

Abstract
Administration of a combination of yeast-derived β-glucan with antitumor monoclonal antibodies (mAb) has significant therapeutic efficacy in a variety of syngeneic murine tumor models. We have now tested this strategy using human carcinomas implanted in immunocompromised severe combined immunodeficient mice. Combined immunotherapy was therapeutically effective in vivo against NCI-H23 human non–small-cell lung carcinomas, but this modality was surprisingly ineffective against SKOV-3 human ovarian carcinomas. Whereas NCI-H23 tumors responded to this combination therapy with increased intratumoral neutrophil infiltration and C5a production, these responses were lacking in treated SKOV-3 tumors. Further results suggested that SKOV-3 tumors were protected by up-regulation of the membrane complement regulatory protein CD55 (decay-accelerating factor). Blockade of CD55 in vitro led to enhanced deposition of C activation product C3b and increased cytotoxicity mediated by β-glucan–primed neutrophils. In vivo, administration of anti-CD55 mAb along with β-glucan and anti–Her-2/neu mAb caused tumor regression and greatly improved long-term survival in animals bearing the previously resistant SKOV-3 tumors. This was accompanied by increased intratumoral neutrophil accumulation and C5a production. We conclude that CD55 suppresses tumor killing by antitumor mAb plus β-glucan therapy (and, perhaps, in other circumstances). These results suggest a critical role for CD55 to regulate iC3b and C5a release and in turn to influence the recruitment of β-glucan–primed neutrophils eliciting killing activity.
Introduction
Antitumor monoclonal antibody (mAb) therapy holds great promise as a targeted anticancer therapeutic approach and has become more widely used in clinical practice (1, 2). The mechanisms by which antitumor mAbs inhibit or kill tumor cells are diverse and may include inhibition of growth factor receptor function, antibody-mediated cellular cytotoxicity, and complement-dependent cytotoxicity. Antitumor mAbs can also effect the delivery of cytotoxic payloads such as radioisotopes. Complement-dependent cytotoxicity has not been thought to play a crucial role in the antitumor effect elicited by most antitumor mAbs due to overexpression of membrane C regulatory proteins (mCRP) on most tumor cells. However, most therapeutic chimeric or humanized mAbs are of the immunoglobulin G1 (IgG1) isotype and can effectively activate C, resulting in C3b deposition and subsequent formation of the opsonin iC3b on the surface of tumor cells. Furthermore, iC3b on tumor cells engages complement receptor 3 (CR3; CD11b/CD18, αMβ2 integrin, Mac-1) on the surface of effector cells, eliciting CR3-dependent cellular cytotoxicity in the presence of the yeast cell wall β-glucan (3, 4). Our previous studies have shown that dual occupancy of CR3 by iC3b and β-glucan leads to the activation of Syk and phosphatidylinositol 3-kinase pathway in phagocytic cells (5). Moreover, C activation results in the release of the chemotactic factors such as C3a and C5a, which can recruit effector cells including natural killer (NK) cells and granulocytes into the tumor.
β-Glucans are glucose polymers derived from a variety of plants and microorganisms. Yeast-derived β-glucans are long polymers of β(1,3) glucose, with 3% to 6% of the backbone glucose units possessing a β(1,6) branch (6). Previous studies have shown significant therapeutic efficacy of yeast-derived β-glucan when it is coadministered with antitumor mAbs or naturally occurring antitumor antibodies in a variety of syngeneic murine tumor models (5, 7–10). In addition, barley β-glucan synergizes with humanized antitumor mAbs for cancer therapy in xenograft models (11–13). These emerging data clearly show that β-glucans can enhance the efficacy of antitumor mAb therapy and suggest that the mAb/β-glucan combination might be clinically effective.
In animal models, tumor regression and enhanced survival mediated by β-glucan immunotherapy require serum C3 and granulocyte CR3 (7, 8). There is also evidence that in the antitumor effects, neutrophils are the predominant effector cells (8). Moreover, neutrophil recruitment was shown to be dependent on leukotriene B4–amplified C5a-mediated chemotaxis (10). In addition, poly-(1,6)-β-D-glucopyranosyl-(1,3)-β-D-glucopyranose (PGG) β-glucan has shown direct effect on neutrophil chemotaxis in vivo and also up-regulates neutrophil chemotaxis toward C5a (14, 15). Together, these studies support a pivotal role for C activation and neutrophil chemotaxis in combined mAb/β-glucan immunotherapy.
Membrane complement regulatory proteins inhibit C activity at different stages such as inhibition of C3 or C5 convertase formation or blockade of membrane attack complex formation. Up-regulation of mCRPs on most human carcinomas indicates that circumvention of C-mediated tumoricidal activity or tumor surveillance may be one of the mechanisms of tumor evasion (16). These molecules include CD46 (membrane cofactor protein), CD55 (decay-accelerating factor), and CD59. CD46 promotes C3b and C4b inactivation by factor I whereas CD59 prevents formation of the membrane attack complex. CD55 is a glycosylphosphatidylinositol-anchored membrane protein and plays a critical role in adaptive T-cell immunity (17, 18). CD55 inhibits C activation via displacement of C2a from C4b and of Bb from C3b, thereby interfering with the function of C3 and C5 convertase in both the classic and alternative pathways (19). This activity has a significant effect not only on C3b/C5b-initiated progression of C activation but also on the local release of chemotactic factors C3a and C5a.
Here, we hypothesized that inhibition of mCRPs could add to the efficacy of combined β-glucan and antitumor mAb therapy because both C activation and C-dependent neutrophil chemotaxis are required for its therapeutic efficacy. This study reaffirms a critical role for mCRPs in limiting C-dependent antitumor effector mechanisms and has identified mCRP inhibition as a means to enhance β-glucan and antitumor mAb immunotherapy. Moreover, this study identifies that blockade of CD55 enhances β-glucan–mediated CR3-dependent cellular cytotoxicity at two stages: iC3b deposition on tumor cells and recruitment of CR3+ neutrophils primed with β-glucan.
Materials and Methods
Antibodies and therapeutic β-glucan
Antihuman CD46-FITC (E4.3) was purchased from GeneTex, Inc. Antihuman CD55-FITC (1A10), biotin-labeled antimouse C5a (I52-1486), purified anti-C5a antibody, and appropriately labeled isotype controls were purchased from BD PharMingen. Antimouse and antihuman C3-FITC antibodies were purchased from Cappel. Biotin-labeled antimouse Gr-1 mAb (RB6-8C5) was purchased from eBioscience (San Diego, CA). Blocking antihuman CD46 mAb (J4.48) was purchased from Chemicon. Anti-CD55 hybridoma (HD1A) was generously provided by Dr. Harris (Cardiff University Complement Biology Group, Cardiff, United Kingdom; ref. 20). The humanized mAb against Her-2 (Herceptin) was produced by Genentech and anti–epidermal growth factor receptor (EGFR) antibody was produced by ImClone Systems, Inc. Therapeutic soluble PGG β-glucan was obtained from Biothera, Inc.
Preparation of F(ab′)2 fragment of anti-CD55 mAb (HD1A)
The F(ab′)2 fragment of anti-CD55 mAb was prepared with Pierce ImmunoPure* F(ab′)2 Preparation Kit. In brief, anti-CD55 mAb was incubated with immobilized pepsin and undigested fragments were removed by protein A chromatography. The products were confirmed by reduced and nonreduced PAGE gel.
