

Abstract
The structure of Escherichia coli leucine-responsive regulatory protein (Lrp) cocrystallized with a short duplex oligodeoxynucleotide reveals a novel quaternary assembly in which the protein octamer forms an open, linear array of four dimers. In contrast, structures of the Lrp homologs LrpA, LrpC and AsnC crystallized in the absence of DNA show that these proteins instead form highly symmetrical octamers in which the four dimers form a closed ring. Although the DNA is disordered within the Lrp crystal, comparative analyses suggest that the observed differences in quaternary state may arise from DNA interactions during crystallization. Interconversion of these conformations, possibly in response to DNA or leucine binding, provides an underlying mechanism to alter the relative spatial orientation of the DNA-binding domains. Breaking of the closed octamer symmetry may be a common essential step in the formation of active DNA complexes by all members of the Lrp/AsnC family of transcriptional regulatory proteins.
Keywords: transcriptional regulation, pap operon, protein-DNA interactions
Introduction
The 18.8 kDa E. coli leucine-responsive regulatory protein (Lrp) is a global transcriptional regulator that controls expression of at least 10% of the genes within the E. coli genome.1 Its homologs are widely distributed throughout the bacterial and archaeal domains. A large proportion of the regulated genes function in amino acid metabolism and transport, while a smaller number are dedicated to pili synthesis or to other cellular functions.2-4 The action of Lrp upon its target genes is sometimes regulated by binding of the small molecule effector leucine, which may either further activate or repress transcription, depending on the particular operon. Due to its high abundance in E. coli, it has been proposed that Lrp may also function as a nonspecific DNA packaging protein,5 though this role most likely is not extended to other homologs of the protein.6 In vitro, at concentrations comparable to those in E. coli (3000 dimers per cell),7 Lrp self-associates to form a mixture of octamers and hexadecamers.8 These higher order structures could potentially play an important functional role, because Lrp-regulated promoters commonly contain multiple adjacent binding sites for the protein.
Crystal structures of four Lrp homolog proteins have previously been reported. Two of these are of archaeal origin: LrpA from Pyrococcus furiosus and FL11 from Pyrococcus OT3.9,10 Very recently, crystal structures of the Escherichia coli AsnC and Bacillus subtilis LrpC homologs were also determined.11 Except for FL11, all of these Lrp homolog structures reveal an octameric organization of the protein, in which subunits are arranged as a ring of dimers around a central hole. The monomer chain contains two domains: (i) an N-terminal helix-turnhelix motif and (ii) a C-terminal α/β domain, which forms a large proportion of the dimer interface. In contrast, the Pyrococcus OT3 FL11 structure, while revealing very similar monomeric and dimeric forms of the protein, is instead organized as a cylindrical helix with 12 dimers per turn. However, cryo-electron microscopy of FL11 revealed disk-shaped moieties comparable in size to an LrpA octamer, rather than helical fibers,10 suggesting that FL11 may also organize as an octamer. None of these crystal structures were crystallized or solved in the presence of DNA. Therefore, a structural basis for Lrp binding to its target DNA sites has not been elucidated.
The pap (pyelonephritis-associated pilus) operon is one of the best-studied members of the Lrp regulon. Its regulatory region consists of a 400 base-pair DNA segment containing two divergent promoters that control genes producing the structural components and regulators of Pap pili (Figure 1A). Expression of Pap pili is regulated by a phase-variation mechanism in which bacteria switch between a phase ON state when the pili genes are expressed, and a phase OFF state when transcription is repressed.12 Lrp recognizes six binding sites within the pap regulatory region, to which it binds cooperatively to a set of three sites at any one time: either to the pBA promoter-proximal sites 1-2-3 during phase OFF (where it blocks binding of RNA polymerase), or to the pBA promoter-distal sites 4-5-6 during phase ON.13 Lrp is known to bind as a dimer to each site.14 Movement of Lrp dimers from promoter-proximal sites 1-2-3 to promoter-distal sites 4-5-6 during activation is triggered by binding of the small coregulator protein PapI, which is expressed from the divergent pI promoter (Figure 1A). Activation also depends on the methylation of DNA by DNA adenine methylase (Dam): methylation of the GATC sequence on both strands of the promoter-distal Lrp site 5 prevents activation, while methylation of GATC at the promoter-proximal Lrp site 2 is required for activation.15-18 Binding of catabolite activator protein (CAP) at a far upstream position (-215) is also required to initiate transcription19. Because the DNA elements for activation are spaced so far apart, it appears that substantial protein-induced DNA curvature and wrapping may be required to bring the required factors into juxtaposition with RNA polymerase.
Figure 1.
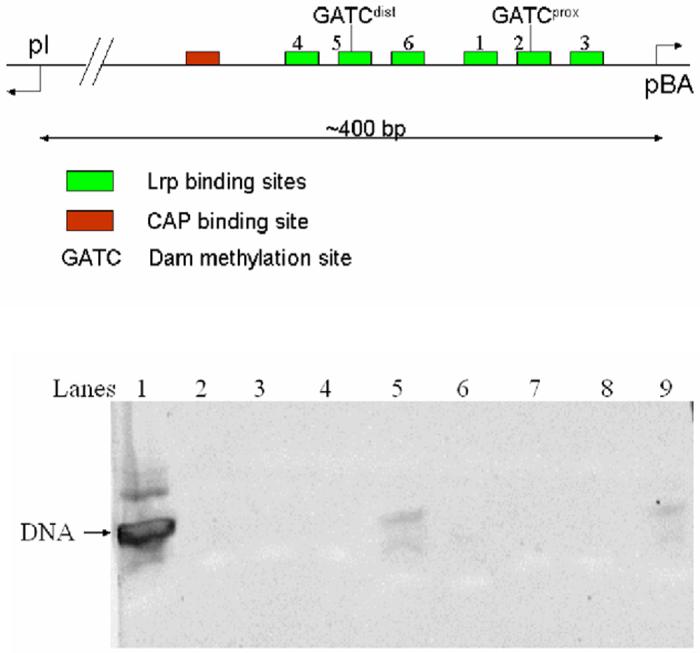
A.) Organization of the E. coli pap operon regulatory region. The six Lrp binding sites are marked in green, with the site of Dam activity at the target GATC sites indicated wthin sites 2 and 5. Site 3 directly overlaps the RNA polymerase binding site for the downstream pBA operon that encodes the structural pili genes. Divergent transcription from pI to produce the papI coregulator is indicated at left. B.) Gel electrophoresis of dissolved crystals. Lane 1: DNA used for cocrystallization. Lanes 2-4: wash solution; Lane 5: dissolved crystal. Lanes 6-9 are the repeat steps from a second independent crystal. The gel was stained with SYBR Gold for 15 minutes using a 1:10,000 dilution of the stock solution in 40 mM Tris-acetate, 1 mM EDTA. The SYBR Gold was purchased from Molecular Probes, Inc.,
Here we report the structure of E. coli Lrp cocrystallized with a 19-bp target DNA sequence from Lrp binding site 2 of the pap operon. Although the DNA within the structure is disordered, comparisons with the Lrp homolog structures show that the E. coli protein adopts a strikingly distinct quaternary organization. Most notably, one of the four octamer interfaces present in the other structures is completely disrupted in the E. coli Lrp octamer, breaking the closed and presumably lower-energy symmetrical assembly observed in the homologs. Using the pap operon as a model, this structure permits informed speculation as to how Lrp can simultaneously bind multiple DNA sites to facilitate transcription.
Results and Discussion
Structure of the E. coli Lrp dimer
The 164 amino acid E. coli leucine-responsive regulatory protein (Lrp) crystallizes as an octamer assembled as a tetramer of dimers (Figure 2). The model contains four monomers (two dimers) per asymmetric unit that were built separately during refinement (see Experimental Procedures). A 19 base-pair DNA oligomer containing the pap operon binding site 2 sequence TGTGGGTTAAAAGATCGTT was included in the crystallization milieu. No ordered electron density for this molecule could be located; all of the observed electron density in the asymmetric unit is assigned to protein. We also were not able to detect binding of this 19-mer to Lrp by gel mobility shift at concentrations up to 0.8 μM DNA. However, the much higher concentrations of DNA present in the crystallization drop, and the observation that duplexes as short as 21 base-pairs do efficiently bind,14 suggested that the 19-mer likely retained the capacity to interact with Lrp. Indeed, thorough washing of crystals followed by dissolution and electrophoresis demonstrates that DNA is present in the crystal and was not excluded from the lattice during crystallization (Figure 1B). Exhaustive crystallization experiments with longer DNA duplexes failed to produce crystals suitable for Xray analysis. Crystals were not obtained in the absence of DNA.
