

Abstract
There is a rapidly growing interest in the family of transient receptor potential (TRP) channels because TRP channels are not only important for many sensory systems, but they are crucial components of the function of neurons, epithelial, blood and smooth muscle cells. These facts make TRP channels important targets for treatment of diseases arising from the malfunction of these channels in the above cells and for treatment of inflammatory pain. TRP channels are also important for a growing number of genetic diseases arising from mutations in various types of TRP channels. The Minerva-Gentner Symposium on TRP channels and Ca2+ signaling, which took place in Eilat, Israel (February 24–28, 2006) has clearly demonstrated that the study of TRP channels is a newly emerging field of biomedicine with prime importance. In the Eilat symposium, investigators who have contributed seminal publications and insight into the TRP field presented their most recent, and in many cases still unpublished, studies. The excellent presentations and excitement generated by them demonstrated that much progress has been achieved. Nevertheless, it was also evident that the field of TRP channels is still in its infancy in comparison to other fields of ion channels, and even the fundamental knowledge of the gating mechanism of TRP channels is still unsolved. The beautiful location of the symposium, together with informal intensive discussions among the participants, contributed to the success of this meeting.
Keywords: TRP channel, PLC, Ca2+ signaling
1. Introduction
In 1969 Cosens and Manning isolated a spontaneous Drosophila mutant with abnormal electroretinogram (ERG), having transient rather than sustained response to prolonged illumination [1]. They designated this strain the A-type mutant and attributed its phenotype to failure in photopigment regeneration [2]. In 1975, Minke et al. analyzed this mutant in great detail and found that its photopigment cycle is normal while the defect is in later stage of the transduction cascade. Minke et al. further suggested designating this mutant transient receptor potential (trp) because of its unique phenotype [3]. Sixteen years later Minke and Selinger proposed that the TRP protein is a Ca2+ channel/transporter [4]. An outline on the history of the Drosophila TRP channels presented by Zvi Selinger (Jerusalem, Israel), clearly showed that the current high interest in TRP channels has developed very slowly and confined for three decades to the Drosophila trp mutation and its physiological implications to the photoreceptor cell. Scientists outside the field of Drosophila phototransduction have become interested in TRP channels only recently. The high interest in TRP appeared only after it was cloned and sequenced by Montell and Rubin [5] and after a patch clamp study by Hardie and Minke [6] and analysis of the protein sequence by Kelly and co-workers (who also discovered the TRP homolog TRPL) [7] provided solid evidence that TRP is most likely a new type of Ca2+ permeable cation channel [6,7]. The final evidence that TRP and TRPL are the light activated channels of the photoreceptor cells came from the studies of Zuker and co-workers who isolated the trpl mutant and showed that the double mutant trpl;trp is blind [8,9]. The identification of the Drosophila TRP as a phosphoinositide-mediated Ca2+ permeable channel [10–12] increased the interest of investigators of Ca2+ signaling in TRP, leading to discovery of mammalian homologues of the Drosophila TRP channels [13–15]. Independent studies on variety biological mechanisms finally revealed the TRP superfamily (for recent reviews see [16–21], Fig. 1). The TRP field has blossomed in the past few years leading to the organization of the Minerva-Gentner Symposium on TRP channels and Ca2+ signaling.
Fig. 1.
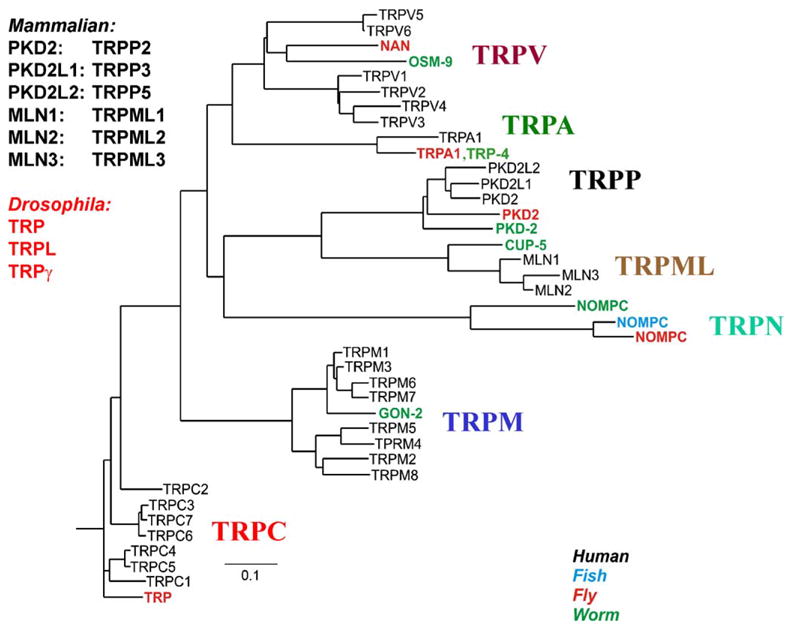
The phylogenetic tree of TRP channels. The figure shows the seven subfamilies that constitute the TRP family. The four different species are indicated by different colors. Only some of the Drosophila and C. elegans (worm) members are included. For more details see [19–21] (from Nilius and Mahieu [21a]) (for interpretation of the references to color in this figure legend, the reader is referred to the web version of the article).