Mice and tumor models
Fox Chase ICR severe combined immunodeficient (SCID) mice were purchased from Taconic. Pilot experiment showed that these mice do not have any defect on the complement system. The murine tumor therapy protocols were done in compliance with all guidelines and were approved by the Institutional Animal Care and Use Committee of the University of Louisville. For the SKOV-3 xenograft model, 6- to 8-week-old SCID mice were implanted s.c. in a mammary fatpad with 10 × 106 SKOV-3 cells. When a palpable tumor was observed, animals were divided into groups (n = 7) and given the humanized anti–Her-2 mAb (0.2 mg i.v. every third day) with or without soluble PGG β-glucan (1.2 mg i.v. every third day). PBS-treated and PGG only–treated animals served as controls. For the NCI-H23 xenograft model, the similar protocol described above was used except the therapeutic mAb. In this model, the humanized anti-EGFR mAb (0.15 mg i.v. every third day) was used. In experiments with combined anti-CD55 mAb with anti–Her-2/neu antibody and PGG β-glucan, animals were divided into groups (n = 8 or 9) and received treatment with anti-CD55 (0.2 mg i.v. every third day), with or without anti–Her-2/neu antibody plus PGG β-glucan. Therapy was continued for up to 4 weeks, during which tumor measurements by calipers were calculated as the average of perpendicular diameters twice weekly. Mice were sacrificed when tumors reached 12 mm in diameter. In some experiments, survival was monitored up to 100 or 150 days beyond tumor implantation.
Immunohistochemical staining of tumors for neutrophils and C5a
Tumors were excised and snap-frozen in tissue freezing medium (optimum cutting temperature compound, Sakura Finetechnical Co., Ltd.). Tissue blocks were cut and fixed with cold acetone. To detect tumor-infiltrating neutophils or released C5a, the sections were blocked with 3% bovine serum albumin (BSA) buffer and incubated with an avidin/biotin blocking kit (Vector Laboratories, Inc.) and then stained with anti–Gr-1-biotin or anti-C5a-biotin for 1 h at room temperature. After three washes with blocking buffer, the sections were stained with streptavidin-horseradish peroxidase (Southern Biotechnology Associates) for 1 h at room temperature. After additional washes, horseradish peroxidase substrate (Vector Laboratories) was added for 30 min at room temperature. Following additional three washes, the sections were counterstained with hematoxylin. The number of infiltrating neutrophils was calculated as the mean of the number of Gr-1–positive cells in 10 representative high-power fields (total magnification, ×400).
mCRP expression on tumor cells
For in vitro cell staining, SKOV-3 or NCI-H23 cells were harvested and Fc receptors were blocked by incubation with anti-CD32/CD16 mAb. The cells were then stained with anti-CD46-FITC or anti-CD55-FITC mAbs and then were analyzed by flow cytometry. For tumor staining, SKOV-3 or NCI-H23 tumors were excised and sectioned. After blocking with 3% BSA/PBS, sections were stained with anti-CD46-FITC or anti-CD55-FITC mAbs. Images were acquired by fluorescence microscopy (Nikon Eclipse TE300 confocal cell images).
Detection of C activation
To determine whether the therapeutic antibodies can activate mouse or human C, mouse or human serum was freshly collected and kept in an ice bath. For every million tumor cells, 100-μL volume of diluted mouse (1:4) or human (1:10) serum containing 10 μg/mL working dilution of therapeutic mAbs was used. In some experiments, human C5-depleted sera (Quidel) were used to avoid cell killing during the process. Tumor cells were mixed and incubated at 37°C for 30 min. Cells were washed twice in ice-cold flow cytometry staining buffer and the tumor cell pellet was resuspended in 100 μL of diluted detecting antibody (goat anti-mouse or anti-human C3-FITC). Cells were incubated on ice for 30 min, washed twice as above, and propidium iodide was used to exclude the dead cells. For the C deposition on tumor cells after anti-CD46 and anti-CD55 mAb blockade, tumor cells were incubated with 10 μg/mL anti-CD46 or intact anti-CD55 mAb or the F(ab′)2 fragment of anti-CD55 mAb, respectively, for 1 h on ice before serum was added.
In vitro β-glucan–mediated cellular cytotoxicity assay
In vitro cytotoxicity of SKOV-3 cells by β-glucan–primed human neutrophils was analyzed using a real-time measure of the impedance of electrical current by viable target cells adhered to a conductor on the bottom of wells in a 16-well plate (Acea Biosciences) according to manufacturer’s instruction (21) and our previous publication (5).
Measurement of C5a level in serum
Sera from different groups in SKOV-3 xenograft model were collected after the last treatment and were measured for C5a by ELISA. The purified rat anti-mouse C5a was used as a capture antibody and was paired with biotinylated anti-mouse C5a as the detection antibody. Recombinant mouse C5a protein was used as a standard.
Statistical analysis
Data from mouse therapy protocols were entered into Prism 4.0 (GraphPad Software) to generate graphs of tumor regression or survival and to determine the significance of differences between data sets. Student’s t test was used to compare differences between two tumor regression curves, whereas the log-rank test was used to determine the significance of differences between two survival curves.
Results
Combined β-glucan and antitumor mAb therapy for the treatment of human ovarian carcinoma and human non–small-cell lung carcinoma
To facilitate translation of β-glucan/mAb therapy from preclinical models to clinical application, two human xenograft models were established in SCID mice. SKOV-3 is a human ovarian carcinoma cell line that expresses high levels of Her-2/neu and the human non–small-cell lung carcinoma (NSCLC) cell line NCI-H23 expresses abundant EGFR. Both cell lines form solid tumors when implanted s.c. in SCID mice.
Preliminary in vitro studies showed that both humanized anti–Her-2/neu antibody (trastuzumab) and anti-EGFR antibody (cetuximab) are able to activate mouse C leading to iC3b deposition on tumor cell surfaces (data not shown). Several pilot studies were carried out to titrate the inoculums of s.c. SKOV-3 and NCI-H23 required to produce a palpable tumor within ~7 to 10 days and this was found to be 10 × 106 cells for both lines. Furthermore, in vivo titration of anti–Her-2/neu mAb and anti-EGFR mAb indicated that administration of 0.2 mg (anti–Her-2/neu) or 0.15 mg (anti-EGFR) mAbs i.v. twice a week resulted in detectable iC3b on the surface of excised tumors 6 days later.
Based on these preliminary findings, groups of mice were implanted with 10 × 106 SKOV-3 or NCI-H23 cells and randomized to receive either 0.2 mg trastuzumab or 0.15 mg cetuximab i.v. twice a week or mAb i.v. in combination with 1.2 mg soluble PGG β-glucan i.v. twice a week or 1.2 mg PGG β-glucan only or PBS control. As indicated in Fig. 1A, anti–Her-2/neu monotherapy failed to achieve a significant reduction in tumor burden (P = 0.175, versus no treatment). In addition, combined therapy with soluble PGG β-glucan and anti–Her-2/neu antibody failed to cause significant tumor regression compared with treatment with mAb alone (P = 0.45). However, combined therapy was able to achieve a reduction in tumor growth compared with nontreated animals (P = 0.0025). Despite this reduction in tumor growth rate, the animals receiving combined therapy were not observed to have enhanced survival (Fig. 1B).
