

Abstract
The cardiac sodium channel (SCN5A) is a target for the treatment of arrhythmias. We hypothesized that vulnerability to atrial fibrillation (AF) could be caused by genetic variation in SCN5A. We recruited 157 patients with early-onset AF who lacked traditional risk factors, and 314 matched controls. SCN5A was subject to targeted genotyping of a common loss-of-function H558R polymorphism and comprehensive mutation scanning. Genotype frequencies in the AF cohort vs controls were as follows: HH, 50 vs 63%; HR, 40 vs 33%; and RR, 10 vs 4% (P = 0.008). Additional coding sequence mutations were ruled out. The R558 allele was more common in patients than in controls (30 vs 21%, P = 0.002), conferring an odds ratios for AF of 1.6 (95% confidence interval 1.2-2.2). The SCN5A R558 allele, present in one-third of the population, thus constitutes a risk factor for lone AF and may increase susceptibility to sodium channel blocker-induced proarrhythmia.
Atrial fibrillation (AF) is the most common sustained arrhythmia affecting more than 2.2 million Americans.1 Risk factors for AF include advancing age, hypertension, structural heart disease and congestive heart failure.2-4 In a subset of younger patients (<60 years of age), AF develops in the absence of known risk factors, a condition classified as lone AF.5 Familial forms of lone AF with autosomal-dominant inheritance have been described.6-11 Loci on chromosomes 10q22 and 6q14-16 have been linked to familial AF, and mutations in KCNQ1, KCNE2, KCNJ2, and KCNA5 have been found to cause familial AF.6,8-12 In patients with acquired AF, disease risk has been associated with polymorphisms in KCNE1, GNB3, renin-angiotensin system genes, and KCNE5.13-16 Heritability of acquired AF is further suggested by two recent population-based studies demonstrating that the presence of AF in first-degree relatives was associated with an increased risk of developing AF.17,18
The human cardiac sodium channel (SCN5A) is responsible for the fast depolarization upstroke of the cardiac action potential and serves as a molecular target for antiarrhythmic drugs. Mutations in the human cardiac sodium channel gene have been previously discovered in a spectrum of cardiac rhythm disorders: the long QT internal syndrome, Brugada syndrome, idiopathic ventricular fibrillation, conduction system disease, and sick sinus syndrome.19-23 Gain-of-function mutations in SCN5A cause the long QT syndrome, whereas the other conditions are caused by mutations that reduce sodium channel current. An S1102Y polymorphism in SCN5A, with a minor allele frequency of 7% in African Americans, was subsequently shown to increase the risk of ventricular arrhythmias by eightfold.24 More recently, loss-of-function mutations in SCN5A have been linked to a syndrome of dilated cardiomyopathy (DCM) and AF.25 Moreover, up to 39% of patients with the Brugada syndrome have AF.25 These findings provide a rationale to test SCN5A as a candidate gene for non-syndromic, lone AF.
A comprehensive survey of SCN5A identified 39 non-synonymous polymorphisms.26 Among these genetic variants, H558R was by far the most common with a minor allele frequency of 20% in Caucasians. Moreover, it was the only SCN5A polymorphism present in all four ethnic groups studied: African Americans, Caucasians, Asians, and Hispanics. Of significance, the minor R allele of this polymorphism, present in over one-third of the population,26 has been shown to alter SCN5A function by reducing depolarizing sodium current27 and modulating the biological effects of concomitant SCN5A mutations.28,29
We hypothesized that the R558 allele or mutations in SCN5A could increase vulnerability to AF. To minimize potentially confounding variables, we recruited a cohort of patients with lone AF, lacking traditional risk factors, to test our hypothesis. We report herein that the common polymorphism H558R is associated with susceptibility to lone AF.
RESULTS
Clinical characteristics of lone AF cohort
Baseline characteristics of the 157 unrelated lone AF patients we recruited are shown in Table 1; all were Caucasian. The mean age at enrollment was 52.0±9.8 years, an average of 7.5 years after the initial diagnosis of AF. These patients did not have hypertension, hyperthyroidism, diabetes, history of coronary artery disease, or structural heart disease. Echocardiographic measures of cardiac structure and function were normal: left ventricular ejection fraction, 59.1±8.5%; left ventricular end diastolic dimension, 45.4±6.4 mm; left ventricular end systolic dimension, 31.8±6.8 mm; ventricular septal thickness in diastole, 10.3±2.1 mm; and left ventricular posterior wall thickness in diastole, 10.0±1.7 mm. The only echocardiographic abnormality was mild left atrial enlargement (mean left atrial volume index, 32.3±10.3 cm3/m2; upper limit of normal = 28 cm3/m2).
Table 1.
