

Abstract
Objective:
To report clinical presentation, perioperative outcome, and long-term results of surgical management of congenital intrahepatic bile duct (IHBD) dilatations (including Caroli disease) in a multi-institutional setting.
Summary Background Data:
Congenital IHBD dilatations are a rare congenital disorder predisposing to intrahepatic stones, cholangitis, and cholangiocarcinoma. The management remains difficult and controversial for bilobar forms of the disease or when concurrent congenital hepatic fibrosis is associated.
Methods:
From 1976 to 2004, 33 patients (range 11 to 79 years) were retrospectively enrolled. Disease extent into the liver was unilobar in 26 patients and bilobar in 7 patients (21%). Cholangiocarcinoma, congenital hepatic fibrosis, and intrahepatic stones were present in 2, 10, and 20 patients, respectively. Transplantations or liver resections were performed in 5 and 27 patients, respectively, whereas 1 asymptomatic patient was managed conservatively.
Results:
Postoperative mortality was nil. Postoperative complications occurred in 16 of 32 operated patients (50%) and additional procedures for residual stones were required in 5 patients. During a median follow-up of 80 months (1 patient being lost for follow-up) no patient developed metachronous carcinoma. Six patients (30%) developed recurrent intrahepatic stones but satisfactory late outcome was achieved in 27 patients (87%).
Conclusions:
Partial or total liver resection achieves satisfactory late outcome in congenital IHBD dilatations, when the affection is treated at an early stage and when the extent of liver resection is tailored to intrahepatic disease extent and takes into consideration the presence and severity of underlying chronic liver and renal diseases.
A retrospective study from 5 European surgical centers enrolled 33 patients with congenital intrahepatic bile duct dilatations, who had undergone liver resection or transplantation. Postoperative mortality was nil and during a median follow-up of 80 months, 87% of the patients achieved satisfactory late outcome.
Congenital intrahepatic bile duct (IHBD) dilatations are a rare disorder resulting from abnormal development of the ductal plate responsive to dilatations of the biliary tree limited to the IHBD,1 belonging thus to the spectrum of congenital bile duct cysts (BDC). Congenital IHBD dilatations correspond to type V BDC according to the Todani et al classification.2 This entity was first defined by Caroli et al3 in 1958, distinguishing between a simple and a fibrous type of the disease, the last one being associated with congenital hepatic fibrosis (CHF) as previously described by Grumbach et al.4 The initial description by Caroli et al concerned communicating IHBD dilatations of peripheral bile ducts.3 However, Guntz et al5 reported later as Caroli disease similar fusiform or saccular IHBD dilatations of large IHBD, the initial disease form described by Caroli et al corresponding to type I in their classification (Fig. 1).

FIGURE 1. Schematic representation of Guntz et al subclassification of congenital IHBD dilatations, according to the aspect and extent of intrahepatic bile ducts dilatations. Type 1: grape-bunch-like saccular communicating dilatations of peripheral IHBD; type 2: fusiform dilatations of large IHBD; type 3: saccular dilatations of large IHBD.
Intrahepatic BDC predispose to biliary stasis and intrahepatic stones (IHS) formation leading to cholangitis, liver abscesses, septicemia, and ultimately to secondary biliary cirrhosis. Moreover, the tendency to cholangiocarcinoma development on these abnormal bile ducts is well documented.6–8 Congenital IHBD dilatations may present itself from a localized form limited to 1 hepatic lobe or segment to a bilobar and diffuse form involving the entire intrahepatic biliary tree. When the disease is associated with coexistent CHF, possibly responsible for portal hypertension, and sometimes to inconstant associated renal disease (from tubular ectasia to polycystic kidney disease), the entity is then called Caroli syndrome.9,10 The management of this rare disease is particularly difficult. The degree of disease extension into the liver and the presence of concurrent underlying chronic liver or kidney disease and cancer are decision-making factors influencing the surgical management. Liver resection is reputed to be the treatment of choice for localized unilobar forms of the disease but the management of diffuse bilobar forms remains controversial.11 Moreover, few data concerning long-term postoperative results are reported in the literature.
The purpose of the present series is to report clinical presentation, perioperative outcome, and to focus on long-term results of the surgical management of congenital IHBD dilatations in a multi-institutional setting according to the disease extent into the liver and the presence and severity of concurrent underlying chronic liver or kidney disease.