Figure 1.

The tumoricidal activity of immunotherapy with PGG β-glucan in combination with humanized antitumor mAbs. A and B, ICR SCID mice (n = 7) were implanted s.c. with SKOV-3 cells and tumors were allowed to form over 10 d before therapy. Mice received PBS, humanized anti–Her-2/neu antibody (0.2 mg every third day) with or without PGG β-glucan (1.2 mg twice a week), or β-glucan only for 4 wks. Both tumor growth (A) and survival (B) were monitored. C and D, similar protocol except that ICR SCID mice (n = 8 or 9) were implanted with NCI-H23 cells. Mice received PBS, humanized anti-EGFR antibody (0.15 mg every third day) with or without PGG β-glucan, or β-glucan only for 4 wks. Both tumor growth (C) and survival (D) were monitored. Tumor measurements were made at the indicated time. Mice were sacrificed when the tumors reached 12 mm in diameter. Points, mean; bars, SE.
Mice implanted with NCI-H23 human NSCLC that received anti-EGFR mAb alone also did not exhibit significant tumor regression with respect to nontreated animals (P = 0.177). However, animals receiving combination therapy with PGG β-glucan, in addition to anti-EGFR mAb, displayed significant tumor regression compared with animals receiving mAb alone or untreated PBS control (Fig. 1C). More importantly, animals receiving combined therapy were also observed to have significantly enhanced survival, with 80% of mice surviving >150 days after tumor implantation (Fig. 1D).
Neutrophil infiltration and C5a release in tumors
Our previous studies indicated that neutrophils are the predominant effector cells in β-glucan immunotherapy (8, 10). To determine whether the difference in observed therapeutic efficacy between the two different human carcinomas was due to differences in the presence and/or activity of effector cells, tumors from animals in both protocols were excised for immunohistochemical staining. Immunohistochemistry analysis of tumor-infiltrating neutrophils showed a relative paucity of Gr-1+ cells in SKOV-3 tumors (Fig. 2A). In contrast, massive neutrophil infiltration was observed in NCI-H23 tumors (Fig. 2A). The number of infiltrating Gr-1+ neutrophils in SKOV-3 tumors was much less than that in NCI-H23 tumors (Fig. 2B), suggesting that SKOV-3 tumors had likely established an immunosuppressive mechanism against the influx of phagocytes.
Figure 2.
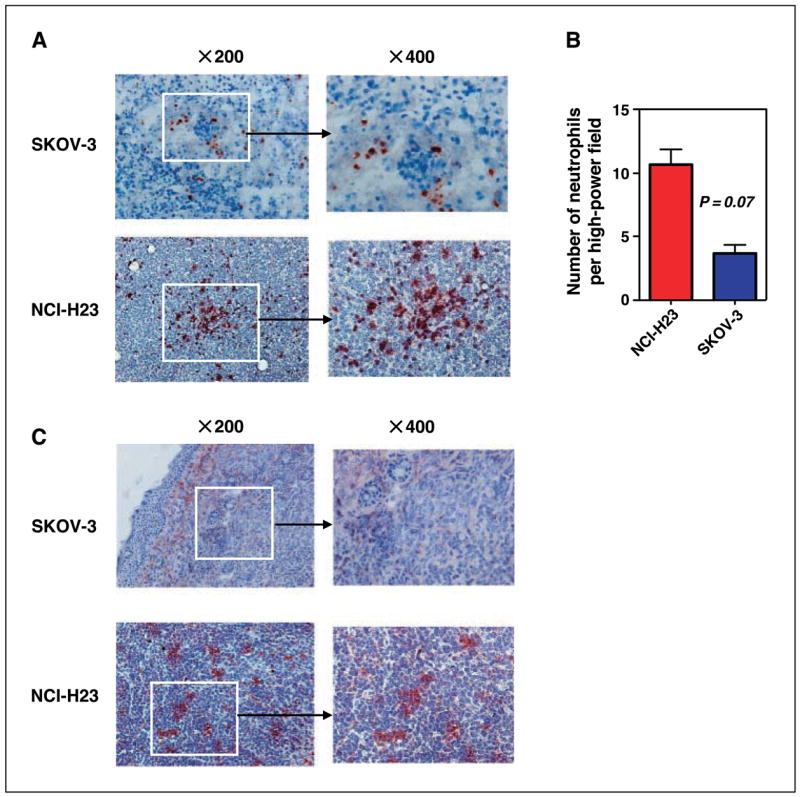
SKOV-3 tumors show a paucity of infiltrating neutrophils and decreased C5a release as compared with NCI-H23 tumors. Tumor masses from SCID tumor-bearing mice treated with humanized anti–Her-2/neu or anti-EGFR antibody, as described in Fig. 1, were cryosectioned and stained with anti–Gr-1 mAb or anti-C5a mAb. A, poor neutrophil infiltration was observed in SKOV-3 tumors and massive neutrophil infiltration was observed in NCI-H23 tumors. B, quantitative summary of the neutrophil infiltrate measured as the mean number of Gr-1+ cells in 10 representative high-power fields. C, a lack of C5a was observed in SKOV-3 tumors whereas abundant C5a was exhibited in NCI-H23 tumors. Representative tumor section of eight or nine total tumor specimens. Original magnification, ×200 or ×400.
C activation by naturally occurring tumor antitumor antibodies or exogenous antitumor mAbs not only targets tumor cells with covalently bound iC3b but also releases the chemotactic factors C3a and C5a. Neutrophils express more C5aR (CD88) than C3aR. Our previous results showed that chemotaxis of granulocytes into tumors is mediated by C5a but not by C3a (10). Because many fewer neutrophils were observed in SKOV-3 tumors with respect to NCI-H23 tumors, we hypothesized that C5a release might be blocked in SKOV-3 tumors. To test this, both SKOV-3 and NCI-H23 tumors were stained for C5a. Indeed, C5a production was significantly lower within SKOV-3 tumors with respect to NCI-H23 tumors (Fig. 2C). Taken together, these data suggest that therapeutic failure of β-glucan and antitumor mAb in SKOV-3 tumors is due to the loss of C5a-mediated neutrophil chemotaxis into the tumors.
Overexpression of mCRPs on SKOV-3 tumors prevents effective β-glucan/mAb immunotherapy
It has been shown that the C activation cascade is regulated by mCRPs in plasma or on the cell surface, functioning in normal conditions to prevent uncontrolled C activation. Overexpression of mCRPs by human tumors has been widely reported (16) and may be an important cause of diminished C5a production in tumors. CD46 can promote inactivation of C3b and C4b by factor I ( forming iC3b and C4d, respectively) whereas CD55 displaces Bb from C3b and C2a from C4b, thereby interfering with C3 and C5 activation. Together, CD46 and CD55 play an important role in controlling C3b deposition, iC3b formation, and C5a release, events which are critical for β-glucan–mediated immunotherapy. SKOV-3 and NCI-H23 cells were assayed for the expression of the mCRPs CD46 and CD55. As shown in Fig. 3A, SKOV-3 cells overexpress both mCRPs and display a 1- to 1.5-log shift in staining intensity by fluorescence-activated cell sorting analysis compared with cells stained with a control mAb. In contrast, NCI-H23 cells express lesser amounts of CD46 and CD55. To confirm this difference, both tumor types were excised and stained with fluorescent-labeled anti-mCRPs. As shown in Fig. 3B, SKOV-3 tumors exhibited much stronger CD46 and CD55 expression compared with NCI-H23 tumors.