Clinical characteristics of lone AF patients with respect to genotype
| Characteristics | Overall (n=157) | HH (n=78) | HR (n=64) | RR (n=15) | P-value* | HR and RR (n=79) | P-value† |
|---|---|---|---|---|---|---|---|
| Age at enrollment (years) | 52.0±9.8 | 53.0±10.8 | 51.4±8.8 | 49±8.1 | 0.31 | 51.0±8.7 | 0.20 |
| Age at AF diagnosis (years) | 44.5±10.3 | 46.0±11.0 | 43.2±10.0 | 42.2±6.2 | 0.16 | 43.0±9.4 | 0.07 |
| Male gender | 119 (75.8%) | 58 (74.4%) | 51 (79.7%) | 10 (66.7%) | 0.52 | 61 (77.2%) | 0.46 |
| Positive family history | 74 (47.1%) | 34 (43.6%) | 33 (51.6%) | 7 (46.7%) | 0.65 | 40 (50.6%) | 0.41 |
| ECG characteristics in sinus rhythm | |||||||
| Ventricular rate (min-1) | 71.9±19.3 | 73.1±20.5 | 71.7±19.4 | 66.4±10.6 | 0.49 | 70.7±18.1 | 0.45 |
| PR interval (ms) | 172.6±31.5 | 170.6±31.4 | 174.7±31.4 | 173.3±34.6 | 0.79 | 174.5±31.7 | 0.51 |
| QRS duration (ms) | 105.0±41.3 | 108.6±54.9 | 100.0±17.3 | 108.3±30.1 | 0.47 | 101.6±20.3 | 0.30 |
| QT interval (ms) | 425.5±29.1 | 424.7±28.7 | 427.8±28.5 | 419.4±34.9 | 0.58 | 426.2±29.8 | 0.75 |
| Pacemaker | 15 (9.6%) | 7 (9.0%) | 7 (10.9%) | 1 (6.7%) | 0.86 | 8 (10.1%) | 0.81 |
| Body mass index (kg/min2) | 28.6±4.0 | 28.5±4.1 | 29.0±4.1 | 26.9±2.9 | 0.24 | 28.6±4.0 | 0.89 |
| Type of AF | 0.12 | 0.15 | |||||
| Paroxysmal | 98 (62.4%) | 44 (56.4%) | 43 (67.2%) | 11 (73.3%) | 54 (68.4%) | ||
| Persistent | 35 (22.3%) | 18 (23.1%) | 13 (20.3%) | 4 (26.7%) | 17 (21.5%) | ||
| Permanent | 24 (15.3%) | 16 (20.5%) | 8 (12.5%) | 0 (0.0%) | 8 (10.1%) |
Abbreviations: AF, atrial fibrillation; ECG, electrocardiogram. Continuous variables are expressed as mean±s.d.; categorical variables are expressed as number (%).
P-value comparing HH, HR, and RR groups.
P-value comparing HH group with HR/RR group.
H558R polymorphism in SCN5A is associated with lone AF
Mutation scanning of all translated exons of SCN5A in the 157 lone AF patients did not reveal any mutation that altered splice junction or amino-acid sequence. Sequencing of samples yielding heteroduplexes on denaturing high-performance liquid chromatography analysis uncovered synonymous single-nucleotide polymorphisms that include A29A (exon 2), E1065E (exon 17), and D1823D (exon 28).
The major finding of this study was a significant difference in frequencies of the three H558R genotypes - HH, HR, and RR (Figure 1) - in lone AF patients compared to normal controls (Figure 2). Fifty percent of patients had at least one R allele compared with 37% of normal controls. The observed genotype frequencies - HH, HR, and RR - were not significantly different from Hardy-Weinberg equilibrium in either the lone AF patients (50, 40, and 10% vs 48, 42, and 10%; P>0.05) or controls (63, 33, and 4% vs 64, 32, and 4%; P>0.05). The minor R allele frequency was 30% in the lone AF patients compared with 21% in the normal controls (P = 0.002; Figure 3). Compared with the HH genotype, the HR genotype conferred an odds ratio (OR) for developing lone AF of 1.5 (95% confidence interval (CI) 1.1-2.3; P = 0.04). The RR genotype conferred a higher OR of 2.9 (95% CI 1.3-6.5; P = 0.008) (Figure 4). In a logistic regression model, the R allele was associated with a 1.6-fold increased risk for lone AF (95% CI 1.2-2.2; P = 0.002).
Figure 1.
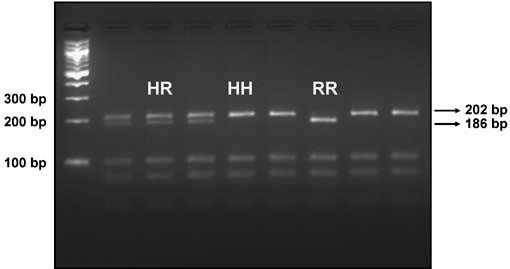
Representative agarose gel, demonstrating separation of H558- and R558-associated alleles by electrophoresis following PCR and restriction enzyme digestion with AciI. The left lane represents fragments from a DNA ladder. The 202 bp fragment identifies the H558-associated allele; the 186 bp fragment is specific to the R558-associated allele.
