

Abstract
The detection of mutant spectra within a population of microorganisms is critical for the management of drug-resistant infections. We performed ultra-deep pyrosequencing to detect minor sequence variants in HIV-1 protease and reverse transcriptase (RT) genes from clinical plasma samples. We estimated empirical error rates from four HIV-1 plasmid clones and used them to develop a statistical approach to distinguish authentic minor variants from sequencing errors in eight clinical samples. Ultra-deep pyrosequencing detected an average of 58 variants per sample compared with an average of eight variants per sample detected by conventional direct-PCR dideoxynucleotide sequencing. In the clinical sample with the largest number of minor sequence variants, all 60 variants present in ≥3% of genomes and 20 of 35 variants present in <3% of genomes were confirmed by limiting dilution sequencing. With appropriate analysis, ultra-deep pyrosequencing is a promising method for characterizing genetic diversity and detecting minor yet clinically relevant variants in biological samples with complex genetic populations.
Dideoxynucleotide (Sanger) sequencing of non-clonal PCR products (direct PCR sequencing) of plasma viral cDNA is widely used to detect more than 50 drug-resistance mutations in the molecular targets of HIV-1 therapy—reverse transcriptase (RT) and protease—in clinical settings (US Department of Health and Human Services Panel on Clinical Practices for Treatment of HIV Infection 2006). A major limitation of direct PCR sequencing, however, is its inability to detect low proportions of drug-resistant variants in the heterogeneous virus population existing in a patient’s plasma sample (Palmer et al. 2005). Several studies have shown that minor drug-resistant variants that are not detected by population-based sequencing are clinically relevant in that they are often responsible for the virological failure of a new antiretroviral treatment regimen (Jourdain et al. 2004; Kapoor et al. 2004; Lecossier et al. 2005; Palmer et al. 2006b). Multiple approaches have been developed to detect minor HIV-1 variants in research settings; however, no single approach has proved useful for clinical settings.
The 454 Life Sciences GS20 sequencing platform allows massively parallel picoliter-scale amplification and pyrosequencing of individual DNA molecules (Margulies et al. 2005). Simons and colleagues described two cases in which ultra-deep pyrosequencing detected minority variant drug-resistance mutations in a previously treated patient in whom mutations were no longer detectable by standard direct PCR sequencing (Simons et al. 2005). Tsibris and colleagues demonstrated that ultra-deep pyrosequencing could accurately quantify a mixture of three HIV-1 envelope variants pooled in defined proportions of 89%, 10%, and 1% (Tsibris et al. 2006). Here, we systematically investigate the potential of ultra-deep pyrosequencing to detect minor variants in the HIV-1 RT and protease genes from clinical plasma samples. We characterize the types of sequence errors associated with such ultra-deep pyrosequencing of HIV-1 virus populations, develop statistical methods for handling these errors, and validate our approach using molecular and limiting dilution clonal Sanger sequencing.
Results
Plasmid DNA clones and clinical RT-PCR products
We performed ultra-deep pyrosequencing on four plasmid DNA clones of cultured HIV-1 isolates and eight RT-PCR products from RNA extracted from cryopreserved plasma samples. Each sequenced sample encompassed all 99 HIV-1 protease codons and the first 241 reverse transcriptase codons. Sample preparation and ultra-deep pyrosequencing are explained in detail in the Methods section and illustrated in Supplemental Figure 1. The plasmid clones included the laboratory strain NL43 (M19921; which was sequenced twice) and two recombinant viruses in which RT codons 24–312 were replaced with cloned cDNA from two multidrug-resistant clinical virus isolates (AY35744, AY351750). The RT-PCR products were obtained from an untreated HIV-1-infected patient in 1992 and from seven antiretroviral-treated patients from 2000 to 2005. The plasma HIV-1 RNA levels in the eight plasma samples were each >100,000 copies/mL. The median number of cDNA copies prior to sequencing was 100 with an inter-quartile range of 75–180.
Sequence data and alignment
The GS20 sequencing platform generated an average of 6827 reads per sample (mean length of 105 nucleotides [nt]) on four HIV-1 plasmid DNA clones and eight RT-PCR products derived from HIV-1-infected plasma samples. For all samples, the consensus sequence generated from the GS20 sequence reads matched the sequence generated using direct-PCR Sanger sequencing. On average, 92% of the GS20 nucleotides mapped onto HIV-1 protease, yielding an average number of 530 sequence reads per position (coverage). Coverage at the ends of amplified cDNA product was lower than at the center of the amplified cDNA product (Supplemental Fig. 2).
As noted in the Methods section, we developed a new method we call the Asymmetric Smith-Waterman (ASW) algorithm, which incorporates the phred-equivalent quality scores into the pairwise alignment between GS20 reads and the sequence generated using direct PCR Sanger sequencing. Supplemental Figure 3 provides an example of how incorporating quality scores can improve the accuracy of an alignment. Supplemental Table 1 summarizes the sequence data obtained by applying the ASW algorithm to each sample. Because the individual GS20 reads were usually similar to the reference sequence, the ASW algorithm did not outperform either BLAST or the Smith-Waterman algorithms (data not shown). However, when we benchmarked the three alignment algorithms by aligning GS20 reads to a more distantly related sequence (e.g., a sequence belonging to a different subtype) as might occur in the case of a virus super-infection with a divergent strain, the ASW algorithm maps a slightly higher percentage of nucleotides and has a slightly lower error rate than both BLAST and Smith-Waterman algorithms (Supplemental Table 2).
Determination of sequence accuracy
To characterize the frequency of GS20 sequencing errors, we compared the GS20 reads to the Sanger sequences of the four, non-heterogeneous plasmid clones. Any differences between the two were considered to be GS20 sequencing errors. The overall error rate for the four plasmid clones was 0.0098. Errors comprised insertions (0.0073), deletions (0.0016), and mismatches (0.0012) (Table 1). Because it has previously been reported that the pyrosequencing error rate is higher in regions with nucleotide repeats (Margulies et al. 2005), we determined the error rates separately in homopolymeric and non-homopolymeric regions. In our analysis, homopolymeric regions were defined as regions containing repeats of three or more identical bases and the flanking non-identical bases. Supplemental Figure 4 shows the distribution of errors according to sequence context for one of the four sequenced plasmid clones, illustrating that sequence errors were more frequent in homopolymeric regions. Overall, for all four plasmid clones, mismatches were six times more frequent in homopolymeric regions (0.0044) than in non-homopolymeric regions (0.0007).