Figure 3.

The expression of CD46 and CD55 on human ovarian and NSCLC carcinomas. A, human SKOV-3 and NCI-H23 cells were stained with anti-CD46 or anti-CD55-FITC mAbs. Gray histogram, staining from an isotype control. Right, columns, mean fluorescence intensity (MFI). Both CD46 and CD55 were overexpressed on SKOV-3 cells with respect to that on NCI-H23 cells (P < 0.05). B, SKOV-3 tumors and NCI-H23 tumors were excised, sectioned, and stained with anti-CD46 or anti-CD55-FITC mAbs. The images were acquired by fluorescent microscopy. Representative tumor section of five total tumor specimens. Original magnification, ×200.
These results suggested that blocking mCRPs on SKOV-3 cells might lead to augmented C3 activation and iC3b deposition on tumors. Neither anti-CD46 nor anti-CD55 mAb activated human or mouse C (Fig. 4A and data not shown), indicating that detected iC3b was the result of C activation by antitumor mAb. Inhibition of CD55 with neutralizing mAb enhanced the deposition of either human or mouse C3b mediated through C activation by anti–Her-2/neu antibody (Fig. 4B). To further confirm that anti-CD55 mAb is a blocking mAb rather than a C-activating mAb, the F(ab′)2 fragment of anti-CD55 mAb was generated. As shown in Fig. 4C, the F(ab′)2 fragment of HD1A mAb and the intact IgG similarly enhanced iC3b-deposition on SKOV-3 tumor cells mediated by anti–Her-2/neu antibody (Fig. 4C). In addition, CD55 neutralization also significantly enhanced cytotoxicity of iC3b-opsonized SKOV-3 cells mediated by β-glucan–primed neutrophil effector cells (Fig. 4D). However, blocking CD46 had minimal effect on C3b deposition on target cells and did not promote β-glucan–primed neutrophil–mediated tumor cell cytotoxicity (Fig. 4D).
Figure 4.
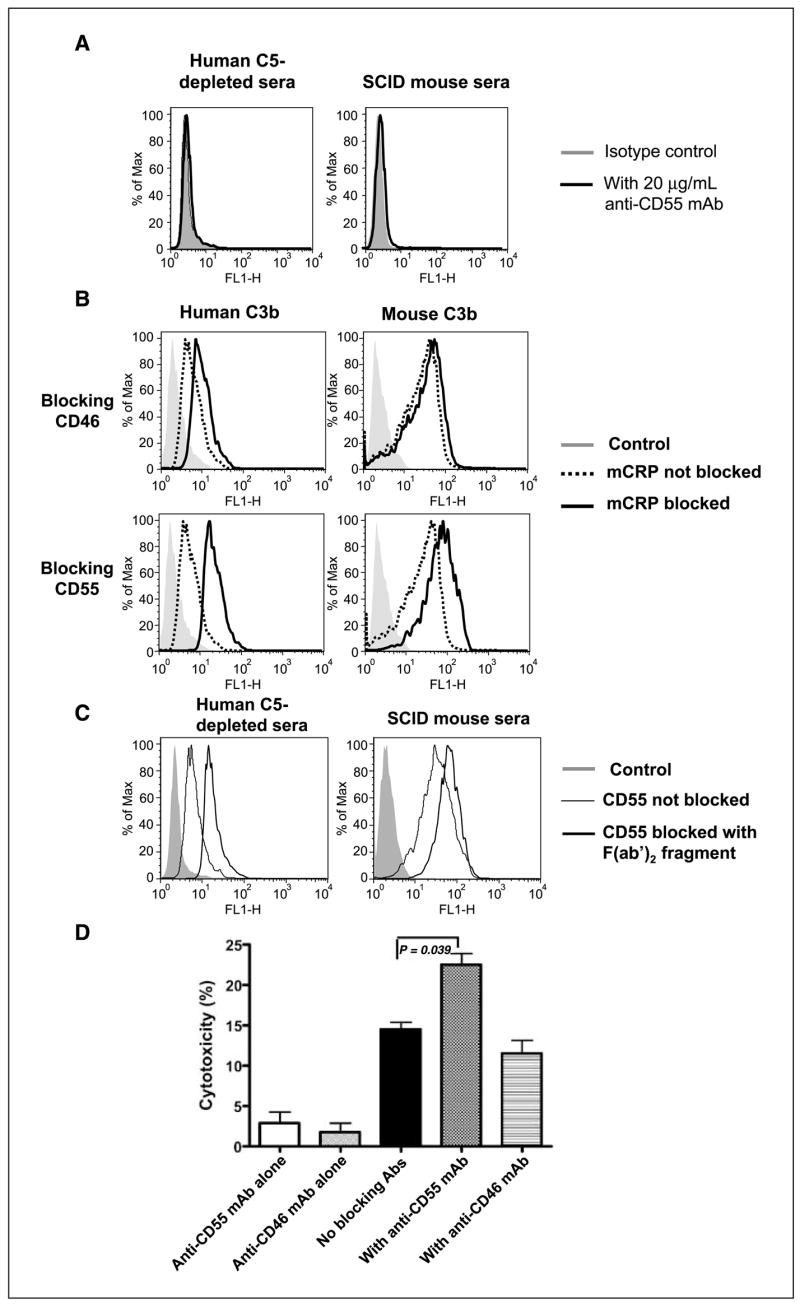
Inhibition of human CD55, but not CD46, significantly enhances the deposition of human and mouse C3b on the surface of SKOV-3 cells as well as β-glucan–mediated CR3-dependent cellular cytotoxicity. A, SKOV-3 cells were incubated with anti-CD55 mAb plus mouse or C5-depleted human sera and stained with anti-mouse or anti-human C3-FITC antibody. Data suggest that anti-CD55 mAb does not activate mouse or human C. B, SKOV-3 cells were incubated with anti–Her-2/neu antibody plus fresh human or mouse serum in the presence or absence of antihuman CD46 or CD55 mAb and stained with anti-C3-FITC antibody for the detection of C3b. Gray histogram, an isotype control. Dotted line, C3b deposition in the absence of mCRP blockade. Bold line, C3b deposition in the presence of mCRP blockade. C, similar protocol was done as described in (B) except that the F(ab′)2 fragment of anti-CD55 mAb was added. Data suggest that the F(ab′)2 fragment of anti-CD55 mAb exhibits comparable levels of enhancement of iC3b deposition on SKOV-3 cells mediated by anti–Her-2/neu antibody. D, in vitro cytotoxicity experiments suggested that inhibition of the mCRP CD55 with an inhibitory mAb could enhance CR3-dependent cellular cytotoxicity.