Figure 2.
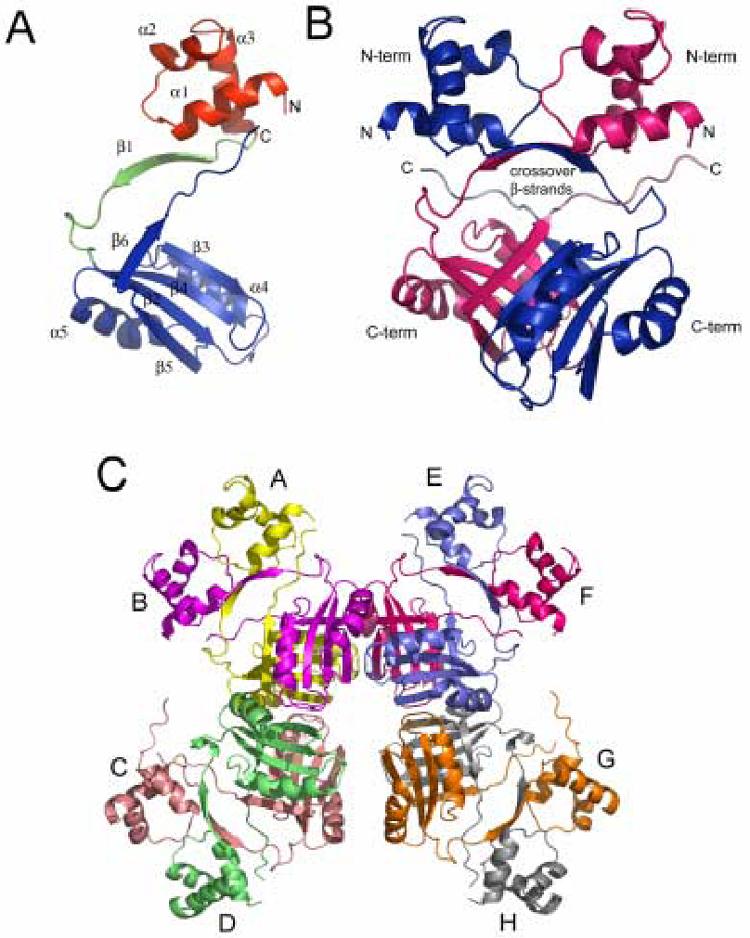
A.) Representation of the E. coli Lrp monomer. The N-terminal HTH domain, the linker peptide, and the C-terminal domain are colored red, green, and blue, respectively. B.) Representation of the E. coli Lrp dimer. Monomer A is colored blue and monomer B is colored red. The N-terminal and C-terminal domains are labeled “N-term” and “C-term”, respectively. C.) Representation of the Lrp octamer. Monomers A through H are shown in yellow, purple, pink, green, blue, red, orange, and gray, respectively.
Each Lrp monomer measures approximately 35 Å × 55 Å × 40 Å, while the dimer measures 55 Å × 55 Å × 50 Å and the octamer roughly 120 Å × 100 Å × 60 Å. The monomer fold consists of two domains: an N-terminal helix-turn-helix (HTH) fold and a C-terminal antiparallel β-sheet flanked on one side by two α-helices (Figure 2a). The HTH motif is a prototypical DNA-binding unit observed in all three domains of life, and consists of three α-helices with a conserved tight turn between the second and third helices. The third “recognition” helix typically binds snugly in the major groove of DNA. The backbone atoms (N, CA, C, O) of the HTH motif of Lrp superimpose on the HTH domains from lambda repressor and Cro with root-mean square deviations (rmsd) of 1.6 Å and 1.76 Å, respectively, demonstrating the close structural correspondence.
The C-terminal domain of Lrp possesses a fold homologous to the “regulation of amino acid metabolism” (RAM) domain involved in the allosteric regulation of amino acid metabolism in prokaryotes.20 Amino acid binding to RAM domains propagates a conformational change that results in regulation of transcription initiation or of enzyme function. RAM domains are composed of a βαββαβ fold that forms a four-stranded anti-parallel β-sheet flanked by two α-helices, as in Lrp (Figure 2B). In Lrp the RAM domain contains the binding site for the effector leucine. Mutational analysis has identified seven residues in the protein that are important for leucine responsiveness, five of which are located at the dimer interface in a highly conserved loop that bridges strands β3 and β4 (Figure 2).21 Together with the high sequence conservation in this region among Lrp homologs, the mutagenesis data suggest that the β3-β4 loop is the effector binding site in all RAM domains. This prediction was confirmed recently by localization of the asparagine binding site in E. coli AsnC by Xray crystallography.11 This structure showed that the asparagine ligand indeed directly contacts a number of the amino acids that are structurally equivalent to those previously identified by mutation of Lrp.
The Lrp monomers adopt a head-to-head orientation in the dimer. Following α3 of the HTH motif, the monomers cross over each other, with one β-strand from each monomer forming a two-stranded anti-parallel β-sheet interaction at the center of the molecule. This crossover places the C-terminal domain of each monomer below the N-terminal domain of the opposing monomer of the dimer (Figure 2B). The crossover β-strand (β1) forms part of a flexible 18 amino acid linker (residues 56 to 73) which connects the N- and C-terminal domains of the Lrp monomer. The flexibility of this linker allows for variation in positioning of the relatively rigid N- and C-terminal domains with respect to each other. Superpositions of the four distinct monomers in the asymmetric unit revealed that the rmsd for polypeptide backbone atoms in the N-terminal core domain (residues 16-55) ranges from 0.73 Å to 1.34 Å, while the C-terminal domains (residues 74-81; 90-154) superimpose with rmsd between 0.54 Å and 0.66 Å. The more highly variable N-terminal subdomain structure correlates with higher B-factors in this portion of the protein. It may exhibit greater flexibility because it does not have a tightly bound DNA partner in the crystal. In contrast, the location of the dimer and octamer interfaces in the C-terminal domain may contribute to its apparently greater rigidity in the crystal.
The C-terminal β-strand (β6) of each monomer makes a second crossover to join the β-sheet in the opposite subunit, where it forms an antiparallel interaction with strand β3 (Figure 2B). The 10 residue C-terminal peptide of Lrp then extends parallel to the flexible linker and to α1 within the HTH, bringing the C and N-termini into close proximity. Leu159 and Ile161 from the C-terminal peptide pack with Arg17, Asn21, and Asn24 of HTH helix α1 to stabilize the structure of the second crossover. Approximately 10 amino acids at the N-terminus of the protein are disordered in each subunit. As this peptide carries a net +4 electrostatic charge and is adjacent to the HTH, a role in DNA binding seems probable (see below).
The two-stranded β-sheet linker forms one portion of the dimer interface between the two monomers. The remaining major portion of the dimer interface is then formed by interactions between the β-sheets of the C-terminal domains, including the crossover strand strand β6 (Figure 2B). Side-chain interactions within the dimer interface are primarily hydrophobic, with potential electrostatic contributions closer to the solvent-exposed portions. A further small contribution to the dimer interface arises from several interdomain interactions made between helix α1 of the N-terminal subdomain, and strand β1 within the flexible linker.
Higher-order architectures of E. coli Lrp and Lrp homologs
With the exception of Pyrococcus OT3 FL11,10 all Lrp homologs that have been studied form octameric assemblies in the crystal.9,11 However, structural comparisons show that the spatial arrangement of the four dimers in E. coli Lrp differs significantly from the other proteins. In contrast to the highly symmetric Pyrococcus LrpA, B. subtilis LrpC and E. coli AsnC octamers, one of the four dimer-dimer interfaces of E. coli Lrp is completely disrupted, such that the closest approach between two of the dimers is 6 Å (Figure 3). The disruption essentially renders the E. coli Lrp octamer a linear structure, in contrast to the closed octameric rings formed by its structural homologs. The other octamers possess strict or approximate 42 symmetry with 90° angles between the two-fold symmetry axes at the center of each dimer. By contrast, the dimers within the Lrp octamer adopt distinct positions, such that the angle between the dimer two-fold axes that span the disrupted interface increases to 110°. The corresponding angle at the opposite side of the Lrp assembly is approximately 80°, while the other two intradimer angles of the octamer measure approximately 85° each (Figure 3). The asymmetric unit in the crystal consists of dimers AB and CD; dimers EF and GH are generated by operation of a crystallographic two-fold axis.
Figure 3.
