial cells. J. Cell. Biol. 1998;143:1329–1339. doi: 10.1083/jcb.143.5.1329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jefford CE, Feki A, Harb J, Krause KH, Irminger-Finger I. Nuclear-cytoplasmic translocation of BARD1 is linked to its apoptotic activity. Oncogene. 2004;23:3509–3520. doi: 10.1038/sj.onc.1207427. [DOI] [PubMed] [Google Scholar]
- Karsai A, Muller S, Platz S, Hauser MT. Evaluation of a homemade SYBR green I reaction mixture for real-time PCR quantification of gene expression. Biotechniques. 2002;32:790–792. doi: 10.2144/02324st05. 794–796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katzenellenbogen BS, Kendra KL, Norman MJ, Berthois Y. Proliferation, hormonal responsiveness, and estrogen receptor content of MCF-7 human breast cancer cells grown in the short-term and long-term absence of estrogens. Cancer Res. 1987;47:4355–4360. [PubMed] [Google Scholar]
- Kleiman FE, Wu-Baer F, Fonseca D, Kaneko S, Baer R, Manley JL. BRCA1/BARD1 inhibition of mRNA 3′ processing involves targeted degradation of RNA polymerase II. Genes Dev. 2005;19:1227–1237. doi: 10.1101/gad.1309505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein-Hitpass L, Ryffel GU, Heitlinger E, Cato ACB. A 13 bp palindrome is a functional estrogen responsive element and interacts specifically with estrogen receptor. Nucleic Acids Res. 1988;16:647–663. doi: 10.1093/nar/16.2.647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klinge CM. Estrogen receptor interaction with estrogen response elements. Nucleic Acids Res. 2001;29:2905–2919. doi: 10.1093/nar/29.14.2905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kushner PJ, Agard DA, Greene GL, Scanlan TS, Shiau AK, Uht RM, Webb P. Estrogen receptor pathways to AP-1. J. Steroid Biochem. Mol. Biol. 2000;74:311–317. doi: 10.1016/s0960-0760(00)00108-4. [DOI] [PubMed] [Google Scholar]
- Liu Y, West SC. Distinct functions of BRCA1 and BRCA2 in double-strand break repair. Breast Cancer Res. 2002;4:9–13. doi: 10.1186/bcr417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loven MA, Wood JA, Nardulli AM. Interaction of estrogen receptors alpha and beta with estrogen response elements. Mol. Cell. Endocrinol. 2001;181:151–163. doi: 10.1016/s0303-7207(01)00491-9. [DOI] [PubMed] [Google Scholar]
- Martini PG, Delage-Mourroux R, Kraichely DM, Katzenellenbogen BS. Prothymosin alpha selectively enhances estrogen receptor transcriptional activity by interacting with a repressor of estrogen receptor activity. Mol. Cell. Biol. 2000;20:6224–6232. doi: 10.1128/mcb.20.17.6224-6232.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neess D, Kiilerich P, Sandberg MB, Helledie T, Nielsen R, Mandrup S. ACBP—a PPAR and SREBP modulated housekeeping gene. Mol. Cell. Biochem. 2006;284:149–157. doi: 10.1007/s11010-005-9039-9. [DOI] [PubMed] [Google Scholar]
- Nilsson S, Makela S, Treuter E, Tujague M, Thomsen J, Andersson G, Enmark E, Pettersson K, Warner M, Gustafsson JA. Mechanisms of estrogen action. Physiol. Rev. 2001;81:1535–1565. doi: 10.1152/physrev.2001.81.4.1535. [DOI] [PubMed] [Google Scholar]
- Petz LN, Nardulli AM. Sp1 binding sites and an estrogen response element half-site are involved in regulation of the human progesterone receptor A promoter. Mol. Endocrinol. 2000;14:972–985. doi: 10.1210/mend.14.7.0493. [DOI] [PubMed] [Google Scholar]
- Petz LN, Ziegler YS, Loven MA, Nardulli AM. Estrogen receptor alpha and activating protein-1 mediate estrogen responsiveness of the progesterone receptor gene in MCF-7 breast cancer cells. Endocrinology. 2002;143:4583–4591. doi: 10.1210/en.2002-220369. [DOI] [PubMed] [Google Scholar]
- Petz LN, Ziegler YS, Schultz JR, Kim H, Kemper JK, Nardulli AM. Differential regulation of the human progesterone receptor gene by an estrogen response element half site and Sp1 sites. J. Steroid Biochem. Mol. Biol. 2004a;88:113–122. doi: 10.1016/j.jsbmb.2003.11.008. [DOI] [PubMed] [Google Scholar]
- Petz LN, Ziegler YS, Schultz JR, Nardulli AM. Fos and Jun inhibit estrogen-induced transcription of the human progesterone receptor gene through an activator protein-1 site. Mol. Endocrinol. 2004b;18:521–532. doi: 10.1210/me.2003-0105. [DOI] [PubMed] [Google Scholar]