PATIENTS AND METHODS
During a 28-year period of time (1976–2004), 142 consecutive patients with BDC were retrospectively enrolled from 5 European academic surgical centers. Liver transplant and pediatric surgery were available in 4 and 2 of these centers, respectively. The type of BDC was classified according to the Todani et al classification,2 based on pre- and intraoperative imaging studies. From this group, 33 patients (23%) issued from 4 of these centers had IHBD dilatations. Four of these patients have already been reported.12,13 IHBD dilatations were defined as the presence of congenital segmental communicating biliary dilatations limited to IHBD.1 The intrahepatic disease extent was defined as being unilobar or bilobar from careful review of imaging studies. The Guntz et al5 subclassification concerning the aspect and location of congenital IHBD dilatations was used, including fusiform or saccular dilatations of large IHBD and “grape-bunch-like” saccular communicating dilatations of peripheral IHBD alternating with normal IHBD (Figs. 1–4). However, when congenital IHBD dilatations were associated with extrahepatic bile duct dilatation, as encountered in 22 of the 142 patients, the disease was not considered as type V BDC but was classified as type IV-A BDC, according to the Todani et al classification. Acquired dilatations of IHBD because of proximal biliary obstruction, such as benign or malignant stricture or obstructive primary IHS, were excluded. Pediatric patients were defined as younger than 15 years. Demographic features, disease, and operative data were collected using standardized questionnaires. Patient operative risk was evaluated according to the American Society of Anesthesiologists (ASA) physical status score.14 The presence of concurrent CHF was confirmed by histologic examination or liver biopsies from both sides of the liver. Radiographic studies included percutaneous ultrasound in 33 patients (100%), computed tomography in 27 patients (82%), magnetic resonance cholangio-pancreatography in 15 patients (45%), and T-tube cholangiography in 2 patients (6%). Invasive imaging studies of the biliary tract, such as endoscopic retrograde cholangio-pancreatography or percutaneous transhepatic cholangiography were used in 9 patients (27%) and 4 patients (12%), respectively. However, the purpose of endoscopic retrograde cholangio-pancreatography and percutaneous transhepatic cholangiography was therapeutic in 8 and 1 patients, respectively. The type of hepatic resection was classified according to the International Hepato-Pancreato-Biliary Association classification reported by Strasberg et al.15 At postoperative follow-up, all patients were assessed by clinical and radiologic examination.
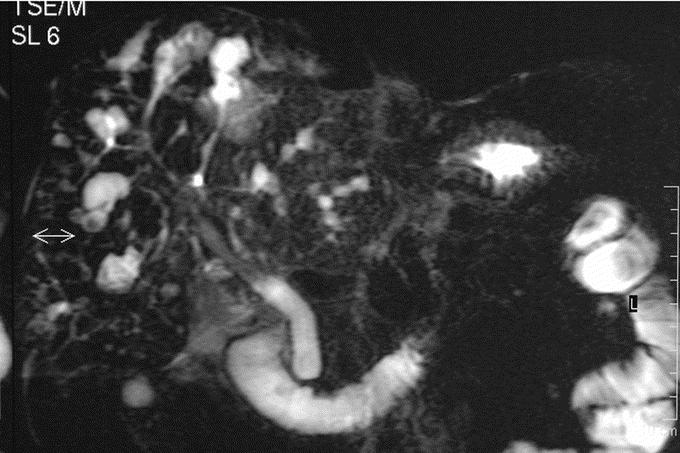
FIGURE 2. Magnetic resonance cholangiography. Communicating IHBD dilatations of peripheral bile ducts corresponding to type I in the Guntz et al classification (Caroli disease).
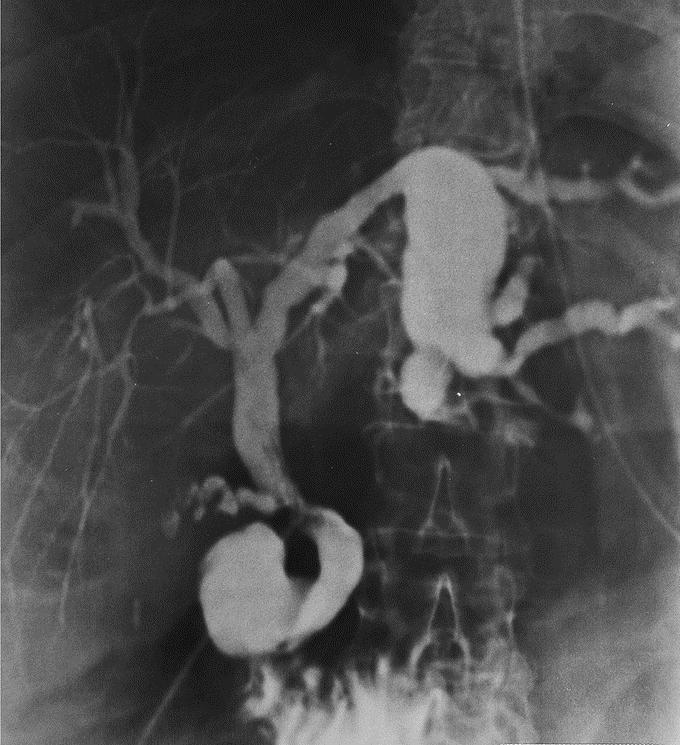
FIGURE 3. Intraoperative cholangiography. Fusiform IHBD dilatations of large IHBD corresponding to type II in the Guntz et al classification.
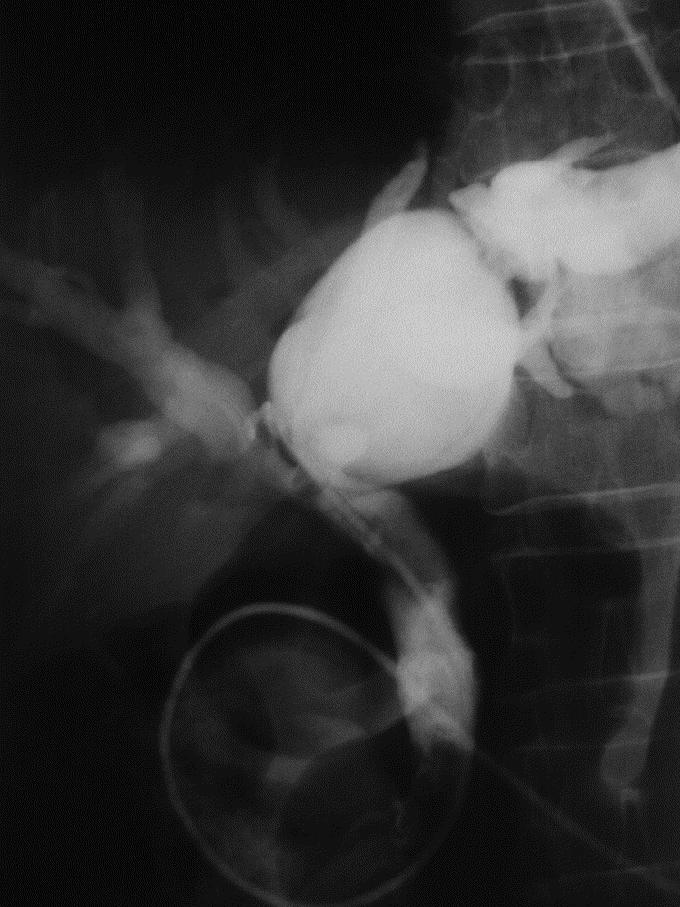
FIGURE 4. Endoscopic retrograde cholangiography: saccular IHBD dilatations of large IHBD corresponding to type III in the Guntz et al classification.