Table 1.
Ultra-deep pyrosequencing error frequency by type and sequence context

Singleton mismatch errors—where only one GS20 read differed from the plasmid clone sequence—predominated in both homopolymeric and non-homopolymeric regions. Mismatch errors occurring in two or more reads were much less common (Supplemental Fig. 5). The distribution of errors approximated a Poisson distribution with μ = 0.0007 in non-homopolymeric and μ = 0.0044 in homopolymeric regions. We used this empirically observed distribution of mismatch error rates to distinguish sequence errors from authentic minor variants in the clinical plasma samples. Only those variants whose frequency of occurrence yielded a P-value of <0.001 according to the Poisson model (see Methods for details) were considered authentic. Figure 1 displays the coverage required to detect minor variants at various thresholds in non-homopolymeric and homopolymeric regions using the Poisson model with the empirically determined error rates in this study.
Figure 1.

Relationship between the number of required ultra-deep pyrosequencing reads per position (Required coverage) and the detection limit of the frequency of a minority sequence variant (Detection threshold). The excess number of reads required to detect minor variants is a result of the Poisson model for handling potential sequencing errors. For example, ∼400 (rather than 100) reads are required to detect a minor variant present at about a 1% level in a non-homopolymeric region. Because homopolymeric regions are more likely to contain sequencing errors, higher numbers of reads are required to obtain similar sensitivities in these regions.
HIV-1 variants in clinical plasma sequences
Direct PCR Sanger sequencing detected an average of eight variants, indicated by the presence of an electrophoretic mixture, per sample over the 1020 nucleotides encompassing all 99 protease codons and the first 241 codons of RT. By comparison, ultra-deep pyrosequencing detected an average of 58 variants per sample: all the variants detected by Sanger sequencing plus an additional 50 variants detected only by ultra-deep sequencing. On average, 32 of the additional variants were synonymous and 18 were non-synonymous (Fig. 2). Overall, ultra-deep pyrosequencing detected 72 variants present at a frequency of ≥20%, and 392 variants at a frequency <20%. Consistent with previous reports, direct PCR Sanger sequencing detected the majority (55/72, 76%) of variants present at a frequency of ≥20% but rarely detected variants present at a frequency of <20% (9/392, 2%).
Figure 2.

Sequence variants detected by ultra-deep pyrosequencing of eight clinical plasma samples including seven samples from antiretroviral-experienced patients (F56272, V10606, V11909, V47683, V63557, V65472, V9878) and one sample from an untreated patient (92_2664). Sequence variants were defined as differences from the consensus population-based sequence. The X-axis represents all 99 protease codon positions followed by the first 240 reverse transcriptase codon positions. Positions are demarcated at intervals of 20 codons. The Y-axis shows the frequency of each minor variant observed by ultra-deep pyrosequencing. Synonymous minor variants are shown in gray; non-synonymous variants are shown in red. The presence of more than one variant at the same position is indicated by superimposing variants of lower frequency onto variants with higher frequency. Drug-resistance mutations detected only by ultra-deep pyrosequencing are indicated with arrows. A horizontal line separates minor variants present at above and below a frequency of 20%. As noted in the text, 55 of 72 variants present in >20% of GS20 reads, but only nine of 392 variants present in <20% of GS20, were detected by conventional Sanger sequencing.
Among samples from seven antiretroviral-experienced patients, 16 drug-resistance mutations were detectable only by ultra-deep sequencing (an average of 2.2 per sample): Five were present in >10% of GS20 reads, six in 2%–10% of GS20 reads, and five in <2% of GS20 reads. We compared the GS20-determined prevalence of three of these mutations with that determined by Sanger sequencing of multiple molecular clones. The protease mutation I47V in sample V10606, present in 22.5% of GS20 reads, was detected in 11 of 55 (20.0%) molecular clones. The RT mutations L74I and V75A, present in 10.0% and 15.4% of GS20 reads in sample V9878, were detected in five of 46 (10.9%) and 16 of 46 (34.8%) clones, respectively.
To estimate the reliability of ultra-deep sequencing at detecting minor variants, we performed limiting dilution Sanger sequencing of an estimated 148 cDNA templates encompassing all 1020 nucleotides of the sample with the greatest number of variants detected by ultra-deep pyrosequencing (V11909 in Fig. 2). Of the 95 nucleotide minor variants in this sample, all 60 variants present in >3% of GS20 reads and 20 of 35 (57%) present in <3% of GS20 reads were present in one or more limiting dilution sequences.
The extensive limiting dilution sequencing of V11909 (148 cDNA templates) is theoretically able to detect minor variants present as low as 2% with 95% certainty. This made it possible to independently assess the effect of the statistical filtering procedure on sensitivity and specificity. Overall, 215 minor variants were detected by ultra-deep pyrosequencing, including all 98 minor variants detected by limiting dilution sequencing. If it is assumed that the 98 variants detected by limiting dilution sequencing represent the complete set of variants in the sample, then the sensitivity and specificity of ultra-deep pyrosequencing prior to filtering were 100% (98/98) and 46% (98/215), respectively. Following statistical filtering, 95 variants were detected by ultra-deep pyrosequencing, of which 80 were detected by limiting dilution sequencing, yielding sensitivities and specificities of 82% (80/98) and 84% (80/95), respectively.
Discussion
In this study, we have shown that ultra-deep pyrosequencing using the GS20 Sequencer can reliably detect minor HIV-1 variants present within a 1000-bp region encompassing the >50 HIV-1 drug-resistance mutations. We have also characterized the error rate of GS20 sequencing using plasmid HIV-1 clones and used this rate to develop a statistical approach for identifying and quantifying authentic minority variants. Our results demonstrate that although the GS20 single-read error rate is not high—substitution errors, which are the main causes of ambiguity in this study, occur at a frequency of 0.1%—the error rate nonetheless influences the sensitivity, cost, and potential clinical utility of ultra-deep pyrosequencing for HIV-1 drug-resistance testing.