Figure 2.
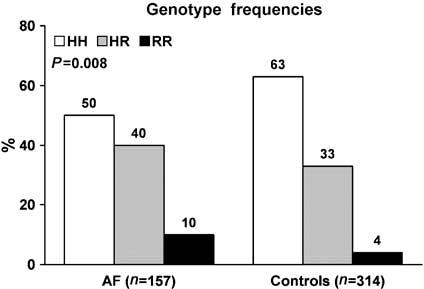
H558R genotype frequencies of lone AF cohort and normal controls.
Figure 3.
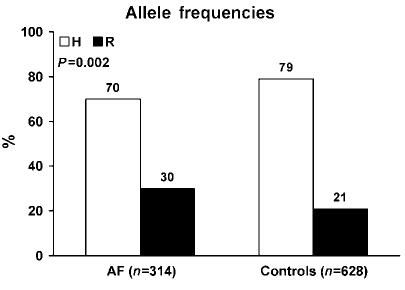
H558R allele frequencies of lone AF cohort and normal controls.
Figure 4.
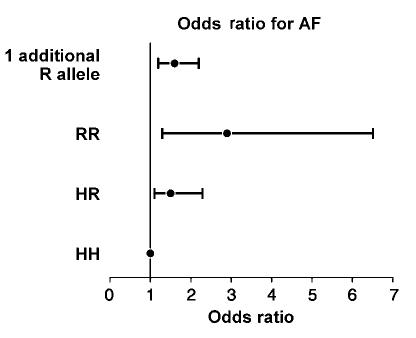
ORs for AF. The error bars represent 95% CI. HH genotype is the reference group. ORs for HR genotype, 1.5 (95% CI 1.1-2.3); RR genotype, 2.9 (95% CI 1.3-6.5); and presence of one R allele, 1.6 (95% CI 1.2-2.2).
H558R polymorphism in SCN5A modifies lone AF phenotype
The distribution of clinical and electrocardiographic variables demonstrated no significant difference among the three genotypes (Table 1). When comparing HH genotype with HR or RR genotype (R allele-carriers), we observed a trend toward younger age at diagnosis in the latter group, 46.0±11.0 vs 43.0±9.4 years (P = 0.07). When the analysis was stratified by gender, we found that male R allele-carriers were diagnosed with AF at a younger age (42.4±9.5 years) compared with male HH patients (46.2±9.2 years; P = 0.03) (Table 2). Consistent with this finding, male R allele-carriers were also enrolled at a younger age (50.0±7.5 years) compared with their HH male counterparts (53.0±8.7 years; Table 2). No other variables were significantly different between HH and R allele-carriers stratified by gender (Table 2).
Table 2.
Clinical characteristics of lone AF patients with respect to genotype and gender
| Males (n=119) |
Females (n=38) |
|||||
|---|---|---|---|---|---|---|
| Characteristics | HH (n=58) | HR/RR (n=61) | P-value* | HH (n=20) | HR/RR (n=18) | P-value† |
| Age at enrollment (years) | 53.0±8.7 | 50.0±7.5 | 0.05 | 53.1±16.0 | 54.7±12.2 | 0.74 |
| Age at AF diagnosis (years) | 46.2±9.8 | 42.4±9.5 | 0.03 | 45.3±14.5 | 45.6±9.2 | 0.94 |
| Positive family history | 24 (41.4%) | 33 (54.1%) | 0.22 | 10 (50%) | 7 (38.9%) | 0.62 |
| ECG characteristics in sinus rhythm | ||||||
| Ventricular rate (min-1) | 72.8±21.7 | 67.7±14.3 | 0.14 | 74.2±16.6 | 83.0±26.0 | 0.25 |
| PR interval (ms) | 176.5±29.1 | 179.7±29.2 | 0.59 | 155.9±32.9 | 143.8±30.1 | 0.36 |
| QRS duration (ms) | 107.6±45.9 | 102.5±20.4 | 0.44 | 111.5±77.1 | 97.7±20.1 | 0.51 |
| QT interval (ms) | 423.1±28.3 | 421.9±28.4 | 0.81 | 430.5±30.0 | 440.6±32.0 | 0.35 |
| Pacemaker | 6 (10.3%) | 5 (8.2%) | 0.66 | 1 (5.0%) | 3 (16.7%) | 0.19 |
| Body mass index (kg/min2) | 28.6±3.8 | 28.6±3.7 | 0.95 | 28.2±5.2 | 28.9±5.5 | 0.74 |
| Type of AF | 0.11 | 0.97 | ||||
| Paroxysmal | 31 (53.4%) | 43 (70.5%) | 13 (68.4%) | 10 (66.7%) | ||
| Persistent | 16 (27.6%) | 13 (21.3%) | 2 (10.5%) | 2 (13.3%) | ||
| Permanent | 11 (19.0%) | 5 (8.2%) | 4 (21.1%) | 3 (20.0%) | ||
Abbreviations: AF, atrial fibrillation; ECG, electrocardiogram. Continuous variables are expressed as mean±s.d.; categorical variables are expressed as number (%).