- Plateroti M, Kress E, Mori JI, Samarut J. Thyroid hormone receptor alpha1 directly controls transcription of the beta-catenin gene in intestinal epithelial cells. Mol. Cell. Biol. 2006;26:3204–3214. doi: 10.1128/MCB.26.8.3204-3214.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez JA, Schuchner S, Au WW, Fabbro M, Henderson BR. Nuclear-cytoplasmic shuttling of BARD 1 contributes to its proapoptotic activity and is regulated by dimerization with BRCA1. Oncogene. 2004;23:1809–1820. doi: 10.1038/sj.onc.1207302. [DOI] [PubMed] [Google Scholar]
- Safe S. Transcriptional activation of genes by 17 beta-estradiol through estrogen receptor-Sp1 interactions. Vitam. Horm. 2001;62:231–252. doi: 10.1016/s0083-6729(01)62006-5. [DOI] [PubMed] [Google Scholar]
- Schuchner S, Tembe V, Rodriguez JA, Henderson BR. Nuclear targeting and cell cycle regulatory function of human BARD1. J. Biol. Chem. 2005;280:8855–8861. doi: 10.1074/jbc.M413741200. [DOI] [PubMed] [Google Scholar]
- Starita LM, Horwitz AA, Keogh MC, Ishioka C, Parvin JD, Chiba N. BRCA1/BARD1 ubiquitinate phosphorylated RNA polymerase II. J. Biol. Chem. 2005;280:24498–24505. doi: 10.1074/jbc.M414020200. [DOI] [PubMed] [Google Scholar]
- Stossi F, Barnett DH, Frasor J, Komm B, Lyttle CR, Katzenellenbogen BS. Transcriptional profiling of estrogen-regulated gene expression via estrogen receptor (ER) alpha or ERbeta in human osteosarcoma cells: distinct and common target genes for these receptors. Endocrinology. 2004;145:3473–3486. doi: 10.1210/en.2003-1682. [DOI] [PubMed] [Google Scholar]
- Thai TH, Du F, Tsan JT, Jin Y, Phung A, Spillman MA, Massa HF, Muller CY, Ashfaq R, Mathis JM, Miller DS, Trask BJ, Baer R, Bowcock AM. Mutations in the BRCA1-associated RING domain (BARD1) gene in primary breast, ovarian and uterine cancers. Hum. Mol. Genet. 1998;7:195–202. doi: 10.1093/hmg/7.2.195. [DOI] [PubMed] [Google Scholar]
- Vega VB, Lin CY, Lai KS, Li Kong S, Xie M, Su X, Teh HF, Thomsen JS, Li Yeo A, Sung WK, Bourque G, Liu ET. Multiplatform genome-wide identification and modeling of functional human estrogen receptor binding sites. Genome Biol. 2006;7:R82. doi: 10.1186/gb-2006-7-9-r82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinmann AS, Farnham PJ. Identification of unknown target genes of human transcription factors using chromatin immunoprecipitation. Methods. 2002;26:37–47. doi: 10.1016/S1046-2023(02)00006-3. [DOI] [PubMed] [Google Scholar]
- Weisz A, Basile W, Scafoglio C, Altucci L, Bresciani F, Facchiano A, Sismondi P, Cicatiello L, De Bortoli M. Molecular identification of ERα-positive breast cancer cells by the expression profile of an intrinsic set of estrogen regulated genes. J. Cell. Physiol. 2004;200:440–450. doi: 10.1002/jcp.20039. [DOI] [PubMed] [Google Scholar]
- Westermark UK, Reyngold M, Olshen AB, Baer R, Jasin M, Moynahan ME. BARD1 participates with BRCA1 in homology-directed repair of chromosome breaks. Mol. Cell. Biol. 2003;23:7926–7936. doi: 10.1128/MCB.23.21.7926-7936.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wise PM, Dubal DB, Wilson ME, Rau SW, Bottner M, Rosewell KL. Estradiol is a protective factor in the adult and aging brain: understanding of mechanisms derived from in vivo and in vitro studies. Brain Res. Rev. 2001;37:313–319. doi: 10.1016/s0165-0173(01)00136-9. [DOI] [PubMed] [Google Scholar]
- Wood JR, Greene GL, Nardulli AM. Estrogen response elements function as allosteric modulators of estrogen receptor conformation. Mol. Cell. Biol. 1998;18:1927–1934. doi: 10.1128/mcb.18.4.1927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood JR, Likhite VS, Loven MA, Nardulli AM. Allosteric modulation of estrogen receptor conformation by different estrogen response elements. Mol. Endocrinol. 2001;15:1114–1126. doi: 10.1210/mend.15.7.0671. [DOI] [PubMed] [Google Scholar]
- Wu LC, Wang ZW, Tsan JT, Spillman MA, Phung A, Xu XL, Yang MC, Hwang LY, Bowcock AM, Baer R. Identification of a RING protein that can interact in vivo with the BRCA1 gene product. Nat. Genet. 1996;14:430–440. doi: 10.1038/ng1296-430. [DOI] [PubMed] [Google Scholar]