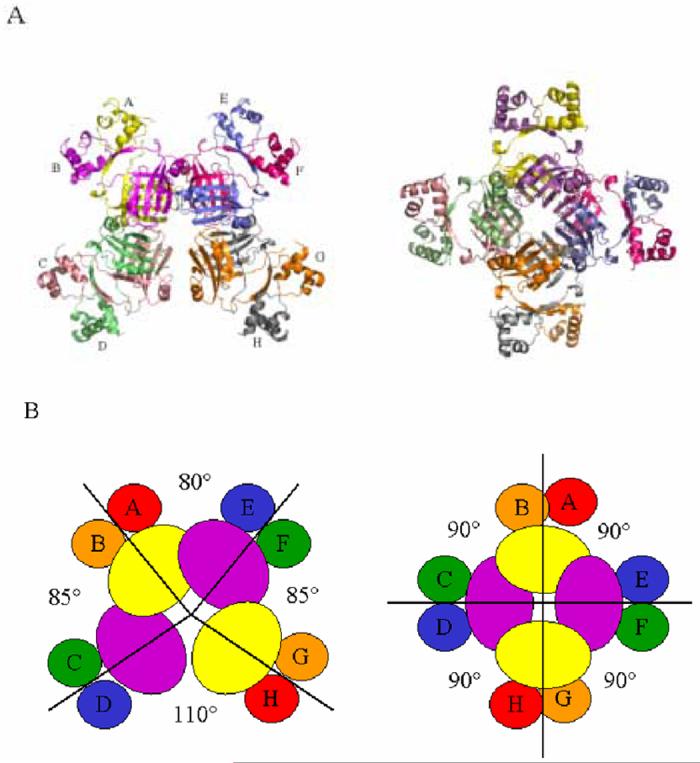
A.) Structures of the E. coli Lrp (left) and P. furiosus LrpA (right) octamers revealing the disruption in symmetry in the former structure. Only three octamer interfaces remain in E. coli Lrp, bridging dimers EF and GH, AB and EF, and AB and CD. The B. subtilis LrpC and AsnC octamers show the same symmetry depicted for P. furiosus LrpA. B) Cartoon representations of the E. coli Lrp octamer (left) and the P. furiosus LrpA octamer (right). The 90° angles are exact for P. furiosus LrpA and E. coli AsnC, for which the asymmetric units are dimers.9,11 Because the asymmetric unit in the B. subtilis LrpC crystal contains a full octamer, the two-fold and four-fold axes are noncrystallographic, and small deviations from the 90° octamer interface angles are present.11
The three remaining octamer interfaces of E. coli Lrp exhibit internal pseudosymmetry and are composed of structural elements arising from the C-terminal domains of all four monomers from two adjacent dimers (Figures 3, 4). The topology of each interface is essentially the same. For example, at the octamer interface between dimers E-F and G-H (see Figure 3 for nomenclature), interactions are made between α5 and β5 of monomer E, with surface loops bridging β2-α4 and β3-β4 emanating from monomer G (Figure 4). In the same E-F/G-H interface, a similar set of pairwise interactions occurs between α5 and β5 of monomer H, with the loops bridging β2-α4 and β3-β4 emanating from monomer F. A number of additional contacts at the center and periphery of the interface are also made - for example, interactions are observed between related surface loops of monomers F and G across the pseudo two-fold axis of the octamer interface (Figure 4).
Figure 4.
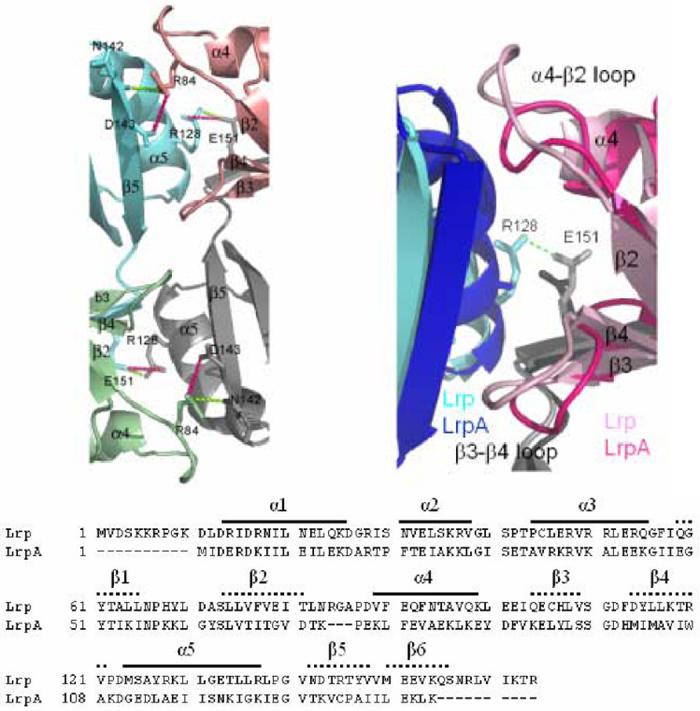
Representation of an E. coli Lrp octamer interface (A) compared with the P. furiosus LrpA octamer interface (B). Monomers A, B, E, and F are colored green, blue, pink, and gray, respectively. The interacting residues that form the interface are shown in sticks with hydrogen bonds shown in dark pink and salt bridges shown in green as dashed lines. C.) Sequence alignment of E. coli Lrp and P. furiosus LrpA
Formation of the octamer interfaces between dimers results in burial of significant additional surface area. Of the roughly 11,000 Å2 accessible surface area associated with an Lrp monomer, about 3,200 Å2 is buried to form the dimer and an additional 1000-1100 Å2 is buried between each pair of dimers. The breaking of the octamer symmetry in E. coli Lrp renders the three remaining octamer interfaces inequivalent: for example, a slightly greater number of van der Waals packing contacts are made at the AB-EF interface as compared with AB-CD, and this arises because the angle between the dimer two-fold axes is smaller for the AB-EF interface. Thus, the AB-CD octamer interface buries 1040 Å2 while 1090 Å2 are buried at the AB-EF octamer interface.
E. coli Lrp is the only protein in the family crystallized in the presence of DNA. Therefore, one possible inference from these observations is that the apoprotein forms the closed octamer assembly observed in the homologs, and that one octamer interface is disrupted by interactions with DNA that occur during crystallization. Alternatively, intrinsic structural features unique to E. coli Lrp may render formation of a closed ring impossible even in the absence of DNA. To evaluate this possibilities, we carried out pairwise superpositions among the E. coli Lrp, P. furiosus LrpA, B. subtilis LrpC and E. coli AsnC octamers, with the aim of identifying unique features in the E. coli Lrp protein that block ring closure in spite of the accompanying favorable additional surface area burial. Such features, if present, might orient the three dimer-dimer interfaces that do form with sufficient rigidity to preclude the close juxtaposition of structural elements necessary to form the fourth dimer-dimer contact.
We first compared the structures at the monomer and dimer levels. All four Lrp homologs possess relatively rigid N-terminal and C-terminal domains joined by a flexible linker, as described above (Figure 2). However, the relative orientations of the N- and C-terminal domains vary significantly (Figure 5). For example, among the four crystallographically independent E. coli Lrp monomers considered pairwise, the N- and C-terminal domains adopt relative orientations that vary from 2.8° - 6.9°. The N- and C-terminal domains of the two independent P. furiosus LrpA monomers similarly deviate by 6° from each other. In contrast, the relative orientations of N- and C-terminal domains compared between Lrp and LrpA vary from 14.9° to 21.5°, suggesting that the interdomain orientation may be an intrinsic feature of the protein structure.
Figure 5.