Definition of Endpoints
Evaluation criteria included type and details of operative procedures, early postoperative course, including complications and reoperation rate, postoperative hospital stay, and late patient outcome. Postoperative mortality and morbidity were defined at 2 months or during hospital stay. The severity of postoperative complications was categorized according to the Clavien et al classification.16 Late clinical results were evaluated according to a modification of the Mayo Clinic score of results evaluation previously reported for congenital BDC17: excellent if the patient remained free of symptoms without further reintervention; good if the patient presented occasional and mild attacks of cholangitis or pancreatitis not impairing the quality of life; fair if the patients had repeated episodes of cholangitis or pancreatitis, or had portal hypertension without further reintervention; and poor, if the patient required later biliary or liver-related reoperative procedures, developed biliary cirrhosis or complications because of portal hypertension (such as variceal bleeding, for example), or died of cyst-related malignancy or liver and biliary-related complications.
Statistical Analysis
Statistical analysis was performed using Fisher exact test or χ2 tests for categorical variables and Mann-Whitney nonparametric rank sum tests for continuous variables, when appropriate. A P value <0.05 was considered statistically significant.
RESULTS
Prevalence
The annual incidence per center of congenital IHBD dilatations over a 28-year period was 0.234 (range 0–0.6) in the present series, indicating an obvious referral bias. Indeed, at the 2 centers where experience in HBP, pediatric, and liver transplant surgery was concentrated, the total number of patients varies from 5 to 18, respectively, totaling 70% of the whole study population.
Demographic Features
Thirty-two adults and 1 child (an 11-year-old girl) with congenital IHBD dilatations were included in the present study. The median age of adults (11 women and 21 men) was 55 years (mean age: 53 years, range: 24–79 years). Six patients (18%) were at high risk, being classified ASA III or IV.
Clinical Presentation
The median delay between first symptoms and surgical treatment was 14 months (mean 54 months, range 1–500 months). This delay was <1 year in 13 patients (39%) but >5 years in only 8 patients (24%). Details of clinical patient presentation are reported in Table 1. Complicated clinical presentation was not correlated with unilobar or bilobar intrahepatic disease extent or to duration of symptoms.
TABLE 1. Clinical Presentation and Coexistent Hepatobiliary and Kidney Diseases in Patients Suffering From Congenital IHBD Dilatations

Coexistent HBP and Renal Diseases
Details of HBP and renal coexistent diseases are given in Table 1. Both hepatobiliary and renal coexistent diseases were associated in 6 patients (18%). Synchronous cholangiocarcinoma was encountered in 2 patients (6%), one of which was located within the left hepatic duct, presenting a 3-cm papillary tumor preoperatively diagnosed on imaging studies and corresponding at final pathology to high-grade dysplasia and focal microinvasive cancer. In the second patient, having undergone a left hemihepatectomy, final pathology discovered large focal areas of high-grade dysplasia within the IHBDs. Duration of symptoms in these patients was 1 and 22 months, respectively. Associated renal disease was significantly more frequent in patients with coexistent CHF (without CHF: 2 of 23 patients vs. with CHF: 5 of 10 patients; P = 0.016).
Biliary Disease
Bilobar and unilobar (left hemiliver: 20 patients, right hemiliver: 6 patients) disease extent was observed in 7 patients (21%) and 26 patients (79%), respectively. There was no difference in terms of patient age, sex ratio, ASA score, severity of clinical presentation, coexistent HBP or renal coexistent disease, and previous treatment modalities between patients with unilobar or bilobar disease extent, and between patients with or without CHF, except for coexistent renal disease and portal hypertension, which were more frequent in CHF patients.
According to the Guntz et al5 classification, type I, II, and III were encountered in 9, 22, and 2 patients, respectively. There was no significant difference among the 3 groups in term of patient age, sex ratio, ASA score, severity of clinical presentation, coexistent HBP (including CHF) or renal coexistent disease, and previous treatment modalities.
Previous Disease-Related Treatments
Details of previous biliary-related treatments are reported in Table 1. One patient with CHF associated with portal hypertension had also undergone a surgical portosystemic shunt. Two of the 5 patients who had undergone common bile duct (CBD) exploration at another institution, allowing adequate disease diagnosis, were referred to the secondary referral treatment center where they underwent liver resection within 1 month after referral.