Although, as we reported, all 60 variants present at the frequency of ≥3% and 20 of 35 variants present at the frequency of <3% in the one most thoroughly investigated clinical plasma sample (V11909) and some low-frequency drug-resistant variants in two additional samples were confirmed, this study does not define the sensitivity of ultra-deep pyrosequencing for detecting minor HIV-1 variants in clinical plasma samples for the following reasons. First, the sensitivity of all methods for detecting minor HIV-1 variants is limited primarily by the number of independent cDNA templates that are isolated from plasma prior to PCR amplification. As plasma HIV-1 RNA levels usually range from 103 to 105 RNA copies/mL (US Department of Health and Human Services Panel on Clinical Practices for Treatment of HIV Infection 2006) and as the efficiency of RNA extraction and reverse transcription of a 1000-bp protease-RT fragment ranges from 1% to 10% (Shafer et al. 1997), it is unusual to isolate >1000 cDNA templates from most clinical plasma samples even if the sample contains a high virus load, the RNA extraction and reverse transcription are highly efficient, and the upstream processing of plasma is nearly complete. Indeed, we isolated a median 100 cDNA templates per sample (inter-quartile range: 75–180) despite confining ourselves to samples with high plasma HIV-1 RNA levels. Improvements that optimize upstream processing of genetic material will make it possible to further exploit the technique’s extreme sensitivity.
Second, the sensitivity of ultra-deep pyrosequencing depends on the coverage or number of reads obtained per base and on the single-read substitution error rate. Our sensitivity was based on a mean coverage of ∼500 and a substitution error rate of 0.0044 in homopolymeric regions and 0.0007 in non-homopolymeric regions. Any increase in sequencing coverage or decrease in error rates would improve sensitivity. For example, if 10,000 reads were obtained per position, then the sensitivity would have been improved to be able to detect minor variants present at as low as 0.67% in homopolymeric regions and 0.17% in non-homopolymeric regions (sensitivities which would of course be meaningful only if the starting material contained well over 100 templates). The GS20 Sequencer that we used for this study is currently being replaced by a second generation sequencer (FLX) that has a longer read-length of 230 nt, a lower error rate, and a higher sequencing capacity. The lower error rate and higher sequencing capacity would both be likely to improve sensitivity.
Finally, although we used high-fidelity enzymes for reverse transcription and PCR amplification, these steps also introduce errors that for reverse transcription—a step necessary for almost all methods of detecting minor variants—the error rate may reach 0.01% (Malet et al. 2003). Those error rates will be significant when detecting very rare variants. Therefore, any study with the capacity of detecting minor variants present at extremely low frequencies must take into account error rates of all upstream enzymes.
Ultra-deep pyrosequencing shares features with the two currently standard approaches for detecting minor drug-resistant HIV-1 variants. Like molecular and limiting dilution clonal sequencing, ultra-deep pyrosequencing provides the sequence at every drug-resistance position encompassed within the region sequenced. However, because the individual read lengths are shorter than those obtained by Sanger sequencing, ultra-deep pyrosequencing does not provide information about the haplotype structure of the viral genomic population within an individual, making it difficult to obtain insights into how drug resistance evolves within individuals.
Ultra-deep pyrosequencing, however, is less laborious and less costly than standard clonal sequencing because cloning is done automatically by a water-in-oil emulsion process that does not require bacterial transformation or numerous dilutions and PCR reactions, and because the cost per base is <10% of the cost of Sanger sequencing, making it possible to sequence each nucleotide hundreds or even thousands of times (Shendure et al. 2004). The GS20 Sequencer has also just been replaced by a second generation machine (FLX Sequencer), which has a read length of 230 nt and which produces twice as many reads at the same cost as the GS20 Sequencer (Langaee and Ronaghi 2005; Leamon et al. 2007).
The second standard approach for detecting minor variants uses allele-specific hybridization to distinguish wild-type from mutant variants at a particular position. Although these methods are less costly than clonal or ultra-deep pyrosequencing they are designed to detect only a small number of drug-resistance mutations. Moreover, their sensitivity is limited by non-hybridization due to heterogeneity surrounding the mutation of interest and their specificity is limited by false-priming. Indeed, the first generation of these tests—the Affymetrix GeneChip HIV PRT440 and the line probe assay (LIPA, Innogenetics)—was unsuccessful for these very reasons, particularly when applied to viruses belonging to non-B subtypes. Subtype B is the most common subtype in the United States, but makes up only ∼10% of subtypes worldwide (Vahey et al. 1999; Tsongalis et al. 2005). Finally, point mutation assays must be optimized for each mutation they are designed to detect to take into account the sequence context in which specific mutations arise.
The Affymetrix GeneChip and line probe assay have since been replaced by a variety of different non-commercial assays. The earliest of these assays, the oligonucleotide ligation assay (OLA), which relied entirely on selective hybridization followed by a confirmatory ligase reaction, has a sensitivity of 5% for minority mutations (Edelstein et al. 1998; Ellis et al. 2004; Halvas et al. 2006). The most recently reported assay, the parallel allele-specific sequencing (PASS), is a minisequencing reaction in which PCR products accumulate around individual DNA templates and the proportion of mutant viruses is inferred from the addition to the PCR products of fluorescent nucleotides complementary to the mutant DNA templates (Cai et al. 2007). The remaining point mutation assays are allele-specific real-time PCR for which quantification of the ratio between the mutant and wild-type allele is based on a standard curve generated using a range of mutant:wild-type mixtures (Hance et al. 2001; Metzner et al. 2003; Flys et al. 2005; Johnson et al. 2005; Moser et al. 2005; Palmer et al. 2006a). The sensitivity of PASS and the real-time PCR assays have generally been reported to be ∼0.1% based on experiments using defined mixtures (Halvas et al. 2006). However, the sensitivity and specificity of point-mutation assays on clinical samples have not been thoroughly investigated because of the impracticality of sequencing thousands of clones to confirm rare minor variants.
The breadth and depth of sequence data obtained by ultra-deep pyrosequencing provide many of the advantages of both clonal sequencing and highly sensitive point mutation assays for HIV-1 and for other chronic viral diseases for which drug resistance has become important including hepatitis B and hepatitis C. Clinical research is required to determine at what point a minor drug-resistant variant becomes clinically relevant. Because HIV-1 RT as well as HBV and HCV polymerase are highly error prone making one error between 1 in 104 to 105 nucleotides, it can be predicted that all HIV-1 mutations naturally occur at a frequency of ∼0.01% (Coffin 1995). Therefore, it is only those variants that occur at higher frequencies and in the right mutation contexts that are likely to be clinically meaningful. In other words, minor mutations are more likely to be significant when they occur within an otherwise viable virus and are linked to mutations associated with immune escape and to mutations associated with resistance to other drugs a patient is receiving. Nonetheless, clinical studies using retrospective plasma samples obtained from patients with well-characterized treatment histories and clinical outcomes will be essential before ultra-deep pyrosequencing can be used as a substitute for direct PCR sequencing.