P-value comparing HH males with HR/RR males.
P-value comparing HH females with HR/RR females.
Influence of the R allele on age at diagnosis was also observed when the analysis was stratified by the type of AF (Table 3). R allele-carriers with paroxysmal AF were diagnosed at a younger age (41.5±8.9 years), compared with HH allele-carriers with paroxysmal AF (47.3±10.9 years; P = 0.005). Moreover, R allele-carriers were enrolled at a younger age (49.5±8.4 years) compared with their HH counterparts (53.6±10.8 years; P = 0.04).
Table 3.
Clinical characteristics of lone AF patients with respect to genotype and AF type
| Paroxysmal AF (n=98) |
Persistent or chronic AF (n=59) |
|||||
|---|---|---|---|---|---|---|
| Characteristics | HH (n=45) | HR/RR (n=53) | P-value* | HH (n=33) | HR/RR (n=26) | P-value† |
| Age at enrollment (years) | 53.6±10.8 | 49.5±8.4 | 0.04 | 52.2±10.9 | 54.4±8.7 | 0.43 |
| Age at AF diagnosis (years) | 47.3±10.9 | 41.5±8.9 | 0.005 | 44.2±11.1 | 46.3±9.8 | 0.46 |
| Male gender | 31 (68.9%) | 43 (81.1%) | 0.22 | 27 (81.8%) | 18 (69.2%) | 0.74 |
| ECG characteristics in sinus rhythm | ||||||
| Ventricular rate (min-1) | 70.0±20.2 | 70.0±17.7 | 0.55 | 77.4±20.4 | 73.0±19.2 | 0.43 |
| PR interval (ms) | 171.7±33.6 | 175.7±28.8 | 0.18 | 168.6±27.0 | 170.9±39.9 | 0.84 |
| QRS duration (ms) | 111.9±68.8 | 100.8±20.2 | 0.27 | 103.8±24.6 | 103.2±20.8 | 0.93 |
| QT interval (ms) | 424.2±27.4 | 420.9±27.3 | 0.56 | 425.5±30.8 | 436.9±32.2 | 0.18 |
| Pacemaker | 4 (8.9%) | 3 (5.7%) | 0.46 | 3 (9.1%) | 5 (19.2%) | 0.16 |
| Body mass index (kg/min2) | 27.9±4.1 | 28.6±3.8 | 0.45 | 29.6±3.9 | 28.8±4.6 | 0.57 |
Abbreviations: AF, atrial fibrillation; ECG, electrocardiogram. Continuous variables are expressed as mean±s.d.; categorical variables are expressed as number (%).
P-value comparing HH paroxysmal AF with HR/RR paroxysmal AF.
P-value comparing HH persistent or chronic AF with HR/RR persistent or chronic AF.
DISCUSSION
In contrast to previous case-control association studies13-16 of patients with AF and underlying structural heart disease, we studied a more homogeneous population of patients with early-onset, lone AF to minimize the potentially confounding effects of acquired risk factors. We established a significant association between the common H558R polymorphism in SCN5A and lone AF. Further evidence for susceptibility to lone AF conferred by the R558 allele is demonstrated by its association with age at diagnosis. Male carriers of the R allele were diagnosed earlier compared with male HH homozygotes. Moreover, the R allele was also associated with a younger age at diagnosis in patients with paroxysmal AF. H558R - the most common polymorphism in SCN5A with an R allele frequency of 20% in Caucasians - is also found in other racial and ethnic groups. Hence, it has significant impact on susceptibility to AF in the general population. Sodium channel blockers, commonly used to treat AF, may worsen the condition in patients harboring the R558 allele by an excess reduction in sodium current.
SCN5A as a candidate gene for lone AF
We have previously shown that mutations in SCN5A can cause a syndrome of DCM and AF.25 A missense mutation was discovered in a large, multigenerational family with a variably expressed phenotype of DCM, AF, sinus node dysfunction, and conduction system disease. Additional SCN5A mutations were identified in four smaller DCM families, three of which also had AF. Among the 37 SCN5A mutation-carriers within these five families, 43% had documented AF. Each of the identified mutations was predicted to cause loss of cardiac sodium channel function. Patients with Brugada syndrome also have an increased incidence of AF.30 Consistent with loss of sodium channel function, these patients had slower atrioventricular conduction, longer atrio-His and His-ventricular intervals, and longer effective refractory period of the atrioventricular node compared with normal controls. Collectively, these observations raise the possibility that mutations in SCN5A could cause non-syndromic, lone AF.