Type of Surgical Procedures
Except for 1 asymptomatic patient with a right unilobar form of congenital IHBD dilatations associated with CHF and with prior kidney transplantation, 32 patients underwent liver resection (27 patients; 84%) or transplantation (5 patients; 16%). The 4 remaining asymptomatic patients who had undergone liver resection had elevated liver function tests related to unilobar or bilobar IHS. No patient underwent emergency operation for control of sepsis. Details of treatment according to unilobar or bilobar disease extension and according to the presence of CHF with or without portal hypertension are reported in Table 2. Transplanted patients with the bilobar form of the disease had associated CHF with portal hypertension, except 2 patients without portal hypertension who were transplanted for repeated attacks of cholangitis from diffuse IHS. Two patients with Caroli disease associated with CHF and chronic renal insufficiency from polycystic kidney disease underwent combined liver and kidney transplantations. In resected patients with unilobar disease extent, a liver resection smaller than formal hemihepatectomy was performed in 10 patients (40%) (Table 2).
TABLE 2. Details on Management of Patients Suffering From Congenital IHBD Dilatations According to Intrahepatic Disease Extent and Presence of Congenital Hepatic Fibrosis (CHF)

Associated procedures were performed in 22 of 32 operated patients (69%), including contralateral IHS extraction (2 patients), CBD exploration (8 patients), cholecystectomy (11 patients), main biliary convergence excision followed by biliodigestive reconstruction with mucosa-to-mucosa hepaticojejunostomy using a 60 cm in length Roux-Y jejunal loop (5 patients) and kidney transplantation or retransplantation (1 patient each). The reason for main biliary convergence excision was not the presence of proximal stricture but of cystic disease involving the liver hilum. The 2 patients with synchronous cholangiocarcinoma were radically treated by left hemihepatectomy and left hemihepatectomy extended to segment 1 associated with extrahepatic bile duct excision and hepaticojejunostomy, respectively.
Postoperative Outcome
Postoperative mortality was nil in the present series. Twenty-two postoperative complications occurred in 16 of the 32 operated patients (50%). The type and severity of postoperative complications according to the extent of liver resection are detailed in Table 3. According to the Clavien et al16 classification, major postoperative complications (namely type 3 and 4) occurred in 7 patients (22%), requiring in all these patients some sort of reoperative procedures. Overall and major postoperative complications occurred in 12 (44%) and 5 patients (19%) after liver resection and in 4 (80%) and 2 patients (40%) after liver transplantation (not significant). Planned postoperative additional therapeutic procedure for residual IHS or CBD stones was required in 5 patients (16%), including endoscopic or radiologic stones extraction in 4 and 1 patient, respectively. Finally, the median postoperative hospital stay was 13 days (mean 18 days; range 8-53 days) in the series.
TABLE 3. Postoperative Complications According to the Extent of Liver Resection

Late Outcome
The not operated patient is alive and free of symptoms and complications after 61 months of follow-up. One patient with left unilobar disease into the liver and without associated CHF having been treated by left lateral sectionectomy was lost to follow-up after an uneventful postoperative course at 2 months. The median follow-up for the 31 remaining patients was 80 months (mean 91 months; range 7–231 months). Twenty-one (67%) and 7 patients (23%) had follow-up longer than 5 and 10 years, respectively. Twenty-eight patients were alive whereas 3 died of nondisease-related events (including the patient presenting with high-grade dysplasia at a follow-up of 144 months). The remaining patient presenting with microinvasive cholangiocarcinoma is alive and disease-free 89 months after surgery.
Patient status during long-term follow-up according to intrahepatic disease extent is reported in Table 4, including symptom-free and disease-free status, details of complications, and reoperative procedures. According to the modified Mayo Clinic evaluation score, excellent, good, fair and poor late results were achieved in 26, 1, 0, and 4 patients, respectively.
TABLE 4. Late Outcome of the 31 Operated Patients According to Intrahepatic Disease Extent and Type of Liver Resection

During the follow-up period, 6 of 20 patients (30%) with initial IHS (one of them harboring postoperative residual IHS) developed recurrent IHS, which led to reoperative procedures in 3 patients. The type of reoperative procedures included segmentectomy 4 after prior left lateral sectionectomy (1 patient), extracorporeal shock-wave lithotripsy of segment 4 stones after prior left lateral sectionectomy (1 patient) and endoscopic extraction procedure in a bilobar form of the disease (1 patient). The 3 remaining patients with recurrent IHS were not surgically treated because of associated secondary biliary cirrhosis with decompensated portal hypertension in 1 patient, the absence of symptoms in 1 patient, and the refusal of surgery by an elderly patient. No patient developed late metachronous carcinoma.
Fair and poor late outcome was observed in 4 patients (13%) in the whole series, which was not significantly different according to intrahepatic disease extent (unilobar: 8%; bilobar: 29%, not significant), the presence of associated CHF (with CHF: 0%; without CHF: 18%, not significant), the type of the disease according to Guntz et al classification (type I: 11%, type II: 14%; type III: 0% (not significant), or the type of surgical resection (liver transplantation: 0%; partial liver resection: 15%, not significant). Fair and poor results were observed in 29% of bilobar disease, 0% of right unilobar disease, and 11% of left unilobar disease (not significant) (Table 4). In unilobar disease extent, fair and poor results were observed in 0% of patients having undergone a hemihepatectomy and in 22% of patients having undergone a liver resection smaller than a hemihepatectomy (not significant) (Table 4).