1.1. The family of TRP channels
At present TRP channels form a novel cation channel family consisting of nearly 30 mammalian members, 13 Drosophila members and 17 C. elegans members (Fig. 1). TRP channels are universal biological sensors that detect changes in the environment. TRP channels gate in response to myriad of stimuli including cold or hot temperatures, natural chemical compounds (menthol, camphor, “hot pepper”), mechanical stimuli, or changes in the composition of the lipid bilayer. TRP channels are crucially involved in physiological processes, e.g. photoreception, pheromone sensing, taste perception, thermosensation, pain perception, mechanosensation, perception of pungent compounds (mustard, garlic), renal Ca2+/Mg2+ handling, smooth muscle tone and blood pressure regulation [16–21].
The field of TRP channels is still in its infancy in comparison to fields of other ion channels. Although the general structure of the trans-membrane domains of six (S1–S6) trans-membrane segments and a pore region between S5 and S6 are common for both voltage gated and TRP channels, the differences of TRP channels from voltage gated channels is large enough to justify considering the TRP channels family as a separate family. The large diversity in the primary amino acid sequence among the seven subfamilies that constitute the TRP family makes one wonder if there is a common denominator among the seven subfamilies that justifies including them into one family. The TRP symposium, which took place in Eilat Israel, has clearly demonstrated that it is legitimate to consider the studies dealing with TRP channels as newly emerging field of biomedicine with a prime importance. In the Eilat symposium, investigators who contributed seminal publications and insight to the TRP field presented their most recent (and in many cases still unpublished) studies. The beautiful location of the symposium together with informal intensive discussions among the participants contributed to the great success of this meeting. It was evident to all participants, including those not in the TRP field, that TRP channels are not only crucial for many sensory system [21], but they are also crucial components of the function of neurons, epithelial, blood and smooth muscle cells. These facts make TRP channels important targets for treatment of diseases arising from the malfunction of these channels in the above cells and for treatment of inflammatory pain. TRP channels are also important for a growing number of genetic diseases arising from mutations in various types of TRP channels [22].
2. TRPC channels
2.1. Functional properties of TRPC channels in Drosophila photoreceptors
Although TRPC channels were the first discovered TRP channels, this symposium shows that they are the least characterized. The physiological function of most TRPC members is still not entirely clear and their gating mechanism is unknown and controversial. The Drosophila TRP and TRP-like (TRPL) channels, which mediates Drosophila phototransduction and the mammalian TRPC2, which most likely mediates pheromone sensing are two exceptions, because their physiological functions are well established. Craig Montell (Baltimore, USA), Roger Hardie (Cambridge, UK), Baruch Minke (Jerusalem, Israel, who organized this symposium together with Veit Flockerzi, Homburg, Germany) and Armin Huber (Stuttgart, Germany) presented recent studies on the Drosophila TRP and TRPL channels.
It has been well established that phospholipase Cβ (PLC) is necessary for light activation of the TRP and TRPL channels [10,12,23]. Furthermore, even a small reduction in PLC level has profound effects on the temporal and intensity resolution (the ability to distinguish between different light intensities) of fly vision, because PLC is also essential for response termination acting as GTPase activating protein (GAP [24]). Diacylglycerol (DAG) production following phosphatidylinositol-4,5-bis-phosphate (PIP2) hydrolysis by PLC has emerged as an important stage in excitation [25]. Nevertheless, the mechanism by which DAG affects the gating of Drosophila TRP and TRPL channels is still an enigma for at least two reasons: first, the control of DAG level by DAG kinase (RDGA [26,27]) is problematic as a mechanism for generating a second messenger of excitation because DAG kinase is localized to the endoplasmic reticulum [28] (i.e. at a different compartment from that of the channels which are localized to the rhabdomere). Second, in spite of many attempts exogenous application of DAG failed to activate the TRP and TRPL channels [29]. DAGs putative role in excitation is not limited to Drosophila phototransduction, but it is also a likely messenger of excitation in several mammalian TRPC channels [30] (see also below).
The presentation of Montell mainly covered two topics: (i) DAG production and its effect on phototransduction and (ii) retinal degeneration induced by defects in phototransduction. In addition to its production by light activated PLC, DAG could potentially be produced through a second light independent pathway involving the combined activities of a phospholipase D (PLD) [31] and a phosphatidic acid (PA) phosphatase (PAP), the latter of which converts PA to DAG [31]. Two laboratories generated mutations in an eye-enriched PAP, referred to as Lazaro (Laza): the lab of Padingat Raguh (Cambridge UK, who presented a poster in this symposium, see [32]) and Craig Montell [33]. The Laza mutation caused a reduction in the light response measured by the electroretinogram (ERG, [33]). Mutation of laza also suppressed the severity of the phenotype caused by loss of the DAG kinase, RDGA, indicating that Laza functions in opposition to RDGA, consistent with its predicted biochemical function. Montell and co-workers also showed that the retinal degeneration resulting from overexpression of the PLD is suppressed by elimination of LAZA. The data demonstrate a requirement for a PLD/PAP dependent pathway for achieving the maximal light response. Moreover, the genetic interactions with both rdgA and Pld indicate that Laza functions in the convergence of both PLC and PLD coupled signaling in vivo [33]. The conundrum arising from these studies is that the PLD/PAP dependent pathway produces the putative excitation messenger in the dark, a fact that does not fit with the extreme sensitivity of the photoreceptor cell to single photons and the extremely small noise level in the dark [34].