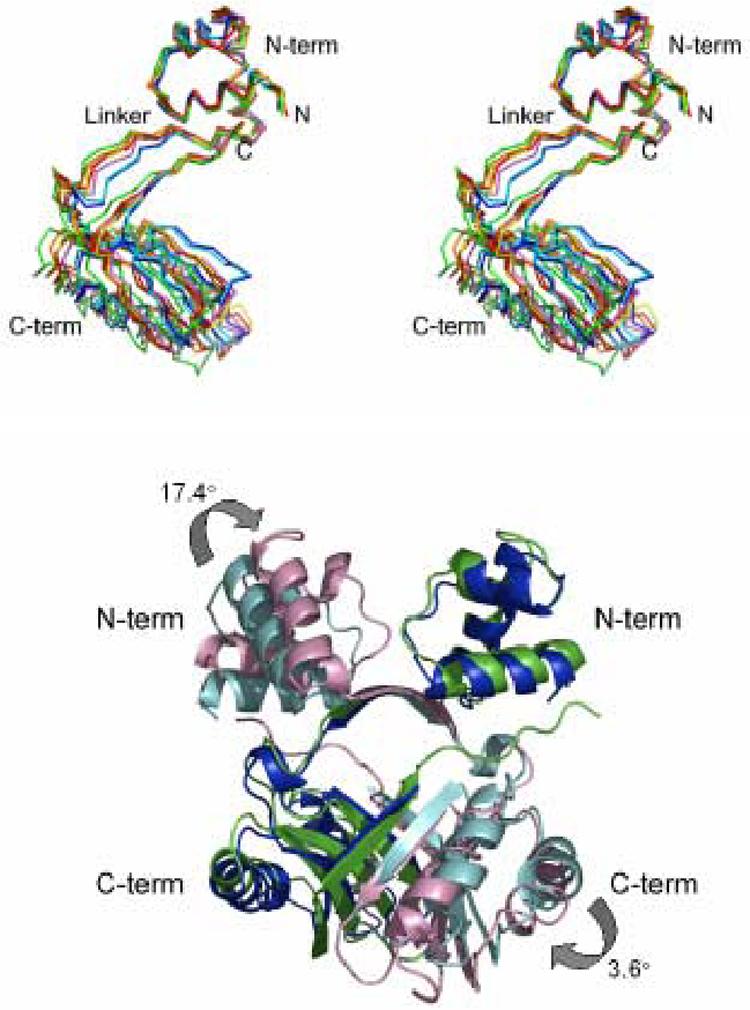
A) Stereo view of a superposition of Lrp monomers based on the N-terminal HTH domain (“N-term” see Experimental Procedures). The four separately built E. coli Lrp monomers A, B, C, and D are colored red, orange, yellow, and green, respectively. The two separately built P. furiosus LrpA monomers A and C are blue and cyan, respectively. The Pyrococcus OT3 FL11 monomer is depicted in purple. The distinct relative orientation of the LrpA C-terminal domain is evident in the backbone trace of the linker region as well. B) Superposition of the N-terminal and C-terminal core regions of monomer A of Lrp on the equivalent residues of LrpA. Lrp monomers A and B are colored green and pink, respectively. LrpA monomers A and B are colored blue and cyan, respectively.
The C-terminus of E. coli Lrp extends 10 amino acids beyond that of LrpA. It appears that interactions made by this C-terminal peptide extension with helix α1 of the N-terminal subdomain force the flexible linker to shift by approximately 3 Å, as compared with the linker’s positoin in LrpA. Further, the difference in N- and C-terminal domain positions also appears at the dimer level. Superposition of the conserved portions of both subdomains of one monomer of Lrp and LrpA (rmsd = 1.88 Å), reveals a 17.4° orientational difference between the N-terminal subdomains of the opposing monomer (Figure 5B). However, while these differences appear to originate in the unique C-terminal interactions of E. coli Lrp, it is not necessarily clear that the consequent differences in domain positions form a basis for excluding symmetric octamer formation in E. coli Lrp. Indeed, extension of this analysis to include AsnC and LrpC, which retain six of the ten C-terminal amino acids present in E. coli Lrp, does not suggest that the respective positions of the N- and C-terminal domains of E. coli Lrp are unusual. Moreover, direct pairwise comparison of N- and C-terminal domain positions in AsnC versus LrpC revealed a 20° difference in relative orientations with concomitant large effects at the dimer level, yet both proteins form symmetric closed octamers in their respective crystals.11
We also examined the interactions forming the octamer interfaces among the four proteins. Although substantial differences exist in the detailed contacts formed, in all structures the central and largest area of the octamer interface is composed of well-packed hydrophobic contacts. The two loops at the octamer interface, bridging β2-α4 and β3-β4, respectively, do adopt somewhat different conformations among the structures (Figure 4). Indeed, the β2-α4 loop is three residues larger in E. coli Lrp than in the other three proteins, suggesting a possible structural deviation that might in principle drive the formation of the unique open quaternary structure. However, detailed superpositions show that the additional amino acids are accommodated by extrusion at the periphery of the interface, and do not appear to be driving formation of a unique molecular surface that would necessarily exclude the possibility of symmetric closure (Figure 4).
Analysis of the E. coli Lrp structure at the monomer, dimer and octamer levels therefore does not suggest any particular reason why it alone, among the homologs, should be unable to form the higher symmetry assembly. Given the expected free-energy benefit from formation of such a closed ring structure, it appears likely that the disruption of one interface is indeed driven by the presence of DNA in the crystal. Of course, this conjecture requires experimental verification by structure determination in the absence of included DNA. The formal possibility that crystal packing forces might drive the opening of one of the octamer interfaces is unlikely, because we have also determined the crystal structure under identical crystallization conditions in a hexagonal lattice at 3.6 Å resolution (data not shown). Although this crystal diffracts more weakly, the structure determined by molecular replacement and refined to Rfree of 33.3% shows an identical quaternary structure with the same disruption to the octameric symmetry that is observed in the better-ordered orthorhombic lattice.
It is relevant to note that there is precedent in other protein-DNA systems for the notion that inclusion of DNA in the crystallization milieu drives the adoption of a specific protein conformation, even if the DNA itself is disordered in the crystal. The crystal structure of HhaI methyltransferase cocrystallized with S-adenosylmethionine (SAM) and a nonspecific 14-mer duplex DNA oligonucleotide showed that the protein structure was rearranged and the orientation of SAM binding altered, as compared with a crystal structure of the binary HhaI - SAM complex determined from crystals grown in the absence of DNA.22 In the binary complex determined from crystals grown with DNA, the SAM adopted a new binding mode consistent with biological activity, even though no DNA was observed in electron density maps. Thus, the protein-DNA interactions that occur in the crystallization drop during nucleation and growth stabilize a particular protein conformer, and such stabilization does not necessarily require formation of the well-ordered protein-DNA interface that is needed to observe discrete DNA electron density in the final formed crystal. We suggest that a similar mechanism accounts for the opening of the E. coli Lrp octamer in the structure reported here.
DNA binding by E. coli Lrp
The presence of the canonical helix-turn-helix motif at the N-terminus of E. coli Lrp suggests a model for DNA binding in which the duplex is wrapped around the outside surface of the octamer, where it can interact with the N-termini of successive monomers (Figure 6). This wrapping suggests the possibility that Lrp may form a higher-order solenoidal structure on DNA in vivo, perhaps with significant similarities to the nucleosome structure formed in eukaryotic chromatin. This would provide a structural rationale for the hypothesis that Lrp functions as a DNA packaging protein under some conditions.5 Indeed, electron microscopy and atomic-force microscopy measurements of the LrpC homolog reveal wrapping of 300 - 600 base-pair DNAs about a protein assembly, to form nucleosome-like structures.23 A model of LrpC wrapped into a solenoid has also been presented based on the crystal structure of that protein.11
Figure 6.
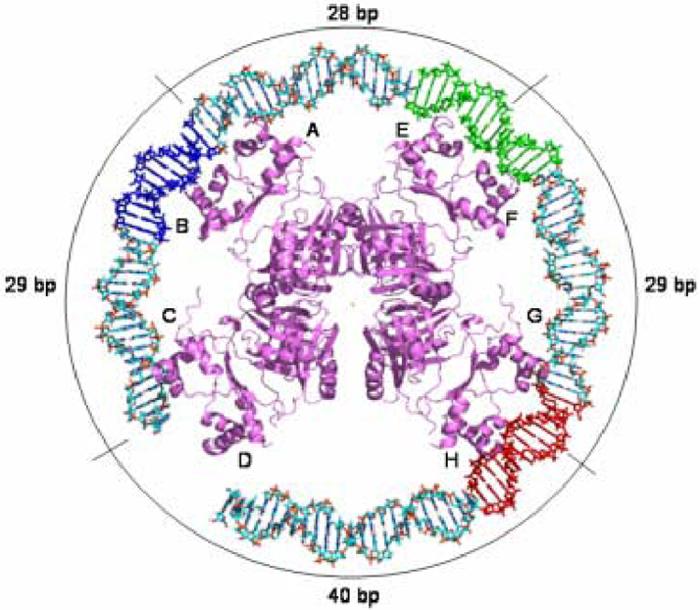
Model of the E. coli Lrp octamer bound to Lrp binding sites 1, 2, and 3 of the pap reguglatory region. Lrp binding site 3 is colored red, site 2 is depicted in green, and site 1 is shown in blue. The labels on the periphery indicate the number of base-pairs that separate the center two-fold axes of the individual dimers from each other.