Inhibition of CD55 in conjunction with combined β-glucan and antitumor mAb immunotherapy significantly reduces tumor burden and leads to long-term survival
Having shown that CD55 suppresses both C3 activation and β-glucan–mediated CR3-dependent cellular cytotoxicity in vitro, we explored the therapeutic efficacy of in vivo blockade of CD55 with neutralizing mAb along with β-glucan/mAb therapy in SKOV-3 tumors. In this xenograft model, groups of mice with palpable SKOV-3 tumors received (a) no treatment (PBS injections), (b) anti–Her-2/neu alone, (c) anti-CD55 mAb alone, (d) both mAbs, (e) anti-CD55 mAb with PGG β-glucan, (f ) anti–Her-2/neu antibody with PGG β-glucan, or (g) anti–Her-2/neu antibody + anti-CD55 mAb + PGG β-glucan. After 3 weeks of therapy, mice with anti–Her-2/neu antibody, PGG β-glucan alone, anti-CD55 mAb alone, anti-CD55 mAb + anti–Her-2/neu antibody, or anti-CD55 mAb + PGG β-glucan did not have statistically smaller tumors than PBS-treated control mice (Fig. 5A and data not shown). However, mice receiving PGG β-glucan in addition to anti–Her-2/neu antibody exhibited a significantly reduced tumor burden compared with untreated animals but not statistically significant compared with anti–Her-2/neu–treated animals, comparable to the data presented in Fig. 1. Strikingly, mice receiving anti-CD55 mAb in addition to combined β-glucan therapy had significantly smaller tumors compared with combined β-glucan therapy. More importantly, 80% of these mice achieved long-term survival (Fig. 5B). These data suggest that the addition of anti-CD55 mAb to combined β-glucan immunotherapy in SKOV-3 tumors significantly enhances the regression of the SKOV-3 tumors and long-term survival compared with treatment with β-glucan and anti–Her-2/neu antibody alone.
Figure 5.
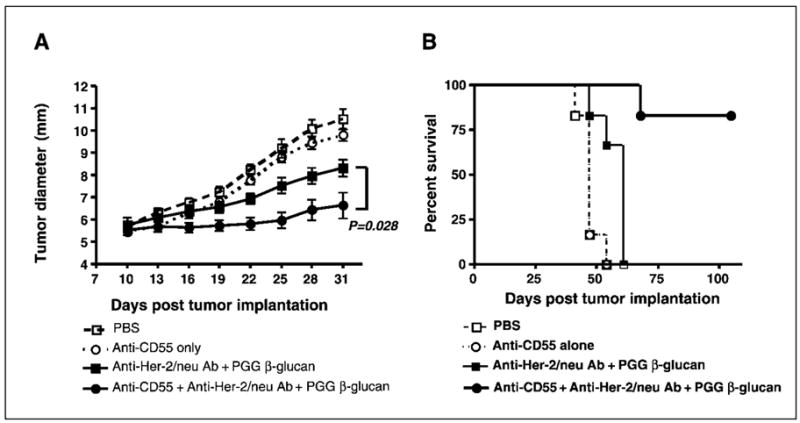
Blockade of mCRP CD55 significantly enhances combined β-glucan with humanized anti–Her-2/neu antibody therapy on SKOV-3 tumors. Having shown the importance of mCRP CD55 in iC3b deposition and CR3-dependent cellular cytotoxicity in vitro, anti-CD55 mAb was added into anti–Her-2/neu antibody plus PGG β-glucan regimens to treat SKOV-3 tumors, as described in Fig. 1. As these results indicate, the addition of anti-CD55 mAb induced significant tumor regression (A) and long-term survival (B) in animals receiving anti–Her-2/neu antibody and PGG β-glucan with respect to animals receiving anti–Her-2/neu antibody plus PGG β-glucan.
Increased neutrophil accumulation and C5a release in SKOV-3 tumors by inhibition of CD55
As shown in Fig. 2, SKOV-3 tumors had markedly decreased neutrophil infiltration and detectable C5a within tumors. To determine whether inhibition of CD55 would enhance C5a production thereby stimulating neutrophil influx into tumors, SKOV-3 tumors treated with different regimens were excised and immunohistochemical analysis was done. Consistent with previous observations, there was a marked absence of infiltrating neutrophils in animals treated with PBS, anti-CD55 only, anti–Her-2/neu antibody only, or combined PGG β-glucan and anti–Her-2/neu antibody (Fig. 6A). However, massive neutrophil infiltration was observed in animals receiving anti-CD55 mAb in addition to combined β-glucan/antitumor mAb therapy (Fig. 6A and B). Similarly, significantly more C5a was detected in the animals treated with anti-CD55 in addition to combined β-glucan immunotherapy (Fig. 6C). Interestingly, the serum C5a level was significantly lower in animals receiving anti-CD55 mAb treatment compared with animals without anti-CD55 mAb therapy (Fig. 6D), perhaps reflecting enhanced intratumoral C activation. Thus, blockade of CD55 by neutralizing mAb overcomes the immunosuppressive microenvironment established by SKOV-3 tumors, leading to phagocyte influx to tumors.
Figure 6.

Increased neutrophil infiltration and C5a release in SKOV-3 tumors in the presence of blocking anti-CD55 mAb. A, SKOV-3 tumors from animals receiving different treatment regimens as described in Materials and Methods were sectioned and stained with anti–Gr-1 mAb. The addition of anti-CD55 mAb to β-glucan immunotherapy significantly increased neutrophil infiltration within SKOV-3 tumors. Left, low-power fields (×200); right, representative high-power field (×400). B, quantitative summary of the neutrophil infiltrate measured as the mean number of Gr-1+ cells in 10 representative high-power fields. Representative tumor section of eight or nine total tumor specimens. C, SKOV-3 tumors from animals receiving different treatment regimens were sectioned and stained with anti-C5a mAb. The addition of CD55 mAb to combined β-glucan and anti–Her-2/neu antibody therapy led to enhanced C5a release within SKOV-3 tumors. Left, low-power fields (×200); right, representative high-power field (×400). Representative tumor section of eight or nine total tumor specimens. D, C5a level in serum as measured by ELISA was decreased when anti-CD55 mAb was administered.
Discussion
Using two human xenograft tumor models, we have found that mCRP (in particular, CD55) is capable of suppressing effective immunotherapy involving administration of β-glucan and anti-tumor mAbs. Whereas this regimen was quite effective in treatment of NCI-H23 human NSCLC xenografts, similar xenografts of SKOV-3 human ovarian carcinoma cells were very resistant to therapy. The latter seems to be due to elevated expression of CD55 on SKOV-3 cells, thus inhibiting C3 and C5 convertase activity and interfering with C3a and C5a release. Ultimately, decreased C5a results in decreased infiltration of β-glucan–primed neutrophils into the tumor microenvironment. In support, we find that inhibition of CD55 with neutralizing mAb significantly enhances iC3b deposition on SKOV-3 cells and elicits strong β-glucan–mediated CR3-dependent cellular cytotoxicity in vitro. Significant therapeutic efficacy of β-glucan/mAb administration in SKOV-3 tumors was achieved on coadministration of anti-CD55 mAb. Blockade of CD55 leads to enhanced intratumoral C5a deposition, increased neutrophil accumulation, tumor regression, and improved long-term survival.