The HIV-1 viral population existing within an individual is a prototypical biological system in which a consensus sequence represents only a fraction of the biologically and clinically relevant genomic information. Additional similarly complex, medically relevant genetic populations include other RNA viruses, heteroplasmic mitochondrial DNA populations (Loeb et al. 2005), and mixtures of cells from cancerous tissues (Shendure et al. 2004; Thomas et al. 2006). Ultra-deep pyrosequencing, coupled with the analytic methods that we describe here, provides an unprecedented opportunity to characterize diversity in these complex genetic populations. Although we have outlined some of the hurdles that must be overcome for the use of this technology in clinical settings, we are optimistic that ultra-deep pyrosequencing provides a wide range of opportunities for translational research and will have a major impact on the management of chronic viral diseases for which drug resistance limits the benefits of therapy.
Methods
Sample preparation and ultra-deep pyrosequencing
HIV-1 plasmid DNA clones were amplified directly using the enzyme Pfu (Stratagene). Clinical RT-PCR products were created by plasma virus ultracentrifugation followed by RNA extraction, reverse transcription using a high fidelity Superscript III Reverse Transcriptase (Invitrogen), and nested PCR amplification using enzyme Pfu. The plasmid clones were amplified with primers that yielded a 1632-nt amplicon (HXB2 positions 2147–3779) whereas the RT-PCR products were amplified with primers that yielded a 1068-bp product (HXB2 positions 2211–3279).
Plasmid DNA clones and RT-PCR products were sequenced using both direct-PCR Sanger sequencing and ultra-deep pyrosequencing with the 454 Life Sciences GS20 platform. Direct PCR cycle sequencing was performed using AmpliTaq DNA FS Polymerase and dRhodamine terminators (Applied Biosystems). Five sequencing reactions per sample were performed to ensure complete bidirectional sequencing. Base calling was done using Sequencher 4.7 DNA Sequence Analysis Software (Gene Cods Co.) and by manual inspection to identify nucleotide variants.
To perform ultra-deep pyrosequencing, PCR products were nebulized, ligated to adaptors, clonally amplified on capture beads in water-in-oil emulsion micro-reactors, and pyrosequenced using one of eight lanes of a 40 × 75 mm PicoTiterPlate, the standard approach for the “shotgun” sequencing PCR amplicons that are too long to sequence completely with one sequence read (>110 bp for the GS20 Sequencer). Ultra-deep sequencing of eight and four samples was performed at 454 Life Sciences and at the Stanford Genome Technology Center, respectively. For each sample, we obtained an SFF file from which nucleotide sequence data and phred-like quality scores were extracted. On average, 92.0% of nucleotides were mapped onto the corresponding direct PCR population-based sequence.
Two approaches were used to confirm the authenticity of minor variants detected by ultra-deep pyrosequencing. For two samples (V9878, V10606) containing drug-resistant mutations detected only by ultra-deep pyrosequencing, we used the Sanger method to sequence 46 and 55 plasmid subclones (Zero Blunt Cloning Kit, Invitrogen) per sample, respectively. For the one sample with the greatest number of minor variants (V11909), the unamplified cDNA product was serially diluted 1/10, 1/30, 1/100, and 1/300 prior to PCR amplification. Bidirectional sequencing was performed directly on 37 amplicons derived from the 1/30 cDNA dilutions and 31 amplicons derived from the 1/100 cDNA dilutions. This sequencing approach is estimated to have sampled about 148 cDNA molecules (1.2 × 31 + 3 × 37 = 148), which, theoretically, should detect minor variants present at 2% with ∼95% confidence. In total, 133 mixed bases were detected from 30 out of 37 amplicons derived from the 1/30 cDNA dilutions, and 45 mixed bases were detected from 12 out of 31 amplicons derived from the 1/100 cDNA dilutions.
Sequence alignment
Each GS20 Sequencer read was mapped onto the sequence using a modification of the Smith-Waterman algorithm—Asymmetric Smith Waterman (ASW)—that we developed to incorporate the phred-equivalent quality value for each GS20 base call. The phred-equivalent quality value (q) is given by the log-transformed probability p of the base call being incorrect according to the equation: q = −10 ⋅ log10p. Thus, a base call with a quality value of q will have a probability of 10−q/10of being incorrect. To apply this algorithm, we transformed the phred scores into reliability weights, w = 1.0 − 10−q/10, and applied these weights asymmetrically (i.e., to the GS20 reads but not to the reference sequence):
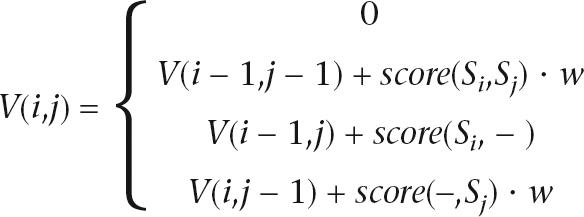 |
The parameters for alignment are as follows: match: 1, mismatch: −3, gap open: 5, gap extension: 2.
Statistical analysis
Ultra-deep sequence errors in both homopolymeric and non-homopolymeric regions were determined by comparing GS20 reads with the known sequences of plasmid clones. The distribution in the frequency of these errors was approximated using the Poisson distribution (Supplemental Fig. 5). For minor variants with n occurrences in N reads, we calculated the probability that such a variant would occur n or more times if it were a sequencing error, using the following formula:
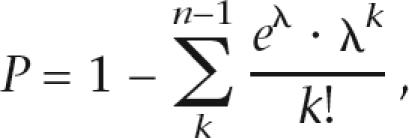 |
where λ is the expected number of errors given N reads and is computed byλ = N ⋅ μ and μ is the error rate per site estimated from the sequences of plasmid clones. Variants that yielded P < 0.001 were considered highly unlikely to be sequencing errors.
The GS20 sequencing system generated sequence reads from both strands of the PCR product. Because authentic variants should exhibit similar frequencies on each strand, we applied a two-tailed Fisher's exact test to identify variants detected disproportionately in one direction after applying a correction for multiple comparisons. Minor variants with P < 0.001 were considered biased to one strand and were not considered to be authentic variants.