We generated a model of E. coli Lrp bound to a 118 base-pair DNA sequence containing Lrp binding sites 1, 2, and 3 of the pap operon (Figure 6). The model was created by placing the α3 recognition helix of the HTH domain from one Lrp monomer in the major groove of the DNA at site 2, and the DNA was then smoothly wrapped about the octamer with no attempt to modify the structures to optimize the docking of other dimers. Several features of this proposed DNA complex are immediately apparent upon inspection of the model. First, the spacing of octamers on the duplex reveals that the Lrp intradimer two-fold axes (between dimers that interact to form the octamer) are separated by 28-29 base-pairs, or roughly three helical turns of the DNA. This is similar to the 30 base-pair spacing between Lrp binding sites in the pap operon (Figure 6). Second, the model requires Lrp-induced bending or curvature in the same direction with respect to the DNA grooves. Indeed, Lrp has been reported to induce a bend of 52° at a single binding site and of at least 135° in a DNA molecule containing two adjacent sites.24 These bending angles are in approximate correspondence with the model, which suggests a bend of 180° or more distributed over three binding sites. Many other Lrp-regulated promoters, such as dadAX and ilvIH,25,26 also possess a number of adjacent binding sites separated by three helical turns, suggesting that some features of the protein-DNA complex are shared among these regulons.
The model also suggests that the HTH domains of successive Lrp dimers do not directly fit into the DNA major groove in successive turns of the duplex, indicating that either the protein octamer, the DNA or (most likely) both macromolecules must undergo conformational adaptations during the DNA wrapping process. Docking of a single Lrp dimer reveals that the HTH recognition helices from each subunit bind nine base-pairs apart along the DNA rather than ten, and this provides a fundamental architectural constraint that strongly predicts significant induced-fit conformational change upon DNA binding. Given the likelihood of such rearrangements, we also considered whether the regular wrapping of the DNA might be modified to allow binding inside the unique interface between dimers created by the failure of the octamer to fully close. However, calculation of the electrostatic potential at the interface shows that it is highly negatively charged, suggesting that DNA binding is unlikely in this region (Figure 7).
Figure 7.
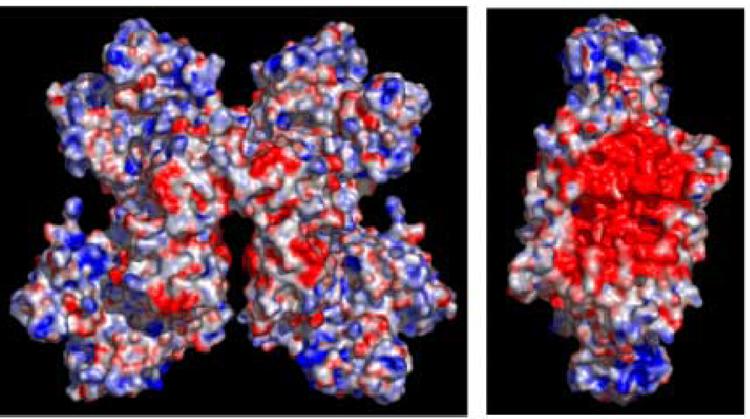
Representation of the electrostatic surface charge potential for the Lrp octamer, calculated using the APBS plug-in in PyMol. Panel A shows a surface representation of the full Lrp octamer. Panel B shows Lrp rotated 90° to the left about the y-axis, with the tetramer depicted on the right side of panel A removed for clarity. This panel reveals the strongly negative potential in the central region of the octamer. Positively charged regions are indicated in blue and negatively charged regions are in red.
The structure of the Lrp dimer brings the N- and C-termini of the protein together adjacent to the DNA-binding N-terminal HTH domain (Figures 2, 6). This suggests that these peptides may play an important role in modulating either DNA binding affinity, assembly of the Lrp dimers on DNA, or interactions with other regulatory proteins. The C-terminal peptide provides a connection from the leucine-binding domain, and hence offers a structural basis for signal propagation to the HTH reading heads that could modulate DNA binding.11 Indeed, a variant of Lrp lacking the C-terminal 11 amino acids was unable to bind to ilvIH promoter DNA, suggesting the importance of this terminal peptide.27
The lengths of both the N- and C-termini vary significantly among members of the Lrp family. For example, the P. furiosus LrpA lacks the ten amino acid peptides present at both termini of E. coli Lrp (Figure 4). Interestingly, the net +4 charge on the N-terminal peptide of E. coli Lrp, and the likely proximity of the N-terminus to the DNA, suggests the possibility that this portion of the protein may also make DNA interactions. To test this hypothesis we generated an N-terminal truncation mutant of the protein and assayed its ability to bind to a 118 base-pair duplex DNA segment containing binding sites 4, 5 and 6 of the pap operon (Figures 1, 8). We found that LrpΔN was able to bind DNA only very weakly when compared with the wild-type protein, suggesting that the N-terminus indeed is important to DNA binding in this system. To confirm the previous findings we also generated a ten amino-acid truncation of the C-terminus of Lrp (LrpΔC). Although the previous report found that an 11 amino-acid C-terminal deletion was inactive in DNA binding,27 we find that the 10 amino acid truncation does retain very weak binding activity near the limit of detectability (Figure 8).
Figure 8.
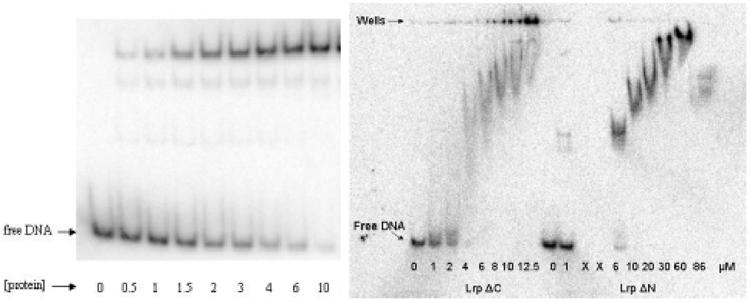
Gel mobility shift analysis for the binding of wild-type Lrp (left) and the Lrp truncation mutants LrpΔN and LrpΔC (right). The concentrations of wild-type Lrp given in the left-hand panel are in nM units. The binding affinity for wild-type Lrp was found to be 2.2 nM, in good agreement with previously reported values using a similar DNA molecule containing pap operon binding sites 4, 5 and 6.29
The model for Lrp binding to DNA reveals the presence of large open gaps between the dimers, which is not consistent with the high degree of protein-DNA complementarity expected in a gene regulatory complex. Part of the open space may be filled by the ten amino acid N-termini from adjacent dimers. Indeed, binding of the N-terminal Lrp peptides may resemble the binding of extended linker regions of the Pax6 paired DNA-binding domain, where a protein linker bridges HTH units spaced one helical turn apart.28 Further, the small 73 amino acid pap-specific coregulator protein PapI, which enhances Lrp binding affinity to pap regulatory sites 2 and 5,29 may also bind in the intervening gaps between the protein and DNA. Nuclear magnetic resonance studies of PapI show that the protein is independently able to bind DNA (F. Dahlquist, personal communication), suggesting that its function in enhancing Lrp binding may involve both protein-protein and protein-DNA interactions.
Our finding that the Lrp octamer can adopt an open linear form offers new perspectives on how the protein regulates transcription. Earlier experiments showed that Lrp can bind to DNA containing a single binding site as a dimer, and that dimer, octamer and hexadecamer forms exist in equilibrium.14,30 This structure now suggests that the octamer form is further able to transition between open and closed states, providing an underlying mechanism by which the relative orientations of the DNA-binding HTH motifs may be changed. Because dimers of Lrp form only in the nanomolar concentration range, it is not likely that further disassembly of the open octamer to form dimers (with concomitant formation of hexamers or tetramers) will occur in vivo. Instead, closed and open octamer states are more likely to exist in equilibrium, with the closed state probably able to open at any of the four symmetrically equivalent interface regions.
The proposed equilibrium between closed and open octamers offers a mechanism for regulating the activity of the protein. Our observation that the open octamer forms only when Lrp (or an Lrp homolog) is crystallized in the presence of DNA suggests that DNA binding provides one means by which rearrangement of the protein assembly is facilitated. We speculate that stabilization of either the closed ring or open linear octamer may also be promoted by binding of leucine to the C-terminal effector domain, although the structural pathway by which the dissociation is triggered will presumably differ in this case.
Two further considerations suggest that the open, linear form of the Lrp octamer observed here is likely to be of biological importance. First, the potential role of Lrp in packaging DNA suggests the need to form a solenoidal arrangement where the DNA wraps around protein octamers. Neither the open nor closed ring forms of Lrp and its homologs exhibit a helical pitch to facilitate wrapping of DNA, but the open form should be much more easily distorted to accommodate a continuous DNA supercoil that extends over many octamers. Second, breaking of the symmetry in the open form moves two dimers approximately 40 base-pairs apart on the DNA (Figure 6). With respect to pap operon regulation, binding of three dimers to sites 1-2-3 in the phase OFF state (Figure 1A) would cause the disrupted interface to adopt a specific spatial orientation with respect to the adjacent and similarly spaced sites 4-5-6, but would prevent binding of the fourth dimer to the adjacent site 6. PapI binding might then trigger the translocation of the trimer from sites 1-2-3 to sites 4-5-6, possibly by a mechanism that might require closing and reopening of the octamer. Interestingly, in vitro experiments using supercoiled DNA have revealed a mutual exclusion mechanism, whereby Lrp is able to bind to either sites 1-2-3 or to sites 4-5-6, but is disfavored from occupying both sets of sites simultaneously.31 On linear DNA, however, all six sites are much more readily occupied. Thus, the constraints imposed by negative supercoiling in vivo appear likely to play a significant role in modulating the required movement from downstream to upstream sites.