DISCUSSION
The present study shows that satisfactory long-term results can be achieved in patients with congenital IHBD dilatations when the disease is treated at an early stage and when extent of liver resection is tailored to intrahepatic disease extent and takes into consideration the presence and severity of underlying chronic liver and kidney diseases.
Congenital IHBD dilatations, including Caroli disease, are a rare congenital disorder with an estimated incidence of 1 in 1,000,000 in the whole population.9 Because the original description by Caroli et al3 in 1958, an increasing number of case reports have been reported in the literature. However, very few series exceeding 4 patients treated by liver resection or transplantation are reported11,18–29 (Table 5). In the present series, the mean annual experience per center in treating such patients was less than 0.5. Although the prevalence of congenital IHBD dilatations was estimated by Nagorney30 to be less than 1% of BDC patients, congenital IHBD dilatations represent 23% of all BDC patients in the present series over a 28-year study period. The prevalence observed in our patient population is thus clearly higher than expected and may be the result of referral bias to tertiary referral centers, concentrating experience in pediatric, HBP and liver transplant surgery.
TABLE 5. Reported Series Exceeding 4 Patients Suffering From Congenital IHBD Dilatations Treated by Liver Resection or Liver Transplantation

We sticked to the definition of congenital communicating intrahepatic BDC whatever the level of affected IHBD, adopting the Guntz et al classification,5 even if only type I is corresponding to the initial description of Caroli et al. Indeed, the spectrum of shape and location of IHBD dilatations that has been categorized by Guntz et al5 may be explained by the time and the degree of embryological malformation of the ductal plate.1
A striking particularity of the present series is that 15% of the patients were completely asymptomatic, thus undergoing prophylactic surgery, and that 39% of the patients had a clinical presentation not yet complicated by biliary infection. This feature is in contradiction with other reported series in which most patients with severe septic complications.11,29
Patients with congenital IHBD dilatations have a predisposition for synchronous cholangiocarcinoma with an estimated risk of 100 times greater than that of the general population.31 The prevalence is reported to be 7% by Dayton et al31 in a large literature review of 142 patients, but possibly reaching 24 and 25%25,29 (Table 5). In the present series, the prevalence was relatively low (6%), but both patients had early-stage cholangiocarcinoma leading to long-term cure at 89 and 144 months after liver resection, respectively. The early disease diagnosis, illustrated by the relatively short delay between first symptoms and surgical treatment and the aggressive surgical treatment, even in asymptomatic patients, may concur to the relatively low incidence of synchronous cholangiocarcinoma encountered in the present series. The same features may explain why metachronous cholangiocarcinoma was not encountered in the present series during a mean follow-up of 91 months.
Congenital IHBD dilatations may involve the entire intrahepatic biliary tree diffusely or be confined to 1 lobe or segment. Unilobar extension of the disease is more frequent, especially on the left side of the liver (Table 5).5 Our results are consistent with these findings with 79% of our patients with unilobar extent and 61% from left unilobar disease extent. However, as for the rest of the literature, this feature is clearly the result in the present surgical series of referral bias in patient's selection to surgical centers, with a false overrepresentation of unilobar disease forms. The prevalence of bilobar disease forms is also underestimated because of the lack of referral by physicians for potential surgical treatment.
The treatment of congenital IHBD dilatations is particularly difficult and remains a challenge because of the rareness and the various presentations of the disease, leading to a lack of experience for most of the centers. Various endoscopic or surgical drainage procedures have been reported, such as biliodigestive anastomoses with or without permanent access stoma using a Roux-Y jejunal loop brought to the skin, to allow retrograde access to the intrahepatic biliary tree for further treatments.32,33 Furthermore, in case of proximal intrahepatic ductal strictures, intrahepatic bilioenteric anastomosis was also reported to provide proximal biliary drainage.19,34,35 However, all these drainage procedures are palliative treatment options, which are ineffective in the long term and associated with recurrent septic complications.19,36 They should thus clearly be abandoned.
Indeed, the only potential surgical treatment remains resection of the diseased liver and aggressive endoscopic biliary drainage procedures should be currently strictly limited to inoperable patients.37 The disease extent within IHBD, the presence and severity of concurrent underlying chronic liver and renal diseases and the presence of cholangiocarcinoma are key factors for decision making when curative surgical treatment is considered.
In case of unilobar form of the disease without any concurrent liver fibrosis or cirrhosis, partial hepatectomy achieves satisfactory results by providing a radical solution to the recurrent problems of cholangitis, stone formation, and intrahepatic cholangiocarcinoma.11,18–20,27,29,38–41 In the present series satisfactory long-term results were achieved in 21 of 23 patients (91%) with right or left unilobar forms of the disease after partial liver resection. These findings are in accordance with the recent report by Kassahun et al11 and Bockhorn et al.29 However, attention should be paid to the clearance of contralateral IHS, requiring additional postoperative radiologic or endoscopic procedures in nearly 20% of the patients in the present series. A percutaneous transhepatic approach for flexible choledochoscopic stone clearance with or without intracorporeal lithotripsy could be useful for treating residual IHS.42,43 Additionally, the rate of late complications and reoperation after left lateral sectionectomy in left unilobar congenital IHBD dilatation in the present series emphasized the probable need for performing initially at least hemihepatectomy to remove the whole biliary tree of the affected hemiliver. Moreover, extrahepatic bile duct resection followed by biliodigestive reconstruction may be indicated in up to 25% of the patients,11 when the disease is involving the main biliary convergence.