Acknowledgments
C.W., Y.M., and R.W.S. were supported in part by grants from the National Institutes of Allergy and Infectious Diseases (AI46148, AI-068581). The work was made possible in part by a High End Instrumentation award from the National Center for Research Resources (1S10RR022982). We thank Roxana Jalili and Shadi Shokralla for the excellent technical assistance.
Footnotes
[Supplemental material is available online at www.genome.org. The raw data from this study are available online at http://dbpartners.stanford.edu/454/pub.]
Article published online before print. Article and publication date are online at http://www.genome.org/cgi/doi/10.1101/gr.6468307
References
- Cai F., Chen H., Hicks C.B., Bartlett J.A., Zhu J., Gao F., Chen H., Hicks C.B., Bartlett J.A., Zhu J., Gao F., Hicks C.B., Bartlett J.A., Zhu J., Gao F., Bartlett J.A., Zhu J., Gao F., Zhu J., Gao F., Gao F. Detection of minor drug-resistant populations by parallel allele-specific sequencing. Nat. Methods. 2007;4:123–125. doi: 10.1038/nmeth995. [DOI] [PubMed] [Google Scholar]
- Coffin J.M. HIV population dynamics in vivo: Implications for genetic variation, pathogenesis, and therapy. Science. 1995;267:483–489. doi: 10.1126/science.7824947. [DOI] [PubMed] [Google Scholar]
- Edelstein R.E., Nickerson D.A., Tobe V.O., Manns-Arcuino L.A., Frenkel L.M., Nickerson D.A., Tobe V.O., Manns-Arcuino L.A., Frenkel L.M., Tobe V.O., Manns-Arcuino L.A., Frenkel L.M., Manns-Arcuino L.A., Frenkel L.M., Frenkel L.M. Oligonucleotide ligation assay for detecting mutations in the human immunodeficiency virus type 1 pol gene that are associated with resistance to zidovudine, didanosine, and lamivudine. J. Clin. Microbiol. 1998;36:569–572. doi: 10.1128/jcm.36.2.569-572.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellis G.M., Mahalanabis M., Beck I.A., Pepper G., Wright A., Hamilton S., Holte S., Naugler W.E., Pawluk D.M., Li C.C., Mahalanabis M., Beck I.A., Pepper G., Wright A., Hamilton S., Holte S., Naugler W.E., Pawluk D.M., Li C.C., Beck I.A., Pepper G., Wright A., Hamilton S., Holte S., Naugler W.E., Pawluk D.M., Li C.C., Pepper G., Wright A., Hamilton S., Holte S., Naugler W.E., Pawluk D.M., Li C.C., Wright A., Hamilton S., Holte S., Naugler W.E., Pawluk D.M., Li C.C., Hamilton S., Holte S., Naugler W.E., Pawluk D.M., Li C.C., Holte S., Naugler W.E., Pawluk D.M., Li C.C., Naugler W.E., Pawluk D.M., Li C.C., Pawluk D.M., Li C.C., Li C.C., et al. Comparison of oligonucleotide ligation assay and consensus sequencing for detection of drug-resistant mutants of human immunodeficiency virus type 1 in peripheral blood mononuclear cells and plasma. J. Clin. Microbiol. 2004;42:3670–3674. doi: 10.1128/JCM.42.8.3670-3674.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flys T., Nissley D.V., Claasen C.W., Jones D., Shi C., Guay L.A., Musoke P., Mmiro F., Strathern J.N., Jackson J.B., Nissley D.V., Claasen C.W., Jones D., Shi C., Guay L.A., Musoke P., Mmiro F., Strathern J.N., Jackson J.B., Claasen C.W., Jones D., Shi C., Guay L.A., Musoke P., Mmiro F., Strathern J.N., Jackson J.B., Jones D., Shi C., Guay L.A., Musoke P., Mmiro F., Strathern J.N., Jackson J.B., Shi C., Guay L.A., Musoke P., Mmiro F., Strathern J.N., Jackson J.B., Guay L.A., Musoke P., Mmiro F., Strathern J.N., Jackson J.B., Musoke P., Mmiro F., Strathern J.N., Jackson J.B., Mmiro F., Strathern J.N., Jackson J.B., Strathern J.N., Jackson J.B., Jackson J.B., et al. Sensitive drug-resistance assays reveal long-term persistence of HIV-1 variants with the K103N nevirapine (NVP) resistance mutation in some women and infants after the administration of single-dose NVP: HIVNET 012. J. Infect. Dis. 2005;192:24–29. doi: 10.1086/430742. [DOI] [PubMed] [Google Scholar]
- Halvas E.K., Aldrovandi G.M., Balfe P., Beck I.A., Boltz V.F., Coffin J.M., Frenkel L.M., Hazelwood J.D., Johnson V.A., Kearney M., Aldrovandi G.M., Balfe P., Beck I.A., Boltz V.F., Coffin J.M., Frenkel L.M., Hazelwood J.D., Johnson V.A., Kearney M., Balfe P., Beck I.A., Boltz V.F., Coffin J.M., Frenkel L.M., Hazelwood J.D., Johnson V.A., Kearney M., Beck I.A., Boltz V.F., Coffin J.M., Frenkel L.M., Hazelwood J.D., Johnson V.A., Kearney M., Boltz V.F., Coffin J.M., Frenkel L.M., Hazelwood J.D., Johnson V.A., Kearney M., Coffin J.M., Frenkel L.M., Hazelwood J.D., Johnson V.A., Kearney M., Frenkel L.M., Hazelwood J.D., Johnson V.A., Kearney M., Hazelwood J.D., Johnson V.A., Kearney M., Johnson V.A., Kearney M., Kearney M., et al. Blinded, multicenter comparison of methods to detect a drug-resistant mutant of human immunodeficiency virus type 1 at low frequency. J. Clin. Microbiol. 