E. coli Lrp is the canonical member of a large family of transcriptional regulatory proteins (the Lrp/AsnC family) that includes representatives from a large number of bacterial and archaeal species. The sequence and structural similarities among these proteins, and their common roles in DNA binding, suggest that the quaternary octamer transition described here may feature prominently in the function of all proteins in the family. Such a rearrangement has the potential to function as a structural mechanism that could underlie both the DNA packaging and gene regulatory roles of these proteins. The quaternary structure rearrangement may be of particular importance in complex regulons such as the E. coli pap operon, where a DNA methylation-driven transition occurs in the context of the binding of several other regulatory proteins, to generate ultimately a molecular surface that can be recognized by RNA polymerase. Although a full elucidation of the stereochemical origins of transcriptional regulation in the pap system remains as a challenge for future research, the octamer transition proposed here may offer an important handle on the complex macromolecular dynamics that appear likely to occur in the shift between regulatory states.
Materials and Methods
Protein expression and purification
E. coli Lrp was expressed in E. coli strain JM105 containing the plasmid pJWD).32 Cells grown in LB media were induced with 0.5 mM IPTG at OD600 = 0.8 and shaken for 2.5 hours at 37°C. Cells were pelleted, resuspended in 50 mM potassium phosphate (pH 7.4), 0.1 M NaCl, 0.1 mM EDTA, 10 mM βME, 10% glycerol (PC buffer), and then lysed by French press. A protease inhibitor tablet (Roche) and 0.05 mM PMSF were added to the lysate. The cleared lysate was loaded onto a P-11 phosphocellulose column (Whatman) and washed with PC buffer. Lrp was eluted with a linear NaCl gradient from 0.1 - 1 M. Fractions containing Lrp were pooled and dialyzed into 10 mM Tris (pH 7.4), 100 mM NaCl, 0.1 mM EDTA, 10 mM β-ME, 10% glycerol (Buffer A). The dialyzed sample was loaded onto a Macroprep High S Support cation exchange column (Biorad), washed with Buffer A, and again eluted with a linear NaCl gradient from 0.1 - 1 M. Fractions containing Lrp were pooled, concentrated, and dialyzed into 10 mM Tris (pH 7.4), 100 mM NaCl, 0.1 mM EDTA, 10 mM βME, 50% glycerol. The final yield of purified protein (estimated at greater then 99% purity from SDS gel electrophoresis) is 50 mg from 6 liters of cell culture.
DNA purification
Oligodeoxynucleotide sequences corresponding to Lrp-binding site 2 of the pap operon were used for crystallization trials. Oligonucleotides were obtained from the Biomolecular Resource Center at the University of California, San Francisco, and from Integrated DNA Technologies, Inc. The DNA was synthesized as single strands and was shipped with the trityl group still attached. Each oligonucleotide was resuspended in 500 μL TE buffer (pH 8.0), and was purified by HPLC on a Rainin Pure DNA column, as described.33 After purification, the single-stranded complementary oligonucleotides were mixed in a 1:1 molar ratio, heated to 90°C for 10 minutes, and then allowed to cool slowly back to room temperature. The annealed oligonucleotides were stored at -20°C prior to their use in crystallization experiments. The sequences used to obtain crystals used for structure determination were: 5’-TGTGGGTTAAAAGATCGTT and 5’-AAACGATCTTTTAACCCAC. The 19-bp duplex contains two one-base 5’-overhangs.
Construction and assay of deletion mutants
Lrp truncation mutants containing deletions of either the first ten (LrpΔN) or last ten amino acids (LrpΔC) were constructed by QuickChange mutagenesis (Stratagene) as previously described,34 with primers designed to loop out 30 base-pairs of DNA from the wild-type gene. PCR products from mutagenesis reactions were purified by extraction from agarose gels and transformed into E.coli XL-1 Blue cells (Stratagene). Deletion constructs were sequenced in their entirety and then transformed into the E. coli Lrp- deletion strain MC4100 (gift from D. Low). The Lrp truncation mutants were expressed, purified, and stored as described for wildtype Lrp.
Gel mobility shift assays were performed using a 118 base-pair DNA duplex corresponding to Lrp binding sites 4, 5, and 6 from the pap operon regulatory region (Figure 1).29 The DNA was 5’-end-labeled with γ-32P-ATP using polynucleotide kinase. The binding buffer for gel shift assays contained 40 mM Tris (pH 7.5), 60 mM KCl, 0.1 mM EDTA, 1 mM DTT, 5% glycerol, 80 mM NaCl, 18 μg/ml herring sperm DNA, 20 μg/ml BSA, and 0.05 fmol/μl of the labelled DNA. Dilutions of Lrp were made in 40 mM Tris (pH 7.5), 60 mM KCl, 0.1 mM EDTA, 1 mM DTT, 1 mg/ml BSA. Varying concentrations of Lrp were added to the binding buffer containing DNA, and the mixture incubated at 37°C for 20 minutes. The binding reactions were then loaded onto 6% native acrylamide gels and run in 0.5X TBE at ambient temperature for 2 hr at 250 V. Gels were developed by phosphorimaging and quantified with the program Imagequant. Data were fit to a hyperbolic binding curve using Kaleidagraph.
Crystallization and diffraction data collection
Crystallization experiments were performed at 17°C by hanging drop vapor diffusion. The Lrp stock solution stored as described above was dialyzed into 10 mM HEPES (pH 7.4), 200 mM NaCl, 0.1 mM EDTA and 1 mM DTT. Dialyzed Lrp (5-10 mg/ml) was mixed with the site 2 oligonucleotide at a DNA:protein molar ratio of 1.5:1, and a 1.5 μl volume of the protein-DNA complex was then mixed with 1 μL of a buffer containing 50 mM MES (pH 5.6), 10 mM MgCl2, 0.4 M KCl, and 8.5% PEG 8000 in a conventional vapor diffusion experiment. Single crystals of thin rectangular shape were obtained in about 1 week with maximum dimensions 0.5 μm × 0.3 μm × 0.05 μm. No crystals were obtained in the presence of leucine, under any conditions. To assess whether the DNA in the crystallization solution formed part of the crystal, individual crystals were isolated on a cover slip and repeatedly washed with successive small volumes of mother liquor. These crystals were then directly dissolved in water prior to electrophoresis through a denaturing polyacrylamide gel.
Crystals were soaked in a cryoprotectant solution containing 50 mM MES (pH 5.6), 10 mM MgCl2, 0.4 M KCl, 10% PEG 8000, and 25% glycerol prior to data collection. The crystals were then flash frozen at 100°K. Native diffraction amplitudes were measured to 3.2 Å resolution at SSRL beamline 7-1; data were processed with MOSFLM and SCALA.35,36 The crystals belong to the orthorhombic space group C2221 with unit cell dimensions a=100.1 Å, b=237.0 Å, c=75.6 Å, and contain four monomers of Lrp per asymmetric unit. Heavy atom derivatives were prepared by soaking crystals in a stabilizing solution containing 10% PEG 8000, 0.05 M MES (pH 5.6), 0.01 M MgCl2, and 0.4 M KCl. Diffraction amplitudes for the two heavy atom derivatives were measured at SSRL beamline 11-3. The EMTS derivative was prepared by soaking in a solution containing 1 mM EMTS for 1 hour, and the NaI derivative was prepared by soaking in a 1 mM NaI solution for 1 minute. Crystals obtained using selenomethionine-modified Lrp exhibited weak diffraction and were not useful in the structure determination.