The failure of combined β-glucan–mediated immunotherapy in SKOV-3 tumors was initially thought to be due to loss of Her-2/neu expression in vivo. However, freshly isolated SKOV-3 cells uniformly expressed similar levels of Her-2/neu as compared with in vitro cultured cells (data not shown). Both anti–Her-2/neu antibody and anti-EGFR antibody are chimeric antibodies that have been engineered in the human IgG1 framework, and therefore should activate C. However, the reported mechanism of action of trastuzumab is the inhibition of the formation of Her-2 heterodimers and internalization of the receptor (22, 23). Thus, blockade of Her-2 heterodimer formation via anti–Her-2/neu antibody is associated with decreased cell proliferation. There was no marginal benefit observed in animals treated with PGG β-glucan alone or β-glucan in combination with anti–Her-2/neu antibody with respect to antibody alone–treated animals. These data seem to confirm previous reports that the mechanism of action of trastuzumab is independent of immune effector functions including complement-dependent cytotoxicity and antibody-mediated cellular cytotoxicity. In a similar manner, cetuximab blocks its ligand binding to EGFR, Her-1, inhibiting cancer cell cycle progression and inducing tumor cell apoptosis (24). The shown therapeutic efficacy in NCI-H23 carcinoma using β-glucan in combination with anti-EGFR mAb suggests that the C system can be manipulated in such a way as to elicit effective antitumor immune responses. In addition, both anti–Her-2/neu and anti-EGFR mAbs are capable of activating mouse and human C, leading to iC3b deposition on tumor cells (data not shown). Therefore, the observed failure of β-glucan–and anti–Her-2/neu antibody–mediated immunotherapy for Her-2–overexpressing xenografts was not hypothesized to be an intrinsic fault of the mAb or β-glucan. Rather, it was hypothesized that the tumor microenvironment was inhibiting immunotherapy with β-glucan and antitumor mAb. Indeed, infiltration of neutrophils, the most important effector cells in β-glucan immunotherapy, was drastically decreased in SKOV-3 xenografts. In contrast, massive neutrophil infiltration was observed in NCI-H23 tumors. Thus, a fundamental difference between these two xenograft models was the potency of immunosuppression mediated by these two different human tumors.
To address the role of immune suppression mediated by the SKOV-3 tumor microenvironment, we sought to explore possible mechanisms by which SKOV-3 tumors block the influx of neutrophil to the tumor. C5a is a potent neutrophil chemotactic factor (25). Thus, it is not surprising to observe decreased C5a within SKOV-3 tumors, given the paucity of infiltrating neutrophils relative to the NCI-H23 tumors. C activation is regulated by a number of regulatory proteins that prevent unchecked C activation and possible autoimmunity. Indeed, suppression of mCRPs is known to exacerbate the development of autoimmune diseases (26–28). Therefore, it was hypothesized that C regulatory proteins may play a critical role in the regulation of C5a release in tumor microenvironment. Most tumor cells overexpress variable levels of mCRPS including CD46, CD55, and CD59 (29, 30). The overexpression of some of these mCRPs is regarded as a poor prognostic factor, whereas increased iC3b observed on tumors is regarded as a favorable prognostic factor (29, 31, 32). These observations may suggest an in situ role for C-mediated immunosurveillance of tumors. SKOV-3 cells were observed to overexpress CD46 and CD55. Up-regulation of CD55 on SKOV-3 cells protected SKOV-3 cells from C-mediated lysis (33). In addition, up-regulation of CD46 and CD55 would also be expected to limit the activity of the C3/C5 convertase and therefore result in decreased opsonization of tumor cells with C3b. Interestingly, the inhibition of human CD55 in SKOV-3 cells resulted in enhanced deposition of both human and mouse C3b whereas inhibition of CD46 only marginally increased C3b deposition on tumor cells. It is worth noting that in the presence of human C, a limited amount of C3b was deposited, whereas in the presence of mouse C, a large amount of C3b was deposited on SKOV-3 cells (Fig. 4B). This may suggest that human CD55 works less well against the mouse C system. Nevertheless, blocking human CD55 enhanced mouse C3b deposition on SKOV-3 cells, confirming previous reports that human and rodent CD55 are not absolutely species restricted in their C-inhibiting activities (34, 35). In addition, in vitro cytotoxicity experiments suggested that inhibition of CD55 would promote β-glucan–mediated CR3-dependent cellular cytotoxicity whereas inhibition of CD46 would provide marginal protection to tumor cells. The mechanism for this observation may be, in part, due to the role of CD46 as a cofactor for serum factor I that inactivates C3b and yields cell-bound iC3b. Because iC3b, and not C3b, is the high-affinity ligand for the I domain of CR3, it is likely that inhibition of CD46 activity results in the deposition of more C3b but in a net decrease in the amount of cell-bound iC3b, thus ameliorating β-glucan–mediated CR3-dependent cellular cytotoxicity. In addition, recent studies suggest that CD46 acts preferentially to inhibit the alternative pathway of C activation (36, 37). Nevertheless, it was of great interest to explore the role of in vivo inhibition of mCRPs in the setting of antitumor mAb and β-glucan immunotherapy, particularly in the SKOV-3 tumor model that had shown failure previously.
Therefore, blockade of CD55 with neutralizing anti-CD55 mAb was used in SKOV-3 tumor therapy, in addition to β-glucan and anti–Her-2/neu antibody therapy, in xenograft models. Although mCRPs need to work across the species boundary in xenograft models (29, 38), our data clearly showed regulatory function of human CD55 for mouse complement system in vitro. Furthermore, we showed that the addition of anti-CD55 mAb to the combined β-glucan and anti–Her-2/neu antibody therapy in SKOV-3 cells yielded significant enhanced tumor regression and long-term survival with respect to the β-glucan– and anti–Her-2/neu antibody–treated animals. The enhanced therapeutic efficacy is associated with increased influx of β-glucan–primed neutrophils into SKOV-3 tumors. In addition, locally produced C5a was observed in tumors. The enhanced C activation in SKOV-3 tumors leads to locally produced C5a thereby recruiting β-glucan–primed neutrophils to the tumor microenvironment. Thus, blockade of mCRPs, especially CD55, may synergize with antitumor mAbs to increase β-glucan–mediated CR3-dependent cellular cytotoxicity, thereby enhancing tumor immunotherapy. Indeed, both anti-CD55 mAb and CD55 small interfering RNA enhance complement-dependent cytotoxicity mediated by antitumor mAb in vitro (39–41). Interestingly, the chemotherapeutic drug fludarabine down-regulates CD55 expression on tumor cells (42). This may well explain the synergistic cytotoxicity of fludarabine and anti-CD20 mAb (rituximab) in a follicular lymphoma cell line (42). However, one concern on the use of CD55 mAb blockade in vivo is expression of CD55 on normal tissues or cells such as RBC (19). This could potentially lead to hemolytic or vascular disease as a result of increased C activation on normal cells or targeting by antibody-mediated cellular cytotoxicity. This drawback may be overcome by using bispecific mAb against tumor antigen with higher affinity and CD55 with lower affinity. A previous study has shown that this strategy could specifically target tumor cells with minimal binding to normal cells and increase β-glucan–mediated CR3-dependent cellular cytotoxicity (43). Indeed, bispecific mAb to epithelial cell adhesion molecule and Crry in rat has shown a significant therapeutic efficacy for a rat colorectal cancer lung metastasis model in vivo (44). Moreover, a recent study showed that CD55 is highly expressed on tumor cells but not on nonneoplastic epithelia, suggesting that it is feasible to predominately target tumor CD55 (45).