2006;44:2612–2614. doi: 10.1128/JCM.00449-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hance A.J., Lemiale V., Izopet J., Lecossier D., Joly V., Massip P., Mammano F., Descamps D., Brun-Vezinet F., Clavel F., Lemiale V., Izopet J., Lecossier D., Joly V., Massip P., Mammano F., Descamps D., Brun-Vezinet F., Clavel F., Izopet J., Lecossier D., Joly V., Massip P., Mammano F., Descamps D., Brun-Vezinet F., Clavel F., Lecossier D., Joly V., Massip P., Mammano F., Descamps D., Brun-Vezinet F., Clavel F., Joly V., Massip P., Mammano F., Descamps D., Brun-Vezinet F., Clavel F., Massip P., Mammano F., Descamps D., Brun-Vezinet F., Clavel F., Mammano F., Descamps D., Brun-Vezinet F., Clavel F., Descamps D., Brun-Vezinet F., Clavel F., Brun-Vezinet F., Clavel F., Clavel F. Changes in human immunodeficiency virus type 1 populations after treatment interruption in patients failing antiretroviral therapy. J. Virol. 2001;75:6410–6417. doi: 10.1128/JVI.75.14.6410-6417.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson J.A., Li J.F., Morris L., Martinson N., Gray G., McIntyre J., Heneine W., Li J.F., Morris L., Martinson N., Gray G., McIntyre J., Heneine W., Morris L., Martinson N., Gray G., McIntyre J., Heneine W., Martinson N., Gray G., McIntyre J., Heneine W., Gray G., McIntyre J., Heneine W., McIntyre J., Heneine W., Heneine W. Emergence of drug-resistant HIV-1 after intrapartum administration of single-dose nevirapine is substantially underestimated. J. Infect. Dis. 2005;192:16–23. doi: 10.1086/430741. [DOI] [PubMed] [Google Scholar]
- Jourdain G., Ngo-Giang-Huong N., Le Coeur S., Bowonwatanuwong C., Kantipong P., Leechanachai P., Ariyadej S., Leenasirimakul P., Hammer S., Lallemant M., Ngo-Giang-Huong N., Le Coeur S., Bowonwatanuwong C., Kantipong P., Leechanachai P., Ariyadej S., Leenasirimakul P., Hammer S., Lallemant M., Le Coeur S., Bowonwatanuwong C., Kantipong P., Leechanachai P., Ariyadej S., Leenasirimakul P., Hammer S., Lallemant M., Bowonwatanuwong C., Kantipong P., Leechanachai P., Ariyadej S., Leenasirimakul P., Hammer S., Lallemant M., Kantipong P., Leechanachai P., Ariyadej S., Leenasirimakul P., Hammer S., Lallemant M., Leechanachai P., Ariyadej S., Leenasirimakul P., Hammer S., Lallemant M., Ariyadej S., Leenasirimakul P., Hammer S., Lallemant M., Leenasirimakul P., Hammer S., Lallemant M., Hammer S., Lallemant M., Lallemant M. Intrapartum exposure to nevirapine and subsequent maternal responses to nevirapine-based antiretroviral therapy. N. Engl. J. Med. 2004;351:229–240. doi: 10.1056/NEJMoa041305. [DOI] [PubMed] [Google Scholar]
- Kapoor A., Jones M., Shafer R.W., Rhee S.Y., Kazanjian P., Delwart E.L., Jones M., Shafer R.W., Rhee S.Y., Kazanjian P., Delwart E.L., Shafer R.W., Rhee S.Y., Kazanjian P., Delwart E.L., Rhee S.Y., Kazanjian P., Delwart E.L., Kazanjian P., Delwart E.L., Delwart E.L. Sequencing-based detection of low-frequency human immunodeficiency virus type 1 drug-resistant mutants by an RNA/DNA heteroduplex generator-tracking assay. J. Virol. 2004;78:7112–7123. doi: 10.1128/JVI.78.13.7112-7123.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langaee T., Ronaghi M., Ronaghi M. Genetic variation analyses by Pyrosequencing. Mutat. Res. 2005;573:96–102. doi: 10.1016/j.mrfmmm.2004.07.023. [DOI] [PubMed] [Google Scholar]
- Leamon J.H., Braverman M.S., Rothberg J.M., Braverman M.S., Rothberg J.M., Rothberg J.M. High-throughput, massively parallel DNA sequencing technology for the era of personalized medicine. Gene Ther. Regul. 2007;3:15–31. [Google Scholar]
- Lecossier D., Shulman N.S., Morand-Joubert L., Shafer R.W., Joly V., Zolopa A.R., Clavel F., Hance A.J., Shulman N.S., Morand-Joubert L., Shafer R.W., Joly V., Zolopa A.R., Clavel F., Hance A.J., Morand-Joubert L., Shafer R.W., Joly V., Zolopa A.R., Clavel F., Hance A.J., Shafer R.W., Joly V., Zolopa A.R., Clavel F., Hance A.J., Joly V., Zolopa A.R., Clavel F., Hance A.J., Zolopa A.R., Clavel F., Hance A.J., Clavel F., Hance A.J., Hance A.J. Detection of minority populations of HIV-1 expressing the K103N resistance mutation in patients failing nevirapine. J. Acquir. Immune Defic. Syndr. 2005;38:37–42. doi: 10.1097/00126334-200501010-00007. [DOI] [PubMed] [Google Scholar]
- Loeb L.A., Wallace D.C., Martin G.M., Wallace D.C., Martin G.M., Martin G.M. The mitochondrial theory of aging and its relationship to reactive oxygen species damage and somatic mtDNA mutations. Proc. Natl. Acad. Sci. 2005;102:18769–18770. doi: 10.1073/pnas.0509776102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malet I., Belnard M., Agut H., Cahour A., Belnard M., Agut H., Cahour A., Agut H., Cahour A., Cahour A. From RNA to quasispecies: A DNA polymerase with proofreading activity is highly recommended for accurate assessment of viral diversity. J. Virol. Methods. 2003;109:161–170. doi: 10.1016/s0166-0934(03)00067-3. [DOI] [PubMed] [Google Scholar]