In case of unilobar form of the disease with concurrent liver fibrosis or cirrhosis, treatment of the coexistent chronic liver disease by liver transplantation has emerged as a valuable option of curative treatment. Indeed, liver transplantation allows radical treatment of both IHBD dilatations (responsible for infectious and malignant complications) and underlying chronic liver disease (responsible for portal hypertension and/or hepatic insufficiency).11,24,26 In the present series, only one such patient with Caroli syndrome with right unilobar disease extent (associated with renal insufficiency and concurrent CHF with variceal bleeding because of the portal hypertension) has benefited from combined kidney-liver transplantation, with excellent long-term results during a postoperative follow-up of 18 years.
In case of bilobar congenital IHBD dilatations without liver fibrosis or cirrhosis, the radical surgical options included extended liver resection or liver transplantation, according to intrahepatic disease extent. When the contralateral disease extent was limited or localized and extirpable, extended liver resection was performed in 3 selected patients, achieving long-term symptom-free and disease-free status in 2 and 1 patient, respectively, during a median follow-up period of 74 months (range: 28–80 months). The limited literature experience with such difficult subgroup of patients confirmed that unsatisfactory long-term outcome is usually achieved after such major hepatectomies.19,24,26,42,44 On the contrary, in case of diffuse bilobar forms of the disease being complicated by cholangitis from diffuse IHS, the sole curative option in our opinion is complete removal of IHBD dilatations by liver transplantation, achieving, good results in the literature45–47 (Table 5). Two patients in the present series underwent a liver transplant in this setting with good late outcome, despite the need for retransplantation for acute humoral rejection in 1 patient. However, Habib et al28 have recently reported a significant poor impact on a 1-year-old patient's survival in the presence of preoperative cholangitis before transplantation for Caroli disease.
In case of bilobar congenital IHBD dilatations complicated by liver fibrosis or cirrhosis, liver transplantation seems to be the treatment of choice.11,23,24,26,28 Two such patients have benefited from this therapeutic option in the present series with excellent long-term results.
Although the surgical management of bilobar forms of congenital IHBD dilatations remains a difficult challenge and controversial, and despite the operative risk in septic patients and consequences of immunosuppressive therapy, we believe that liver transplantation represents the ultimate successful treatment option for complicated forms of the disease (septic complications from diffuse extent of the disease into the liver and fibrosis or cirrhosis responsive to portal hypertension). De Kerckhove et al13 have recently reviewed the reported literature experience concerning 19 patients and the European Liver Transplant Registry (ELTR) experiences concerning 110 patients who had undergone liver transplant for Caroli disease or syndrome, representing 0.2% of the whole number of liver transplants in the ELTR. In the ELTR experience, the median follow-up was 27 months and the 5- and 10-year actuarial survival rates were 86% and 76%, respectively. In case of chronic renal insufficiency because of the coexistent polycystic kidney disease, combined liver-kidney transplantation should be favored, as illustrated in 2 patients in the present series and 14.5% in the ELTR experience. In the recent Habib et al series of 30 transplanted patients for Caroli disease,28 the patient survival rates at 1, 5, and 10 years were 76%, 65%, and 56%, respectively, comparable to survival after liver transplantation for other aetiologies of chronic liver disease. However, the 1-year survival rate was worse in patients with Caroli syndrome whereas there was no difference in long-term survival between Caroli disease and Caroli syndrome patients. However, the appropriate timing for transplantation remains difficult to appreciate in this instance. It should also be emphasized that early disease diagnosis and avoidance of multiple ineffective endoscopic or surgical drainage procedures will undoubtedly reduce the technical and septic risk of liver transplantation.9,28 Finally, the management of invasive cholangiocarcinoma developed on congenital IHBD dilatations is also controversial. Despite the theoretical advantage of liver transplantation to allow radical resection of the carcinoma and of the whole intrahepatic biliary tree at risk for metachronous malignancy,6,8 a high rate of postoperative recurrence has been reported, favored by immunosuppressive therapy. The introduction of new medication combining immunosuppressive and anticancerous effects might resolve this problem. In localized carcinoma on unilobar disease extent, radical liver resection seems to be an appropriate option, when oncologically possible.
Finally, aggressive surgical management of congenital IHBD dilatations was safe in the present series, which was associated with no mortality but with major postoperative complication and reoperation rates of 19% and 6%, respectively. Kassahun et al11 reported a mortality rate of 6% after partial liver resections and 1 patient with aborted right hemihepatectomy for intraoperative septic shock after liver mobilization. Early disease diagnosis, treatment at an early disease stage (including 5 patients at a preclinical stage), strict avoidance of endoscopic diagnostic approach and treatment and avoidance of emergency operation on uncontrollable septic patients without adequate percutaneous transhepatic biliary drainage are concurrent factors to explain the low morbidity rate achieved in the present series. In contradiction with Kassahun et al,11 we promote noninvasive imaging studies, such as magnetic resonance cholangio-pancreatography, to achieve accurate disease diagnosis and staging with no risk of superinfection. Additionally, intraoperative anterior approach using the hanging maneuver48 may be suggested in right-sided congenital IHBD dilatations when septic presentation was initially severe or when superinfection is not completely controlled by percutaneous transhepatic biliary drainage and wide spectrum antibiotic treatment.
In conclusion, the present series suggests that unilobar forms of congenital IHBD dilatations can be successfully treated by liver resection, whereas bilobar diseases with CHF and portal hypertension or diffuse IHS are only curable by liver transplantation. Treatment at an early stage of the disease, and avoidance of an endoscopic approach and of an emergency operation on septic patients are key factors to reduce the operative risk for the patients. Patients with congenital IHBD dilatations should be referred early for surgical management.