Despite a plausible rationale for SCN5A as a candidate gene for lone AF, we did not identify any SCN5A mutations in our cohort of 157 unrelated patients. There are several possible explanations for this. First, because AF is a genetically heterogeneous disorder, a larger sample may be required to determine whether or not SCN5A mutations can cause lone AF. Notwithstanding, we conclude that mutations in coding regions of SCN5A are not a common cause for lone AF. Second, certain mutations in SCN5A may in fact only cause syndromic AF (in association with DCM or Brugada syndrome) but not lone AF per se. Third, since only exons were scanned for mutations, our methodology does not exclude mutations in introns or promoter elements. Indeed, there is evidence in the literature that intronic mutations in ion channel genes may cause arrhythmia syndromes.31 Feasibility of mutation scanning in introns of SCN5A is limited, however, by the relatively large amount of sequence. Recently, a haplotype of polymorphisms in the SCN5A promoter was found to reduce sodium channel expression in vitro.32 This haplotype, however, is present in Asians but not in Caucasians, the racial group that constitutes our cohort. Finally, certain types of SCN5A mutations would be missed by our denaturing high-performance liquid chromatography methodology owing to <100% sensitivity of detection or deletion of the binding site of a polymerase chain reaction (PCR) primer, resulting in failed amplification of the mutant allele.
Biological effect of the H558R polymorphism
H558R, located in the intracytoplasmic linker connecting the first and second domains of the sodium channel, is the most common polymorphism in SCN5A resulting from a single-nucleotide change (1,673A>G). Its functional significance in modulating the effects of concomitant SCN5A mutations has been shown in previous studies.28,29 The minor allele frequencies in African Americans, Caucasians, and Asians are 29, 20, and 9%, respectively.26 The genotype frequencies (HH, HR, RR) for African Americans and Caucasians are 53, 40, and 7% and 65, 30, and 5%, respectively.27 The biological effect of H558R is further influenced by alternative splicing at a distal site of the gene, resulting in a longer 2,016-amino-acid splice variant containing glutamine at position 1,077 (Q1077), and a shorter 2,015-amino-acid splice variant lacking glutamine at position 1,077 (Q1077del). Preferential splicing favors Q1077del in a ratio of 2:1.27 In a heterologous expression system, presence of R558 and Q1077 on the same allele was associated with a profound reduction in sodium current density.27 By inference, individuals harboring the R558 allele in the heterozygous state would have sodium current density reduced by 17%, and R558 homozygotes would have sodium current density attenuated by 35%. An R558 allele-associated reduction in sodium current, recently linked to Brugada syndrome susceptibility,33 could also provide an electrophysiological substrate for AF.
We propose the following mechanism by which the H558R polymorphism would increase vulnerability to AF. A reduction in sodium current density would lead to decreased conduction velocity in the atria. This would, in turn, shorten the atrial re-entry wavelength and promote re-entry via a single re-entrant circuit or multiple re-entrant circuits.34 Such an electrophysiological effect would provide a substrate for maintaining AF, once triggered by an atrial ectopic beat. Similarly, gain-of-function potassium channel mutations known to cause familial lone AF shorten the atrial re-entry wavelength, albeit by a different mechanism: shortening of the atrial refractory period.9-11
Pharmacologic and therapeutic implications
Class I antiarrhythmics (sodium channel blockers) can be effective in the management of AF, yet they may also paradoxically exacerbate arrhythmia. Use of these drugs can cause 1:1 atrioventricular conduction during AF, owing to conduction delay in the atria and a slower atrial rate.35 If the fibrillation cycle length prolongs sufficiently, 1:1 atrioventricular transmission will result in a paradoxical increase in ventricular response. In patients with loss-of-function SCN5A mutations, sodium channel blockers can unmask an abnormal electrocardiographic phenotype.36 By analogy, individuals carrying the R558 allele, predicted to have a baseline reduction in cardiac sodium current density, may have increased susceptibility to the proarrhythmic effects of pharmacological sodium channel blockade. Further pharmacogenetic studies will be required to determine whether or not R558 allele-carriers, particularly homozygotes, should avoid Class I antiarrhythmics for the management of AF.
In conclusion, we report an association between the common H558R polymorphism in SCN5A and lone AF. The presence of the minor R allele increases susceptibility to lone AF by 1.6-fold, and is associated with younger age at diagnosis in males and in those with paroxysmal disease. This genetic variant, present in one-third of the population, is thus a common risk factor for lone AF. Moreover, the use of sodium channel blockers, by further reducing sodium current, may increase the risk of proarrhythmia in R558 allele-carriers.