Structure determination and refinement
Heavy atom sites were found and initial MIR phases were determined by SOLVE to 3.5 Å resolution.37 Initial phases were improved by solvent flipping as implemented in the program CNS.38 The model was built manually using the program O.39 Initially, the backbone was built guided by the maps and by BONES produced using MAPMAN.40 The initial trace was superimposed on the LrpA homolog structure to help determine the orientation of the molecules. Four monomers (two dimers) were built into the asymmetric unit. Non-crystallographic symmetry (NCS) matrices were calculated between each of the monomers using the program O. A mask for NCS was calculated using MAMA;41 then the NCS matrices were improved using IMP.42 The NCS was used to average the maps using Solomon for density modification, followed by Sfall to calculate structure factors from the resulting map, Sigmaa to combine phases, and Fft to generate a new map.36 This procedure was repeated 10 times. After averaging, side chains for all residues were added to the model. Structure refinement was done using CNS.38 The refinement included rounds of simulated annealing refinement as well as group B-factor refinement, followed by manual rebuilding of the model in O. A random 10% of the starting data was set aside for cross validation (Rfree calculation). Initially, NCS restraints with a weight of 100 were used in the refinement. A composite omit map was calculated; then, side chains for certain residues were removed as required. NCS restraints were then relaxed between the four monomers to accommodate structural differences. After a majority of the model was built, the model was refined against native data to 3.2 Å. The refinement converged to a final R-factor of 26.7% with Rfree of 32.2% (20-3.2 Å range) and very tight stereochemistry. Electron density was not defined for the N-terminal 11 residues of monomers A, B, and D, the N-terminal 7 residues of monomer C, the C-terminal 2 residues of monomer B, nor for the DNA oligonucleotide used in crystallization. Electrospray mass spectroscopy analysis of Lrp used for crystallization confirmed the presence of all residues except the initial methionine in the protein. The quality of the refined model was analyzed with PROCHECK,36 which showed that 80% of the model lies within the core or most favored regions of the Ramachandran plot, with only 1 residue per monomer in a disallowed region (Lys10 of chain C). No solvent molecules were included in the model. A summary of the data processing and refinement statistics is given in Table 1.
Table 1.
Data Processing and Refinement Statistics
| Native | EMTS | NaI | |
|---|---|---|---|
| Data Collection | |||
| Space Group | C2221 | C2221 | C2221 |
| Unit Cell Dimensions (Å) | 100.1, 237.0, 75.6 | 100.5, 236.0, 75.6 | 100.7, 235.2, 74.8 |
| Resolution (Å) | 3.2 | 3.2 | 3.5 |
| Number of observations | 60,492 | 58,917 | 35,834 |
| Number of unique reflections | 15,185 | 14,439 | 9,797 |
| Completeness (outer shell) (%) | 99.9 (99.9) | 99.0 (99.7) | 99.5 (99.9) |
| Redundancy | 4.0 | 4.0 | 3.6 |
| Rsym (outer shell) (%) | 9.9 (47.6) | 7.9 (35.8) | 8.2 (39.5) |
| I/σ(I) (outer shell) | 6.7 (1.6) | 7.4 (1.9) | 8.2 (1.8) |
| Phasing Statistics | |||
|---|---|---|---|
| Number of Sites | 4 | 2 | |
| Phasing power (acentric/centric) | 1.48/1.18 | 0.53/0.47 | |
| FOM (before solvent flattening) | 0.50 | ||
| FOM (after solvent flattening) | 0.71 | ||
| Refinement | |||
|---|---|---|---|
| Rcryst/Rfree (%) | 26.7/32.2 | ||
| Total number of atoms | 4343 | ||
| Average B factor | 41.6 | ||
| Rms deviations | |||
| Bond lengths (Å) | 0.006 | ||
| Bond angles (°) | 1.1 | ||
Structural analysis
Least-squares superpositions of pairs of core subdomains using backbone atoms were carried out in Insight II.43 The four residues at the N-terminus of the HTH domain, the loop between β-strand 2 and α-helix 4 of the C-terminal domain (Figure 2A), and the C-terminal eight amino acids are highly variable in structure; removing these segments allowed definition of the N-terminal subdomain of an Lrp monomer as residues 16-55 and the C-terminal subdomain as residues 74-81 and 90-154. Determination of the rotational and translational differences between superimposed subdomains was carried out in the program GEM.44 Electrostatic surfaces were computed using Pymol with the APBS plug-in.45,46 Models of the pap promoter DNA alone were created using the program NAMOT2,47 while models of the Lrp/DNA complex were created manually using the program O.39 All figures showing cartoon representations of molecular structure were prepared using Pymol.45
Acknowledgments
We thank Professors Norbert Reich, David Low and Rick Dahlquist for helpful discussions. This research was supported by NIH grant GM62630 (to J.J.P.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Tani TH, Khodursky A, Blumenthal RM, Brown PO, Matthews RG. Adaptation to famine: a family of stationary-phase genes revealed by microarray analysis. Proc. Natl. Acad. Sci. USA. 2002;99:13471–13476. doi: 10.1073/pnas.212510999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Calvo JM, Matthews RG. The leucine-responsive regulatory protein, a global regulator of metabolism in Escherichia coli. Microbiol. Rev. 1994;58:466–490. doi: 10.1128/mr.58.3.466-490.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Newman EB, Lin R. Leucine-responsive regulatory protein: a global regulator of gene expression in E. coli. Ann. Rev. Microbiol. 1995;49:747–775. doi: 10.1146/annurev.mi.49.100195.003531. [DOI] [PubMed] [Google Scholar]
- 4.Brinkman AB, Ettema TJG, de Vos WM, van der Oost J. The Lrp family of transcriptional regulators. Mol. Microbiol. 2003;48:287–294. doi: 10.1046/j.1365-2958.2003.03442.x. [DOI] [PubMed] [Google Scholar]
- 5.D’Ari R, Lin RT, Newman EB. The leucine-responsive regulatory protein: more than a regulator? Trends Biochem. Sci. 1993;18:260–263. doi: 10.1016/0968-0004(93)90177-o. [DOI] [PubMed] [Google Scholar]
- 6.Friedberg D, Midkiff M, Calvo JM. Global versus local regulatory roles for Lrp-related proteins: Haemophilus influenzae as a case study. J. Bacteriol. 2001;183:4004–4011. doi: 10.1128/JB.183.13.4004-4011.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Willins DA, Ryan CW, Platko JV, Calvo JM. Characterization of Lrp, an Escherichia coli protein that mediates a global response to leucine. J. Biol. Chem. 1991;266:10768–10774. [PubMed] [Google Scholar]
- 8.Chen S, Rosner MH, Calvo JM. Self-association of leucine-responsive regulatory protein (Lrp) from Escherichia coli. J. Mol. Biol. 2001;312:625–635. doi: 10.1006/jmbi.2001.4955. [DOI] [PubMed] [Google Scholar]
- 9.Leonard PM, Smits SHJ, Seddelnikova SE, Brinkman AB, de Vos WM, van der Oost J, Rice DW, Rafferty JB. Crystal structure of the Lrp-like transcriptional regulator from the archaeon Pyrococcus furiosus. EMBO J. 2001;20:990–997. doi: 10.1093/emboj/20.5.990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Koike H, Ishijima SA, Clowney L, Suzuki M. The archaeal feast/famine regulatory protein: potential roles of its assembly forms for regulating transcription. Proc. Natl. Acad. Sci. USA. 2004;101:2840–2845. doi: 10.1073/pnas.0400109101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Thaw P, Sedelnikova SE, Muranova T, Wiese S, Ayora S, Alonso JC, Brinkman AB, Akerboom J, van der Oost J, Rafferty JB. Structural insight into gene transcriptional regulation and effector binding by the Lrp/AsnC family. Nucl. Acids Res. 2006;34:1439–1449. doi: 10.1093/nar/gkl009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Blyn LB, Braaten BA, White-Ziegler CA, Rolfson DH, Low DA. Phase-variation of pyelonephritis-associated pili in Escherichia coli: evidence for transcriptional regulation. EMBO J. 1989;8:613–620. doi: 10.1002/j.1460-2075.1989.tb03416.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Weyand N, Low DA. Regulation of pap phase variation. Lrp is sufficient for the establishment of the phase off pap DNA methylation pattern and repression of pap transcription in vitro. J. Biol. Chem. 2000;275:3192–3200. doi: 10.1074/jbc.275.5.3192. [DOI] [PubMed] [Google Scholar]