Footnotes
Reprints: Jean-François Gigot, MD, PhD, FRCS, Hepatobiliary and Pancreatic Surgical Division, Department of Abdominal Surgery and Transplantation, Saint-Luc University Hospital, Avenue Hippocrate 10, B 1200 Brussels, Belgium. E-mail: gigot@chir.ucl.ac.be.
REFERENCES
- 1.Desmet VJ. Congenital disease of intrahepatic bile ducts: variations on the ductal plate malformation. Hepatology. 1992;16:1069–1083. [DOI] [PubMed] [Google Scholar]
- 2.Todani T, Watanabe Y, Narusue M, et al. Congenital bile duct cyst: classification, operative procedures, and review of 37 cases including cancer arising from choledochal cyst. Am J Surg. 1977;134:263–269. [DOI] [PubMed] [Google Scholar]
- 3.Caroli J, Couinaud C, Soupault R, et al. Une affection nouvelle, sans doute congénitale, des voies biliaires: la dilatation kystique unilobaire des canaux hépatiques. Sem Hop Paris. 1958;34:136–142. [PubMed] [Google Scholar]
- 4.Grumbach R, Bourillon J, Auvert JP. Maladie fibro-kystique du foie avec hypertension portale chez l'enfant. Sem Hop Arch Anat Pathol. 1954;74:30. [Google Scholar]
- 5.Guntz P, Coppo B, Lorimier G, et al. La maladie de Caroli unilobaire: aspects anatomocliniques. Démarche diagnostique et thérapeutique. A propos de 3 observations personnelles et de 101 observations de la littérature. J Chir. 1991;128:167–181. [PubMed] [Google Scholar]
- 6.Balsells J, Margarit C, Murio E, et al. Adenocarcinoma in Caroli's disease treated by liver transplantation. HPB Surg. 1993;7:81–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Totkas S, Hohenberger P. Cholangiocarcinoma associated with segmental Caroli's disease. Eur J Surg Oncol. 2000;26:520–521. [DOI] [PubMed] [Google Scholar]
- 8.Takatsuki M, Uemoto S, Inomata Y, et al. Living-donor liver transplantation for Caroli's disease with intrahepatic adenocarcinoma. J Hepatobiliary Pancreat Surg. 2001;8:284–286. [DOI] [PubMed] [Google Scholar]
- 9.Nagorney DM. Biliary and liver cysts. In: Blumgart LH, Fong Y, eds. Surgery of the Liver and Biliary Tract. Vol. 2. 3rd ed. London: WB Saunders; 2000:1245–1258. [Google Scholar]
- 10.Summerfield JA, Nagafuchi Y, Sherlock S, et al. Hepatobiliary fibropolycystic diseases. A clinical and histological review of 51 patients. J Hepatol. 1986;2:141–156. [DOI] [PubMed] [Google Scholar]
- 11.Kassahun WT, Kahn T, Wittekind C, et al. Caroli's disease: liver resection and liver transplantation. Experience in 33 patients. Surgery. 2005;138:888–898. [DOI] [PubMed] [Google Scholar]
- 12.Partensky C. Hépatectomie droite pour maladie de Caroli. Lyon Chir. 1996;92:165–166. [Google Scholar]
- 13.De Kerckhove L, De Meyer M, Verbaandert C, et al. The place of liver transplantation in Caroli's disease and syndrome. Transpl Int. 2006;19:381–388. [DOI] [PubMed] [Google Scholar]
- 14.American Society of Anaesthesiologists. New classification of physical status. Anesthesiology. 1963;24:111. [Google Scholar]
- 15.Strasberg SM, Belghiti J, Clavien PA, et al. Modified from the Brisbane 2000 terminology of liver anatomy and resections. Terminology committee of the international HPB association. HPB. 2000;2:333–339. [Google Scholar]
- 16.Clavien PA, Sanabria JR, Strasberg SM. Proposed classification of complications of surgery with examples of utility in cholecystectomy. Surgery. 1992;111:518–526. [PubMed] [Google Scholar]
- 17.Gigot JF, Nagorney DM, Farnell MB, et al. Bile duct cysts: a changing spectrum of presentation. J Hepatobiliary Pancreat Surg. 1996;3:405–411. [Google Scholar]
- 18.Nagasue N. Successful treatment of Caroli's disease by hepatic resection. Report of six patients. Ann Surg. 1984;200:718–723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mercadier M, Chigot JP, Clot JP, et al. Caroli's disease. World J Surg. 1984;8:22–29. [DOI] [PubMed] [Google Scholar]
- 20.Knoop M, Keck H, Langrehr JM, et al. Therapy of unilobar Caroli syndrome by liver resection. Chirurg. 1994;65:861–866. [PubMed] [Google Scholar]
- 21.Dagli U, Atalay F, Sasmaz N, et al. Caroli's disease: 1977–1995 experiences. Eur J Gastroenterol Hepatol. 1998;10:109–112. [PubMed] [Google Scholar]
- 22.Gillet M, Favre S, Fontolliet C, et al. Maladie de Caroli monolobaire. A propos de 12 cas. Chirurgie. 1999;124:13–18. [DOI] [PubMed] [Google Scholar]