METHODS
Clinical evaluation
The study protocol was approved by the Institutional Review Board of the Mayo Clinic and participants were enrolled following informed written consent. Between November 2000 and June 2004, patients referred to our Heart Rhythm Center and diagnosed with lone AF were identified. AF was defined as replacement of sinus P waves by rapid oscillations or fibrillatory waves that varied in size, shape, and timing, and were associated with an irregular ventricular response when atrioventricular conduction was intact. Documentation of AF on an electrocardiogram, rhythm strip, event monitor, or Holter monitor recording was required. Lone AF was defined as AF in individuals <60 years of age without hypertension or overt structural heart disease by clinical examination, electrocardiography, and echocardiography.5
“Paroxysmal AF” was defined as AF lasting more than 30 s that terminated spontaneously. The AF was classified as “persistent” when it lasted more than 7 days and required either pharmacologic therapy or electrical cardioversion for termination. AF that was completely refractory to cardioversion or was allowed to continue was classified as “permanent”.37
Familial AF was defined as the presence of lone AF in one or more first-degree relative of the index case. Family history information was initially obtained from the medical record and was supplemented by a questionnaire. The questionnaire pertained to past medical history, family history, and clinical symptoms. For individuals with a positive family history, a more detailed family history was obtained and medical records of relatives were reviewed.
Normal control individuals were selected from a cross-sectional, population-based cohort of 2,042 individuals from Olmsted County, Minnesota.38 Each subject underwent a comprehensive medical evaluation consisting of medical history, physical examination, echocardiography, and electrocardiography. In a 1:2 ratio (patients: controls), we selected age, gender, and ethnicity matched controls for our study from this population cohort. Control subjects did not have history or clinical evidence of AF, hypertension, hyperthyroidism, diabetes, coronary artery disease, or structural heart disease.
Genetic analyses
Whole blood was collected for genomic DNA extraction and analysis from patients and normal controls. Primer pairs for exon-specific PCR amplification of genomic DNA were designed using OLIGO v6.51 Primer Analysis Software (National Biosciences, Plymouth, MN) and WAVEMAKER version 4.0.32 software (Transgenomic, Omaha, NE). Primer pairs (n = 31) were designed for all 28 translated exons of SCN5A, including an alternatively spliced exon. Heterozygous sequence variants in PCR-amplified DNA fragments were identified by denaturing high-performance liquid chromatography heteroduplex analysis (WAVE DNA Fragment Analysis System, Transgenomic), carried out according to the manufacturer’s instructions. Chromatographic elution profiles of amplified fragments were compared against the wild-type homoduplex pattern. Samples yielding anomalous traces were selected for further analysis by sequencing. Genomic DNA was reamplified by PCR, and products were treated with the PCR Product Pre-sequencing Kit (USB Corporation, Cleveland, OH). Treated products were sequenced by the dye-terminator method in a core facility, using an ABI PRISM 377 DNA Sequencer (Applied Biosystems, Foster City, CA). DNA sequence was viewed and analyzed using the Sequencher computer program (Gene Codes Corporation, Ann Arbor, MI). Numbering of amino acids for SCN5A was based on inclusion of an alternatively spliced residue, Q1077.
Genotyping of the H558R polymorphism was performed by restriction fragment length polymorphism analysis. The corresponding A>G nucleotide transition creates a -CCGC-sequence, which is a recognition site for the restriction enzyme AciI. This variant, located in exon 12 of SCN5A, was first amplified within a 443 base pair (bp) PCR product. Restriction digestion of the PCR product with AciI resulted in the following fragment sizes (bp): H allele (-CCAC-): 202, 96, 80, 43, and 22; R allele (-CCGC-): 186, 96, 80, 43, 22, and 16. Fragments were then separated by agarose gel electrophoresis. Hence, the H and R alleles could be distinguished by the presence of 202 and 186 bp fragments, respectively.
Statistical analyses
The Hardy-Weinberg law was applied to analyze distribution of SCN5A H558R genotypes, where p = H allele frequency, q = R allele frequency, p2 = HH, 2pq = HR, and q2 = RR. Genotype and allele frequencies were compared between patients and controls using the χ2 test. Associations between the three genotypes and continuous variables were tested using one-way analysis of variance; the χ2 test or Fisher’s exact test was used to compare associations between genotypes and categorical variables. Comparisons were also made between HH and HR or RR genotype. Effects of the minor R allele on the risk of developing lone AF and on modifying the phenotype were assessed with logistic regression. To evaluate the impact of the minor R allele, dummy codes 00, 10, and 01 were used to represent the presence of 0 (HH), 1 (HR), and 2 (RR) minor R alleles, respectively, in the logistic regression model. ORs were calculated, together with 95% CI. Statistical significance was accepted for values of P<0.05.
ACKNOWLEDGMENTS
This work was supported by the Mayo Foundation and the US National Institutes of Health.
Footnotes
CONFLICT OF INTEREST
The authors declared that they have no competing financial interests.