- Margulies M., Egholm M., Altman W.E., Attiya S., Bader J.S., Bemben L.A., Berka J., Braverman M.S., Chen Y.J., Chen Z., Egholm M., Altman W.E., Attiya S., Bader J.S., Bemben L.A., Berka J., Braverman M.S., Chen Y.J., Chen Z., Altman W.E., Attiya S., Bader J.S., Bemben L.A., Berka J., Braverman M.S., Chen Y.J., Chen Z., Attiya S., Bader J.S., Bemben L.A., Berka J., Braverman M.S., Chen Y.J., Chen Z., Bader J.S., Bemben L.A., Berka J., Braverman M.S., Chen Y.J., Chen Z., Bemben L.A., Berka J., Braverman M.S., Chen Y.J., Chen Z., Berka J., Braverman M.S., Chen Y.J., Chen Z., Braverman M.S., Chen Y.J., Chen Z., Chen Y.J., Chen Z., Chen Z., et al. Genome sequencing in microfabricated high-density picolitre reactors. Nature. 2005;437:376–380. doi: 10.1038/nature03959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metzner K.J., Bonhoeffer S., Fischer M., Karanicolas R., Allers K., Joos B., Weber R., Hirschel B., Kostrikis L.G., Gunthard H.F., Bonhoeffer S., Fischer M., Karanicolas R., Allers K., Joos B., Weber R., Hirschel B., Kostrikis L.G., Gunthard H.F., Fischer M., Karanicolas R., Allers K., Joos B., Weber R., Hirschel B., Kostrikis L.G., Gunthard H.F., Karanicolas R., Allers K., Joos B., Weber R., Hirschel B., Kostrikis L.G., Gunthard H.F., Allers K., Joos B., Weber R., Hirschel B., Kostrikis L.G., Gunthard H.F., Joos B., Weber R., Hirschel B., Kostrikis L.G., Gunthard H.F., Weber R., Hirschel B., Kostrikis L.G., Gunthard H.F., Hirschel B., Kostrikis L.G., Gunthard H.F., Kostrikis L.G., Gunthard H.F., Gunthard H.F. Emergence of minor populations of human immunodeficiency virus type 1 carrying the M184V and L90M mutations in subjects undergoing structured treatment interruptions. J. Infect. Dis. 2003;188:1433–1443. doi: 10.1086/379215. [DOI] [PubMed] [Google Scholar]
- Moser M.J., Ruckstuhl M., Larsen C.A., Swearingen A.J., Kozlowski M., Bassit L., Sharma P.L., Schinazi R.F., Prudent J.R., Ruckstuhl M., Larsen C.A., Swearingen A.J., Kozlowski M., Bassit L., Sharma P.L., Schinazi R.F., Prudent J.R., Larsen C.A., Swearingen A.J., Kozlowski M., Bassit L., Sharma P.L., Schinazi R.F., Prudent J.R., Swearingen A.J., Kozlowski M., Bassit L., Sharma P.L., Schinazi R.F., Prudent J.R., Kozlowski M., Bassit L., Sharma P.L., Schinazi R.F., Prudent J.R., Bassit L., Sharma P.L., Schinazi R.F., Prudent J.R., Sharma P.L., Schinazi R.F., Prudent J.R., Schinazi R.F., Prudent J.R., Prudent J.R. Quantifying mixed populations of drug-resistant human immunodeficiency virus type 1. Antimicrob. Agents Chemother. 2005;49:3334–3340. doi: 10.1128/AAC.49.8.3334-3340.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer S., Kearney M., Maldarelli F., Halvas E.K., Bixby C.J., Bazmi H., Rock D., Falloon J., Davey R.T., Dewar R.L., Kearney M., Maldarelli F., Halvas E.K., Bixby C.J., Bazmi H., Rock D., Falloon J., Davey R.T., Dewar R.L., Maldarelli F., Halvas E.K., Bixby C.J., Bazmi H., Rock D., Falloon J., Davey R.T., Dewar R.L., Halvas E.K., Bixby C.J., Bazmi H., Rock D., Falloon J., Davey R.T., Dewar R.L., Bixby C.J., Bazmi H., Rock D., Falloon J., Davey R.T., Dewar R.L., Bazmi H., Rock D., Falloon J., Davey R.T., Dewar R.L., Rock D., Falloon J., Davey R.T., Dewar R.L., Falloon J., Davey R.T., Dewar R.L., Davey R.T., Dewar R.L., Dewar R.L., et al. Multiple, linked human immunodeficiency virus type 1 drug resistance mutations in treatment-experienced patients are missed by standard genotype analysis. J. Clin. Microbiol. 2005;43:406–413. doi: 10.1128/JCM.43.1.406-413.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer S., Boltz V., Maldarelli F., Kearney M., Halvas E.K., Rock D., Falloon J., Davey R.T., Dewar R.L., Metcalf J.A., Boltz V., Maldarelli F., Kearney M., Halvas E.K., Rock D., Falloon J., Davey R.T., Dewar R.L., Metcalf J.A., Maldarelli F., Kearney M., Halvas E.K., Rock D., Falloon J., Davey R.T., Dewar R.L., Metcalf J.A., Kearney M., Halvas E.K., Rock D., Falloon J., Davey R.T., Dewar R.L., Metcalf J.A., Halvas E.K., Rock D., Falloon J., Davey R.T., Dewar R.L., Metcalf J.A., Rock D., Falloon J., Davey R.T., Dewar R.L., Metcalf J.A., Falloon J., Davey R.T., Dewar R.L., Metcalf J.A., Davey R.T., Dewar R.L., Metcalf J.A., Dewar R.L., Metcalf J.A., Metcalf J.A., et al. Selection and persistence of non-nucleoside reverse transcriptase inhibitor-resistant HIV-1 in patients starting and stopping non-nucleoside therapy. AIDS. 2006a;20:701–710. doi: 10.1097/01.aids.0000216370.69066.7f. [DOI] [PubMed] [Google Scholar]
- Palmer S., Boltz V., Martinson N., Maldarelli F., Gray G., McIntyre J., Mellors J., Morris L., Coffin J., Boltz V., Martinson N., Maldarelli F., Gray G., McIntyre J., Mellors J., Morris L., Coffin J., Martinson N., Maldarelli F., Gray G., McIntyre J., Mellors J., Morris L., Coffin J., Maldarelli F., Gray G., McIntyre J., Mellors J., Morris L., Coffin J., Gray G., McIntyre J., Mellors J., Morris L., Coffin J., McIntyre J., Mellors J., Morris L., Coffin J., Mellors J., Morris L., Coffin J., Morris L., Coffin J., Coffin J. Persistence of nevirapine-resistant HIV-1 in women after single-dose nevirapine therapy for prevention of maternal-to-fetal HIV-1 transmission. Proc. Natl. Acad. Sci. 2006b;103:7094–7099. doi: 10.1073/pnas.0602033103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shafer R.W., Levee D.J., Winters M.A., Richmond K.L., Huang D., Merigan T.C., Levee D.J., Winters M.A., Richmond K.L., Huang D., Merigan T.C., Winters M.A., Richmond K.L., Huang D., Merigan T.C., Richmond K.L., Huang D., Merigan T.C., Huang D., Merigan T.C., Merigan T.C. Comparison of QIAamp HCV kit spin columns, silica beads, and phenol-chloroform for recovering human immunodeficiency virus type 1 RNA from plasma. J. Clin. Microbiol. 1997;35:520–522. doi: 10.1128/jcm.35.2.520-522.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shendure J., Mitra R.D., Varma C., Church G.M., Mitra R.D., Varma C., Church G.M., Varma C., Church G.M., Church G.M. Advanced sequencing technologies: Methods and goals. Nat. Rev. Genet. 2004;5:335–344. doi: 10.1038/nrg1325. [DOI] [PubMed] [Google Scholar]