- 23.Shi LB, Peng SY, Meng XK, et al. Diagnosis and treatment of congenital choledochal cyst: 20 years’ experience in China. World J Gastroenterol. 2001;7:732–734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ammori BJ, Jenkins BL, Lim PM, et al. Surgical strategy for cystic disease of the liver in a western hepatobiliary center. World J Surg. 2002;26:462–469. [DOI] [PubMed] [Google Scholar]
- 25.Levy AD, Rohrmann CA, Murakata LA, et al. Caroli's disease: radiologic spectrum with pathologic correlation. AJR Am J Roentgenol. 2002;179:1053–1057. [DOI] [PubMed] [Google Scholar]
- 26.Ulrich F, Steinmüller T, Settmacher U, et al. Therapy of Caroli's disease by orthotopic liver transplantation. Transplant Proc. 2002;34:2279–2280. [DOI] [PubMed] [Google Scholar]
- 27.Espinoza R, San Martin S, Court F, et al. Hepatic resection in localized Caroli disease. Rev Med Chil. 2003;131:183–189. [PubMed] [Google Scholar]
- 28.Habib S, Shakil O, Couto OF, et al. Caroli's disease and orthotopic liver transplantation. Liver Transpl. 2006;12:416–421. [DOI] [PubMed] [Google Scholar]
- 29.Bockhorn M, Malago M, Lang H, et al. The role of surgery in Caroli's disease. J Am Coll Surg. 2006;202:928–932. [DOI] [PubMed] [Google Scholar]
- 30.Nagorney DM. Bile duct cysts in adults. In: Blumgart LH, Fong Y, eds. Surgery of the Liver and Biliary Tract. Vol. 2. 3rd ed. London: WB Saunders; 2000:1229–1244. [Google Scholar]
- 31.Dayton MT, Longmire WP, Tomkins R. Caroli's disease: a premalignant conditions. Am J Surg. 1983;145:41–48. [DOI] [PubMed] [Google Scholar]
- 32.Barker EM, Kallideen JM. Caroli's disease: successful management using permanent-access hepaticojejunostomy. Br J Surg. 1985;72:641–643. [DOI] [PubMed] [Google Scholar]
- 33.Pridgen JE, Aust JB, McInnis D. Primary intrahepatic gallstones. Arch Surg. 1977;112:1037–1044. [DOI] [PubMed] [Google Scholar]
- 34.Watts DR, Lorenzo GA, Beal JM. Congenital dilatation of the intrahepatic biliary ducts. Arch Surg. 1974;108:592–597. [DOI] [PubMed] [Google Scholar]
- 35.Glenn F, McSherry C. Congenital segmental cystic dilatation of the biliary ductal system. Ann Surg. 1973;177:705–713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Witlin LT, Gadacz TR, Zuidema GD, et al. Transhepatic decompression of the biliary tree in Caroli's disease. Surgery. 1982;97:205–209. [PubMed] [Google Scholar]
- 37.Caroli-Bosc FX, Demarquay JF, Conio M, et al. The role of therapeutic associated with extracorporal shock-wave lithotripsy and bile acid treatment in the management of Caroli's disease. Endoscopy. 1998;30:559–563. [DOI] [PubMed] [Google Scholar]
- 38.Lipsett PA, Pitt HA. Surgical treatment of choledochal cysts. J Hepatobiliary Pancreat Surg. 2003;10:352–359. [DOI] [PubMed] [Google Scholar]
- 39.Ramond MJ, Huguet C, Danan G, et al. Partial hepatectomy in the treatment of Caroli's disease. Report of a case and review of the literature. Dig Dis Sci. 1984;29:367–370. [DOI] [PubMed] [Google Scholar]
- 40.Madariaga JR, Iwatsuki S, Starzl TE, et al. Hepatic resection for cystic lesions of the liver. Ann Surg. 1993;218:610–614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Merono-Cabajosa EA, Celdran-Uriarte A, Moreno-Caparros A, et al. Caroli's disease: study of six cases, including one with epithelial dysplasia. Int Surg. 1993;78:46–49. [PubMed] [Google Scholar]
- 42.Aeberhard P. Surgical management of Caroli's disease involving both lobes of the liver. Br J Surg. 1985;72:651–652. [DOI] [PubMed] [Google Scholar]
- 43.Pitt HA, Venbrux AC, Coleman J, et al. Intrahepatic stones: the transhepatic team approach. Ann Surg. 1994;219:527–537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Izawa K, Tanaka K, Furui J, et al. Extended right lobectomy for Caroli's disease: report of a case and review of hepatectomized cases in Japan. Surg Today. 1993;23:649–655. [DOI] [PubMed] [Google Scholar]
- 45.Sans M, Rimola A, Navasa M, et al. Liver transplantation in patients with Caroli's disease and recurrent cholangitis. Transpl Int. 1997;10:241–244. [DOI] [PubMed] [Google Scholar]
- 46.Schiano TD, Fiel MI, Miller CM, et al. Adult presentation of Caroli's syndrome treated with orthotopic liver transplantation. Am J Gastroenterol. 1997;92:1938–1940. [PubMed] [Google Scholar]
- 47.Waechter FL, Sampaio JA, Pinto RD, et al. The role of liver transplantation in patients with Caroli's disease. Hepatogastroenterology. 2001;48:672–674. [PubMed] [Google Scholar]
- 48.Belghiti J, Guevara OA, Noun R, et al. Liver hanging maneuver: a safe approach to right hepatectomy without liver mobilization. J Am Coll Surg. 2001;193:109–111. [DOI] [PubMed] [Google Scholar]