References
- 1.Thom T, et al. Heart disease and stroke statistics - 2006 update: a report from the American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Circulation. 2006;113:e85–e151. doi: 10.1161/CIRCULATIONAHA.105.171600. [DOI] [PubMed] [Google Scholar]
- 2.Krahn AD, Manfreda J, Tate RB, Mathewson FA, Cuddy TE. The natural history of atrial fibrillation: incidence, risk factors, and prognosis in the Manitoba Follow-Up Study. Am. J. Med. 1995;98:476–484. doi: 10.1016/S0002-9343(99)80348-9. [DOI] [PubMed] [Google Scholar]
- 3.Benjamin EJ, et al. Independent risk factors for atrial fibrillation in a population-based cohort. The Framingham Heart Study. JAMA. 1994;271:840–844. [PubMed] [Google Scholar]
- 4.Psaty BM, et al. Incidence of and risk factors for atrial fibrillation in older adults. Circulation. 1997;96:2455–2461. doi: 10.1161/01.cir.96.7.2455. [DOI] [PubMed] [Google Scholar]
- 5.Chugh SS, Blackshear JL, Shen WK, Hammill SC, Gersh BJ. Epidemiology and natural history of atrial fibrillation: clinical implications. J. Am. Coll. Cardiol. 2001;37:371–378. doi: 10.1016/s0735-1097(00)01107-4. [DOI] [PubMed] [Google Scholar]
- 6.Brugada R, et al. Identification of a genetic locus for familial atrial fibrillation. N. Engl. J. Med. 1997;336:905–911. doi: 10.1056/NEJM199703273361302. [DOI] [PubMed] [Google Scholar]
- 7.Darbar D, et al. Familial atrial fibrillation is a genetically heterogeneous disorder. J. Am. Coll. Cardiol. 2003;41:2185–2192. doi: 10.1016/s0735-1097(03)00465-0. [DOI] [PubMed] [Google Scholar]
- 8.Ellinor PT, Shin JT, Moore RK, Yoerger DM, MacRae CA. Locus for atrial fibrillation maps to chromosome 6q14-16. Circulation. 2003;107:2880–2883. doi: 10.1161/01.CIR.0000077910.80718.49. [DOI] [PubMed] [Google Scholar]
- 9.Chen YH, et al. KCNQ1 gain-of-function mutation in familial atrial fibrillation. Science. 2003;299:251–254. doi: 10.1126/science.1077771. [DOI] [PubMed] [Google Scholar]
- 10.Yang Y, et al. Identification of a KCNE2 gain-of-function mutation in patients with familial atrial fibrillation. Am. J. Hum. Genet. 2004;75:899–905. doi: 10.1086/425342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Xia M, et al. A Kir2.1 gain-of-function mutation underlies familial atrial fibrillation. Biochem. Biophy. Res. Commun. 2005;332:1012–1019. doi: 10.1016/j.bbrc.2005.05.054. [DOI] [PubMed] [Google Scholar]
- 12.Olson TM, et al. Kv1.5 channelopathy due to KCNA5 loss-of-function mutation causes human atrial fibrillation. Hum. Mol. Genet. 2006;15:2185–2191. doi: 10.1093/hmg/ddl143. [DOI] [PubMed] [Google Scholar]
- 13.Lai LP, et al. Association of the human minK gene 38G allele with atrial fibrillation: evidence of possible genetic control on the pathogenesis of atrial fibrillation. Am. Heart J. 2002;144:485–490. doi: 10.1067/mhj.2002.123573. [DOI] [PubMed] [Google Scholar]
- 14.Schreieck J, et al. C825T polymorphism of the G-protein beta3 subunit gene and atrial fibrillation: association of the TT genotype with a reduced risk for atrial fibrillation. Am. Heart J. 2004;148:545–550. doi: 10.1016/j.ahj.2004.03.024. [DOI] [PubMed] [Google Scholar]
- 15.Tsai CT, et al. Renin-angiotensin system gene polymorphisms and atrial fibrillation. Circulation. 2004;109:1640–1646. doi: 10.1161/01.CIR.0000124487.36586.26. [DOI] [PubMed] [Google Scholar]
- 16.Ravn LS, et al. Relation of 97T polymorphism in KCNE5 to risk of atrial fibrillation. Am. J. Cardiol. 2005;96:405–407. doi: 10.1016/j.amjcard.2005.03.086. [DOI] [PubMed] [Google Scholar]
- 17.Fox CS, et al. Parental atrial fibrillation as a risk factor for atrial fibrillation in offspring. JAMA. 2004;291:2851–2855. doi: 10.1001/jama.291.23.2851. [DOI] [PubMed] [Google Scholar]
- 18.Arnar DO, et al. Familial aggregation of atrial fibrillation in Iceland. Eur. Heart J. 2006;27:708–712. doi: 10.1093/eurheartj/ehi727. [DOI] [PubMed] [Google Scholar]
- 19.Wang Q, et al. SCN5A mutations associated with an inherited cardiac arrhythmia, long QT syndrome. Cell. 1995;80:805–811. doi: 10.1016/0092-8674(95)90359-3. [DOI] [PubMed] [Google Scholar]
- 20.Chen Q, et al. Genetic basis and molecular mechanism for idiopathic ventricular fibrillation. Nature. 1998;392:293–296. doi: 10.1038/32675. [DOI] [PubMed] [Google Scholar]