In summary, these observations underscore the importance of the tumor milieu in the setting of antitumor immunotherapy. Indeed, it seems that a more complete understanding of the tumor microenvironment and its potential for local immunosuppression, such as up-regulation of mCRPs, is required for the ultimate success of tumor immunotherapy strategies, including β-glucan–mediated immunotherapy.
Acknowledgments
Grant support: NIH/National Cancer Institute grant R01 CA86412, the Kentucky Lung Cancer Research Board, the James Graham Brown Cancer Center Pilot Project Program, and a gift fund from Biothera.
We thank Dr. John W. Eaton for his critical reading and editing of the manuscript.
Footnotes
Note: B. Li and D.J. Allendorf contributed equally to this work.
References
- 1.Adams GP, Weiner LM. Monoclonal antibody therapy of cancer. Nat Biotechnol. 2005;23:1147–57. doi: 10.1038/nbt1137. [DOI] [PubMed] [Google Scholar]
- 2.Ross JS, Schenkein DP, Pietrusko R, et al. Targeted therapies for cancer 2004. Am J Clin Pathol. 2004;122:598–609. doi: 10.1309/5CWP-U41A-FR1V-YM3F. [DOI] [PubMed] [Google Scholar]
- 3.Thornton BP, Vetvicka V, Pitman M, Goldman RC, Ross GD. Analysis of the sugar specificity and molecular location of the β-glucan-binding lectin site of complement receptor type 3 (CD11b/CD18) J Immunol. 1996;156:1235–46. [PubMed] [Google Scholar]
- 4.Xia Y, Vetvicka V, Yan J, et al. The β-glucan-binding lectin site of mouse CR3 (CD11b/CD18) and its function in generating a primed state of the receptor that mediates cytotoxic activation in response to iC3b-opsonized target cells. J Immunol. 1999;162:2281–90. [PubMed] [Google Scholar]
- 5.Li B, Allendorf DJ, Hansen R, et al. Yeast β-glucan amplifies phagocyte killing of iC3b-opsonized tumor cells via complement receptor 3-Syk-phosphatidylinositol 3-kinase pathway. J Immunol. 2006;177:1661–9. doi: 10.4049/jimmunol.177.3.1661. [DOI] [PubMed] [Google Scholar]
- 6.Yan J, Allendorf DJ, Brandley B. Yeast whole glucan particle (WGP) β-glucan in conjunction with antitumour monoclonal antibodies to treat cancer. Expert Opin Biol Ther. 2005;5:691–702. doi: 10.1517/14712598.5.5.691. [DOI] [PubMed] [Google Scholar]
- 7.Yan J, Vetvicka V, Xia Y, et al. β-Glucan, a “specific” biologic response modifier that uses antibodies to target tumors for cytotoxic recognition by leukocyte complement receptor type 3 (CD11b/CD18) J Immunol. 1999;163:3045–52. [PubMed] [Google Scholar]
- 8.Hong F, Hansen RD, Yan J, et al. β-Glucan functions as an adjuvant for monoclonal antibody immunotherapy by recruiting tumoricidal granulocytes as killer cells. Cancer Res. 2003;63:9023–31. [PubMed] [Google Scholar]
- 9.Hong F, Yan J, Baran JT, et al. Mechanism by which orally administered β-1,3-glucans enhance the tumoricidal activity of antitumor monoclonal antibodies in murine tumor models. J Immunol. 2004;173:797–806. doi: 10.4049/jimmunol.173.2.797. [DOI] [PubMed] [Google Scholar]
- 10.Allendorf DJ, Yan J, Ross GD, et al. C5a-mediated leukotriene B4-amplified neutrophil chemotaxis is essential in tumor immunotherapy facilitated by anti-tumor monoclonal antibody and β-lucan. J Immunol. 2005;174:7050–6. doi: 10.4049/jimmunol.174.11.7050. [DOI] [PubMed] [Google Scholar]
- 11.Cheung NK, Modak S. Oral (1→3),(1→4)-β-D-glucan synergizes with antiganglioside GD2 monoclonal antibody 3F8 in the therapy of neuroblastoma. Clin Cancer Res. 2002;8:1217–23. [PubMed] [Google Scholar]
- 12.Cheung NK, Modak S, Vickers A, Knuckles B. Orally administered β-glucans enhance anti-tumor effects of monoclonal antibodies. Cancer Immunol Immunother. 2002;51:557–64. doi: 10.1007/s00262-002-0321-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Modak S, Koehne G, Vickers A, O’Reilly RJ, Cheung NK. Rituximab therapy of lymphoma is enhanced by orally administered (1→3),(1→4)-D-β-glucan. Leuk Res. 2005;29:679–83. doi: 10.1016/j.leukres.2004.10.008. [DOI] [PubMed] [Google Scholar]
- 14.LeBlanc BW, Albina JE, Reichner JS. The effect of PGG-β-glucan on neutrophil chemotaxis in vivo. J Leukoc Biol. 2006;79:667–75. doi: 10.1189/jlb.0305150. [DOI] [PubMed] [Google Scholar]
- 15.Tsikitis VL, Albina JE, Reichner JS. β-Glucan affects leukocyte navigation in a complex chemotactic gradient. Surgery. 2004;136:384–9. doi: 10.1016/j.surg.2004.05.014. [DOI] [PubMed] [Google Scholar]
- 16.Fishelson Z, Donin N, Zell S, Schultz S, Kirschfink M. Obstacles to cancer immunotherapy: expression of membrane complement regulatory proteins (mCRPs) in tumors. Mol Immunol. 2003;40:109–23. doi: 10.1016/s0161-5890(03)00112-3. [DOI] [PubMed] [Google Scholar]
- 17.Liu J, Miwa T, Hilliard B, et al. The complement inhibitory protein DAF (CD55) suppresses T cell immunity in vivo. J Exp Med. 2005;201:567–77. doi: 10.1084/jem.20040863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Heeger PS, Lalli PN, Lin F, et al. Decay-accelerating factor modulates induction of T cell immunity. J Exp Med. 2005;201:1523–30. doi: 10.1084/jem.20041967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lublin DM, Atkinson JP. Decay-accelerating factor: biochemistry, molecular biology, and function. Annu Rev Immunol. 1989;7:35–58. doi: 10.1146/annurev.iy.07.040189.000343. [DOI] [PubMed] [Google Scholar]
- 20.Harris CL, Lublin DM, Morgan BP. Efficient generation of monoclonal antibodies for specific protein domains using recombinant immunoglobulin fusion proteins: pitfalls and solutions. J Immunol Methods. 2002;268:245–58. doi: 10.1016/s0022-1759(02)00207-7. [DOI] [PubMed] [Google Scholar]
- 21.Solly K, Wang X, Xu X, Strulovici B, Zheng W. Application of real-time cell electronic sensing (RT-CES) technology to cell-based assays. Assay Drug Dev Technol. 2004;2:363–72. doi: 10.1089/adt.2004.2.363. [DOI] [PubMed] [Google Scholar]
- 22.Gordon MS, Matei D, Aghajanian C, et al. Clinical activity of pertuzumab (rhuMAb 2C4), a HER dimerization inhibitor, in advanced ovarian cancer: potential predictive relationship with tumor HER2 activation status. J Clin Oncol. 2006;24:4324–32. doi: 10.1200/JCO.2005.05.4221. [DOI] [PubMed] [Google Scholar]