- Simons J.F., Egholm M., Lanza J.R., Turenchalk G., Desany B., Ronan M.T., Knight J.R., Du L., Leamon J.H., Rothberg J.M., Egholm M., Lanza J.R., Turenchalk G., Desany B., Ronan M.T., Knight J.R., Du L., Leamon J.H., Rothberg J.M., Lanza J.R., Turenchalk G., Desany B., Ronan M.T., Knight J.R., Du L., Leamon J.H., Rothberg J.M., Turenchalk G., Desany B., Ronan M.T., Knight J.R., Du L., Leamon J.H., Rothberg J.M., Desany B., Ronan M.T., Knight J.R., Du L., Leamon J.H., Rothberg J.M., Ronan M.T., Knight J.R., Du L., Leamon J.H., Rothberg J.M., Knight J.R., Du L., Leamon J.H., Rothberg J.M., Du L., Leamon J.H., Rothberg J.M., Leamon J.H., Rothberg J.M., Rothberg J.M., et al. 14th International HIV Drug Resistance Workshop. Québec City, Canada: 2005. Ultra-deep sequencing of HIV from drug-resistant patients [abstract 142] [Google Scholar]
- Thomas R.K., Nickerson E., Simons J.F., Janne P.A., Tengs T., Yuza Y., Garraway L.A., LaFramboise T., Lee J.C., Shah K., Nickerson E., Simons J.F., Janne P.A., Tengs T., Yuza Y., Garraway L.A., LaFramboise T., Lee J.C., Shah K., Simons J.F., Janne P.A., Tengs T., Yuza Y., Garraway L.A., LaFramboise T., Lee J.C., Shah K., Janne P.A., Tengs T., Yuza Y., Garraway L.A., LaFramboise T., Lee J.C., Shah K., Tengs T., Yuza Y., Garraway L.A., LaFramboise T., Lee J.C., Shah K., Yuza Y., Garraway L.A., LaFramboise T., Lee J.C., Shah K., Garraway L.A., LaFramboise T., Lee J.C., Shah K., LaFramboise T., Lee J.C., Shah K., Lee J.C., Shah K., Shah K., et al. Sensitive mutation detection in heterogeneous cancer specimens by massively parallel picoliter reactor sequencing. Nat. Med. 2006;12:852–855. doi: 10.1038/nm1437. [DOI] [PubMed] [Google Scholar]
- Tsibris A., Russ C., Lee W., Paredes R., Arnaout R., Honan T., Cahill P., Nusbaum C., Kuritzkes D.R., Russ C., Lee W., Paredes R., Arnaout R., Honan T., Cahill P., Nusbaum C., Kuritzkes D.R., Lee W., Paredes R., Arnaout R., Honan T., Cahill P., Nusbaum C., Kuritzkes D.R., Paredes R., Arnaout R., Honan T., Cahill P., Nusbaum C., Kuritzkes D.R., Arnaout R., Honan T., Cahill P., Nusbaum C., Kuritzkes D.R., Honan T., Cahill P., Nusbaum C., Kuritzkes D.R., Cahill P., Nusbaum C., Kuritzkes D.R., Nusbaum C., Kuritzkes D.R., Kuritzkes D.R. 15th International HIV Drug Resistance Workshop. International Medical Press; Sitges, Spain: 2006. Detection and quantification of minority HIV-1 env V3 loop sequences by ultra-deep sequencing: Preliminary results. [Google Scholar]
- Tsongalis G.J., Gleeson T., Rodina M., Anamani D., Ross J., Joanisse I., Tanimoto L., Ziermann R., Gleeson T., Rodina M., Anamani D., Ross J., Joanisse I., Tanimoto L., Ziermann R., Rodina M., Anamani D., Ross J., Joanisse I., Tanimoto L., Ziermann R., Anamani D., Ross J., Joanisse I., Tanimoto L., Ziermann R., Ross J., Joanisse I., Tanimoto L., Ziermann R., Joanisse I., Tanimoto L., Ziermann R., Tanimoto L., Ziermann R., Ziermann R. Comparative performance evaluation of the HIV-1 LiPA protease and reverse transcriptase resistance assay on clinical isolates. J. Clin. Virol. 2005;34:268–271. doi: 10.1016/j.jcv.2005.01.011. [DOI] [PubMed] [Google Scholar]
- US Department of Health and Human Services Panel on Clinical Practices for Treatment of HIV Infection . 2006. [Google Scholar]
- Vahey M., Nau M.E., Barrick S., Cooley J.D., Sawyer R., Sleeker A.A., Vickerman P., Bloor S., Larder B., Michael N.L., Nau M.E., Barrick S., Cooley J.D., Sawyer R., Sleeker A.A., Vickerman P., Bloor S., Larder B., Michael N.L., Barrick S., Cooley J.D., Sawyer R., Sleeker A.A., Vickerman P., Bloor S., Larder B., Michael N.L., Cooley J.D., Sawyer R., Sleeker A.A., Vickerman P., Bloor S., Larder B., Michael N.L., Sawyer R., Sleeker A.A., Vickerman P., Bloor S., Larder B., Michael N.L., Sleeker A.A., Vickerman P., Bloor S., Larder B., Michael N.L., Vickerman P., Bloor S., Larder B., Michael N.L., Bloor S., Larder B., Michael N.L., Larder B., Michael N.L., Michael N.L., et al. Performance of the Affymetrix GeneChip HIV PRT 440 platform for antiretroviral drug resistance genotyping of human immunodeficiency virus type 1 clades and viral isolates with length polymorphisms. J. Clin. Microbiol. 1999;37:2533–2537. doi: 10.1128/jcm.37.8.2533-2537.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]