- 14.Cui Y, Midkiff MA, Wang Q, Calvo JM. The leucine-responsive regulatory protein (Lrp) from Escherichia coli. Stoichiometry and minimal requirements for binding to DNA. J. Biol. Chem. 1996;271:6611–6617. doi: 10.1074/jbc.271.12.6611. [DOI] [PubMed] [Google Scholar]
- 15.Blyn LB, Braaten BA, Low DA. Regulation of pap pilin phase variation by a mechanism involving differential dam methylation states. EMBO J. 1990;9:4045–4054. doi: 10.1002/j.1460-2075.1990.tb07626.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Braaten BA, Blyn LB, Skinner BS, Low DA. Evidence for a methylation-blocking factor (mbf) locus involved in pap pilus expression and phase variation in Escherichia coli. J. Bacteriol. 1991;173:1789–1800. doi: 10.1128/jb.173.5.1789-1800.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nou X, Skinner B, Braaten B, Blyn L, Hirsch D, Low D. Regulation of pyelonephritis-association pili phase-variation in Escherichia coli: binding of the PapI and Lrp regulatory proteins is controlled by DNA methylation. Mol. Microbiol. 1993;7:545–553. doi: 10.1111/j.1365-2958.1993.tb01145.x. [DOI] [PubMed] [Google Scholar]
- 18.Kaltenbach LS, Braaten BA, Low DA. Specific binding of PapI to Lrppap DNA complexes. J. Bacteriol. 1995;177:6449–6455. doi: 10.1128/jb.177.22.6449-6455.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Weyand NJ, Braaten BA, van der Woude M, Tucker J, Low DA. The essential role of the promoter-proximal subunit of CAP in pap phase variation: Lrp- and helical phase-dependent activation of papBA transcription by CAP from -215. Mol. Microbiol. 2001;39:1504–1522. doi: 10.1046/j.1365-2958.2001.02338.x. [DOI] [PubMed] [Google Scholar]
- 20.Ettema TJG, Brinkman AB, Tani TH, Rafferty JB, van der Oost J. A novel ligand-binding domain involved in regulation of amino acid metabolism in prokaryotes. J. Biol. Chem. 2002;277:37464–37468. doi: 10.1074/jbc.M206063200. [DOI] [PubMed] [Google Scholar]
- 21.Platko JV, Calvo JM. Mutation affecting the ability of Escherichia coli Lrp to bind DNA, activate transcription, or respond to leucine. J. Bacteriol. 1993;175:1110–1117. doi: 10.1128/jb.175.4.1110-1117.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.O’Gara M, Zhang X, Roberts RJ, Cheng X. Structure of a binary complex of HhaI methyltransferase with S-adenosyl-L-methionine formed in the presence of a short non-specific DNA oligonucleotide. J. Mol. Biol. 1999;287:201–209. doi: 10.1006/jmbi.1999.2608. [DOI] [PubMed] [Google Scholar]
- 23.Beloin C, Jeusset J, Revet B, Mirambeau G, Le Hegarat F, Le Cam E. Contribution of DNA conformation and topology in right-handed DNA wrapping by the Bacillus subtilis LrpC protein. J. Biol. Chem. 2003;278:5333–5342. doi: 10.1074/jbc.M207489200. [DOI] [PubMed] [Google Scholar]
- 24.Wang Q, Calvo JM. Lrp, a major regulatory protein in Escherichia coli, bends DNA and can organize the assembly of a higher-order nucleoprotein structure. EMBO J. 1993;12:2495–2501. doi: 10.1002/j.1460-2075.1993.tb05904.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhi J, Mathew E, Freundlich M. Lrp binds to two regions in the dadAX promoter region of Escherichia coli to repress and activate transcription directly. Mol. Microbiol. 1999;32:29–40. doi: 10.1046/j.1365-2958.1999.01314.x. [DOI] [PubMed] [Google Scholar]
- 26.Jafri S, Chen S, Calvo JM. ilvIH operon expression in Escherichia coli requires Lrp binding to two distinct regions of DNA. J. Bacteriol. 2002;184:5293–5300. doi: 10.1128/JB.184.19.5293-5300.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chen S, Calvo JM. Leucine-induced dissociation of Escherichia coli Lrp hexadecamers to octamers. J. Mol. Biol. 2002;318:1031–1042. doi: 10.1016/S0022-2836(02)00187-0. [DOI] [PubMed] [Google Scholar]
- 28.Xu HE, Rould MA, Xu W, Epstein JA, Maas RL, Pabo CO. Crystal structure of the human Pax6 paired domain-DNA complex reveals specific roles for the linker region and carboxy-terminal subdomain in DNA binding. Genes Dev. 1999;13:1263–1275. doi: 10.1101/gad.13.10.1263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hernday AD, Braaten BA, Low DA. The mechanism by which DNA adenine methylase and PapI activate the pap epigenetic switch. Mol. Cell. 2003;12:947–957. doi: 10.1016/s1097-2765(03)00383-6. [DOI] [PubMed] [Google Scholar]
- 30.Chen S, Hao Z, Bieniek E, Calvo JM. Modulation of Lrp action in Escherichia coli by leucine: effects on non-specific binding of Lrp to DNA. J. Mol. Biol. 2001;314:1067–1075. doi: 10.1006/jmbi.2000.5209. [DOI] [PubMed] [Google Scholar]
- 31.Hernday A, Krabbe M, Braaten B, Low D. Self-perpetuating epigenetic switches in bacteria. Proc. Natl. Acad. Sci. USA. 2002;99:16470–16476. doi: 10.1073/pnas.182427199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ernsting BR, Denninger JW, Blumental RM, Matthews RG. Regulation of the gltBDF operon of Escherichia coli: how is a leucine-insensitive operon regulated by the leucine-responsive regulatory protein? J. Bacteriol. 1993;175:7160–7169. doi: 10.1128/jb.175.22.7160-7169.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Aggarwal AK. Crystallization of DNA binding proteins with oligodeoxynucleotides. Methods. 1990;1:83–90. doi: 10.1016/S1046-2023(05)80150-1. [DOI] [PubMed] [Google Scholar]
- 34.Sam MD, Horton NC, Nissan TA, Perona JJ. Catalytic efficiency and sequence selectivity of a restriction endonuclease modulated by a distal manganese ion binding site. J. Mol. Biol. 2001;306:851–861. doi: 10.1006/jmbi.2000.4434. [DOI] [PubMed] [Google Scholar]
- 35.Leslie AG. Integration of macromolecular diffraction data. Acta Crystallogr. D Biol. Crystallogr. 1999;55:1696–1702. doi: 10.1107/s090744499900846x. [DOI] [PubMed] [Google Scholar]
- 36.CCP4 The CCP4 suite: Programs for protein crystallography. Acta Crystallogr. D Biol. Crystallogr. 1994;50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 37.Terwilliger TC, Berendzen J. Automated MAD and MIR structure solution. Acta Crystallogr. D Biol. Crystallogr. 1999;55:849–861. doi: 10.1107/S0907444999000839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Brunger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL. Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr. D Biol. Crystallogr. 1998;54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 39.Jones TA, Zou JY, Cowan SW, Kjeldgaard M. Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Crystallogr. A. 1991;47:110–119. doi: 10.1107/s0108767390010224. [DOI] [PubMed] [Google Scholar]
- 40.Kleywegt GJ, Jones TA. xdlMAPMAN and xdlDATAMAN - programs for reformatting, analysis and manipulation of biomacromolecular electron-density maps and reflection data sets. Acta Crystallogr. D Biol. Crystallogr. 1996;52:826–828. doi: 10.1107/S0907444995014983. [DOI] [PubMed] [Google Scholar]
- 41.Kleywegt GJ, Jones TA. Software for handling macromolecular envelopes. Acta Crystallogr. D Biol. Crystallogr. 1999;55:941–944. doi: 10.1107/s0907444999001031. [DOI] [PubMed] [Google Scholar]
- 42.Jones TA. A set of averaging programs. In: Dodson EJ, Glover S, Wolf W, editors. Molecular Replacement. SERC Daresbury Laboratory; Daresbury, U.K.: 1992. pp. 91–105. [Google Scholar]
- 43.Dayringer H, Tramontano A, Sprang S, Fletterick RJ. Interactive program for visualization and modelling of proteins, nucleic acids and small molecules. J. Mol. Graphics. 1986;4:82–91. [Google Scholar]
- 44.Perona JJ, Martin AM. Conformational transitions and structural deformability of EcoRV endonuclease revealed by crystallographic analysis. J. Mol. Biol. 1997;273:207–225. doi: 10.1006/jmbi.1997.1315. [DOI] [PubMed] [Google Scholar]
- 45.DeLano WL. The PyMOL Molecular Graphics System. 2002 ( http://www.pymol.org)
- 46.Baker NA, Sept D, Joseph S, Holst MJ, McCammon JA. Electrostatics of nanosystems: application to microtubules and the ribosome. Proc. Natl. Acad. Sci. USA. 2001;98:10037–10041. doi: 10.1073/pnas.181342398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Carter ES, Tung CS. NAMOT2—a redesigned nucleic acid modeling tool: construction of non-canonical DNA structures. Comput. Appl. Biosci. 1996;12:25–30. doi: 10.1093/bioinformatics/12.1.25. [DOI] [PubMed] [Google Scholar]