- 23.Kumar Pal S, Pegram M. Targeting HER2 epitopes. Semin Oncol. 2006;33:386–91. doi: 10.1053/j.seminoncol.2006.04.004. [DOI] [PubMed] [Google Scholar]
- 24.Govindan R. Cetuximab in advanced non-small cell lung cancer. Clin Cancer Res. 2004;10:4241–4s. doi: 10.1158/1078-0432.CCR-040015. [DOI] [PubMed] [Google Scholar]
- 25.Binder R, Kress A, Kan G, Herrmann K, Kirschfink M. Neutrophil priming by cytokines and vitamin D binding protein (Gc-globulin): impact on C5a-mediated chemotaxis, degranulation and respiratory burst. Mol Immunol. 1999;36:885–92. doi: 10.1016/s0161-5890(99)00110-8. [DOI] [PubMed] [Google Scholar]
- 26.Jha P, Sohn JH, Xu Q, et al. Suppression of complement regulatory proteins (CRPs) exacerbates experimental autoimmune anterior uveitis (EAAU) J Immunol. 2006;176:7221–31. doi: 10.4049/jimmunol.176.12.7221. [DOI] [PubMed] [Google Scholar]
- 27.Lin F, Kaminski HJ, Conti-Fine BM, et al. Markedly enhanced susceptibility to experimental autoimmune myasthenia gravis in the absence of decay-accelerating factor protection. J Clin Invest. 2002;110:1269–74. doi: 10.1172/JCI16086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lin F, Emancipator SN, Salant DJ, Medof ME. Decay-accelerating factor confers protection against complement-mediated podocyte injury in acute nephrotoxic nephritis. Lab Invest. 2002;82:563–9. doi: 10.1038/labinvest.3780451. [DOI] [PubMed] [Google Scholar]
- 29.Gelderman KA, Tomlinson S, Ross GD, Gorter A. Complement function in mAb-mediated cancer immunotherapy. Trends Immunol. 2004;25:158–64. doi: 10.1016/j.it.2004.01.008. [DOI] [PubMed] [Google Scholar]
- 30.Niehans GA, Cherwitz DL, Staley NA, Knapp DJ, Dalmasso AP. Human carcinomas variably express the complement inhibitory proteins CD46 (membrane cofactor protein), CD55 (decay-accelerating factor), and CD59 (protectin) Am J Pathol. 1996;149:129–42. [PMC free article] [PubMed] [Google Scholar]
- 31.Watson NF, Durrant LG, Madjd Z, et al. Expression of the membrane complement regulatory protein CD59 (protectin) is associated with reduced survival in colorectal cancer patients. Cancer Immunol Immunother. 2006;55:973–80. doi: 10.1007/s00262-005-0055-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Madjd Z, Durrant LG, Pinder SE, et al. Do poor-prognosis breast tumours express membrane cofactor proteins (CD46)? Cancer Immunol Immunother. 2005;54:149–56. doi: 10.1007/s00262-004-0590-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bjorge L, Hakulinen J, Wahlstrom T, Matre R, Meri S. Complement-regulatory proteins in ovarian malignancies. Int J Cancer. 1997;70:14–25. doi: 10.1002/(sici)1097-0215(19970106)70:1<14::aid-ijc3>3.0.co;2-9. [DOI] [PubMed] [Google Scholar]
- 34.Harris CL, Spiller OB, Morgan BP. Human and rodent decay-accelerating factors (CD55) are not species restricted in their complement-inhibiting activities. Immunology. 2000;100:462–70. doi: 10.1046/j.1365-2567.2000.00066.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rees MA, Butler AJ, Negus MC, Davies HF, Friend PJ. Classical pathway complement destruction is not responsible for the loss of human erythrocytes during porcine liver perfusion. Transplantation. 2004;77:1416–23. doi: 10.1097/01.tp.0000121135.24688.a3. [DOI] [PubMed] [Google Scholar]
- 36.Liszewski MK, Leung MK, Schraml B, Goodship TH, Atkinson JP. Modeling how CD46 deficiency predisposes to atypical hemolytic uremic syndrome. Mol Immunol. 2007;44:1559–68. doi: 10.1016/j.molimm.2006.08.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Barilla-LaBarca ML, Liszewski MK, Lambris JD, Hourcade D, Atkinson JP. Role of membrane cofactor protein (CD46) in regulation of C4b and C3b deposited on cells. J Immunol. 2002;168:6298–304. doi: 10.4049/jimmunol.168.12.6298. [DOI] [PubMed] [Google Scholar]
- 38.Shin ML, Hansch G, Hu VW, Nicholson-Weller A. Membrane factors responsible for homologous species restriction of complement-mediated lysis: evidence for a factor other than DAF operating at the stage of C8 and C9. J Immunol. 1986;136:1777–82. [PubMed] [Google Scholar]
- 39.Ziller F, Macor P, Bulla R, et al. Controlling complement resistance in cancer by using human monoclonal antibodies that neutralize complement-regulatory proteins CD55 and CD59. Eur J Immunol. 2005;35:2175–83. doi: 10.1002/eji.200425920. [DOI] [PubMed] [Google Scholar]
- 40.Terui Y, Sakurai T, Mishima Y, et al. Blockade of bulky lymphoma-associated CD55 expression by RNA interference overcomes resistance to complement-dependent cytotoxicity with rituximab. Cancer Sci. 2006;97:72–9. doi: 10.1111/j.1349-7006.2006.00139.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cheung NK, Walter EI, Smith-Mensah WH, et al. Decay-accelerating factor protects human tumor cells from complement-mediated cytotoxicity in vitro. J Clin Invest. 1988;81:1122–8. doi: 10.1172/JCI113426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Di Gaetano N, Xiao Y, Erba E, et al. Synergism between fludarabine and rituximab revealed in a follicular lymphoma cell line resistant to the cytotoxic activity of either drug alone. Br J Haematol. 2001;114:800–9. doi: 10.1046/j.1365-2141.2001.03014.x. [DOI] [PubMed] [Google Scholar]
- 43.Gelderman KA, Lam S, Sier CF, Gorter A. Cross-linking tumor cells with effector cells via CD55 with a bispecific mAb induces β-glucan-dependent CR3-dependent cellular cytotoxicity. Eur J Immunol. 2006;36:977–84. doi: 10.1002/eji.200535653. [DOI] [PubMed] [Google Scholar]
- 44.Gelderman KA, Kuppen PJ, Okada N, Fleuren GJ, Gorter A. Tumor-specific inhibition of membrane-bound complement regulatory protein Crry with bispecific monoclonal antibodies prevents tumor outgrowth in a rat colorectal cancer lung metastases model. Cancer Res. 2004;64:4366–72. doi: 10.1158/0008-5472.CAN-03-2131. [DOI] [PubMed] [Google Scholar]
- 45.Ravindranath NM, Shuler C. Expression of complement restriction factors (CD46, CD55 and CD59) in head and neck squamous cell carcinomas. J Oral Pathol Med. 2006;35:560–7. doi: 10.1111/j.1600-0714.2006.00466.x. [DOI] [PubMed] [Google Scholar]