- 21.Akai J, et al. A novel SCN5A mutation associated with idiopathic ventricular fibrillation without typical ECG findings of Brugada syndrome. FEBS Lett. 2000;479:29–34. doi: 10.1016/s0014-5793(00)01875-5. [DOI] [PubMed] [Google Scholar]
- 22.Schott JJ, et al. Cardiac conduction defects associate with mutations in SCN5A. Nat. Genet. 1999;23:20–21. doi: 10.1038/12618. [DOI] [PubMed] [Google Scholar]
- 23.Benson DW, et al. Congenital sick sinus syndrome caused by recessive mutations in the cardiac sodium channel gene (SCN5A) J. Clin. Invest. 2003;112:1019–1028. doi: 10.1172/JCI18062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Splawski I, et al. Variant of SCN5A sodium channel implicated in risk of cardiac arrhythmia. Science. 2002;297:1333–1336. doi: 10.1126/science.1073569. [DOI] [PubMed] [Google Scholar]
- 25.Olson TM, et al. Sodium channel mutations and susceptibility to heart failure and atrial fibrillation. JAMA. 2005;293:447–454. doi: 10.1001/jama.293.4.447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ackerman MJ, et al. Spectrum and prevalence of cardiac sodium channel variants among black, white, Asian, and Hispanic individuals:implications for arrhythmogenic susceptibility and Brugada/long QT syndrome genetic testing. Heart Rhythm. 2004;1:600–607. doi: 10.1016/j.hrthm.2004.07.013. [DOI] [PubMed] [Google Scholar]
- 27.Makielski JC, et al. A ubiquitous splice variant and a common polymorphism affect heterologous expression of recombinant human SCN5A heart sodium channels. Circ. Res. 2003;93:821–828. doi: 10.1161/01.RES.0000096652.14509.96. [DOI] [PubMed] [Google Scholar]
- 28.Viswanathan PC, Benson DW, Balser JR. A common SCN5A polymorphism modulates the biophysical effects of an SCN5A mutation. J. Clin. Invest. 2003;111:341–346. doi: 10.1172/JCI16879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ye B, Valdivia CR, Ackerman MJ, Makielski JC. A common human SCN5A polymorphism modifies expression of an arrhythmia causing mutation. Physiol. Genomics. 2003;12:187–193. doi: 10.1152/physiolgenomics.00117.2002. [DOI] [PubMed] [Google Scholar]
- 30.Morita H, et al. Atrial fibrillation and atrial vulnerability in patients with Brugada syndrome. J. Am. Coll. Cardiol. 2002;40:1437–1444. doi: 10.1016/s0735-1097(02)02167-8. [DOI] [PubMed] [Google Scholar]
- 31.Zhang L, et al. An intronic mutation causes long QT syndrome. J. Am. Coll. Cardiol. 2004;44:1283–1291. doi: 10.1016/j.jacc.2004.06.045. [DOI] [PubMed] [Google Scholar]
- 32.Bezzina CR, et al. Common sodium channel promoter haplotype in Asian subjects underlies variability in cardiac conduction. Circulation. 2006;113:338–344. doi: 10.1161/CIRCULATIONAHA.105.580811. [DOI] [PubMed] [Google Scholar]
- 33.Chen J-Z, et al. Single nucleotide polymorphisms of the SCN5A gene in Han Chinese and their relation with Brugada syndrome. Chin. Med. J. 2004;117:652–656. [PubMed] [Google Scholar]
- 34.Allessie MA, Bonke FI, Schopman FJ. Circus movement in rabbit atrial muscle as a mechanism of tachycardia. III. The “leading circle” concept: a new model of circus movement in cardiac tissue without the involvement of an anatomical obstacle. Circ. Res. 1977;41:9–18. doi: 10.1161/01.res.41.1.9. [DOI] [PubMed] [Google Scholar]
- 35.Roden DM. Mechanisms and management of proarrhythmia. Am. J. Cardiol. 1998;82:49I–57I. doi: 10.1016/s0002-9149(98)00472-x. [DOI] [PubMed] [Google Scholar]
- 36.Brugada R, et al. Sodium channel blockers identify risk for sudden death in patients with ST-segment elevation and right bundle branch block but structurally normal hearts. Circulation. 2000;101:510–515. doi: 10.1161/01.cir.101.5.510. [DOI] [PubMed] [Google Scholar]
- 37.Fuster V, et al. ACC/AHA/ESC Guidelines for the Management of Patients With Atrial Fibrillation: Executive Summary A Report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines and the European Society of Cardiology Committee for Practice Guidelines and Policy Conferences (Committee to Develop Guidelines for the Management of Patients with Atrial Fibrillation) Developed in Collaboration with the North American Society of Pacing and Electrophysiology. Circulation. 2001;104:2118–2150. [PubMed] [Google Scholar]
- 38.Redfield MM, et al. Burden of systolic and diastolic ventricular dysfunction in the community: appreciating the scope of the heart failure epidemic. JAMA. 2003;289:194–202. doi: 10.1001/jama.289.2.194. [DOI] [PubMed] [Google Scholar]