The studies of Montell and co-workers on retinal degeneration concentrated on the effects of mutations of the TRP channel. Functionally null mutation in TRP results in a very slow (>1 week) light-dependent retinal degeneration [35]. Elimination of the structural anchoring function of TRP by a mutation had minor effects on retinal morphology while disruption of channel function caused more profound and faster light-induced cell death [36]. This retinal degeneration was greatly suppressed by elimination of the Na+/Ca2+ exchanger, CalX [37], indicating that the cell death was due primarily to deficient Ca2+ entry rather than disruption of the TRP anchoring function. A previous study by Yoon, Pak, Minke and co-workers revealed that constitutive activation of TRP by F550I mutation results in profound and very fast photoreceptor cell death (occurring already at late embryonic stage) due to constitutive uncontrolled inward current composed of Ca2+ as the main charge carrier, strongly suggesting that toxic increase in cellular Ca2+ underlies the degeneration [38,39]. Montell and co-workers have found that the retinal degeneration associated with constitutive activity of TRP due to the above mutation is greatly suppressed by overexpression of CalX, which enhances Ca2+ extrusion [37]. Interestingly, constitutive activity of TRPs by anoxic conditions found by Minke and co-workers [40] has been proposed to underlie the massive cell death in the mammalian brain that can occur under anoxic conditions, such as occurs as a result of ischemia [41].
Roger Hardie presented new data, which give insight on the mechanism underlying the relatively large Ca2+ selectivity of the TRP channel [6]. The crucial amino acid residue determining the high Ca2+ selectivity of the TRP channel shows a remarkable structural and functional similarity with the pore of the highly Ca2+ selective TRP channels, TRPV5 and TRPV6. Mutation of this crucial site (D621) did not only remove Ca2+ selectivity of TRP, but also reduce Ca2+ influx via TRP. To study the function of Ca2+ influx on channels and PLC activities, the effects of increasing cellular Ca2+ via the Na–Ca2+ exchanger equilibrium were measured [42]. A genetically targeted electrophysiological biosensor (PIP2 sensitive Kir2.1 channel) was used to measure the rate of PIP2 hydrolysis by PLC in order to measure the effect of Ca2+ on PLC activity [43]. The results indicate that light adaptation is primarily mediated by Ca2+ dependent inhibition of TRP by a still unknown mechanism via global Ca2+ levels (<5 μM), while rapid, protein kinase C (PKC)-dependent inhibition of PLC by local Ca2+ transients (>50 μM) is required to terminate the response and to ensures that PIP2 reserve are not depleted during illumination.
A specific mechanism, by which Ca2+ exerts light adaptation was presented by Minke. Light adaptation is the process by which the photoreceptor cell adjusts its sensitivity to light during increases in the ambient background illumination. In invertebrate photoreceptors this process is strongly Ca2+ dependent [44] and the recent study of Hardie and coworkers described above suggests that a rise in cellular Ca2+ mimics light adaptation by effecting late stage in the cascade downstream of phospholipase Cβ (PLC) and possibly on the TRP channel itself. However, the underlying mechanism is still not clear [42]. Minke and co-workers resolved this issue by application of a quantitative kinetic model of TRPL–Ca2+ interactions. They demonstrated that in contrast to the TRPV and TRPM channels [45] the voltage dependence of the Drosophila TRPL channel is not an intrinsic property but depends on Ca2+ open channel block. Single channel analysis of TRPL expressed in Drosophila S2 cells reveals that the outward rectification of the channel in the presence of Ca2+ arises from a voltage dependent Ca2+ block, solely affecting the channels opening frequency. Thus a linearization of the I–V curve is a sensitive measure of removal of light adaptation. Model calculations indicate that the voltage dependence of the channel arises from changes in the rate constant of Ca2+–TRPL dissociation. Studies of the native TRP and TRPL channels in Drosophila photoreceptors reveal that Ca2+ open channel block underlies the hitherto unknown mechanism of light adaptation.
Armin Huber presented the continuation of a collaborative study with Minke, which demonstrate a most intriguing property of TRP channels namely, a stimulus dependent translocation of the channels between the plasma membrane and the cell body [46]. Signal-mediated translocation of TRPC channels emerges as a novel mechanism to fine tune a variety of signaling pathways including neuronal path finding [47] and Drosophila photoreception [46]. Huber, Paulsen, Minke and co-workers have recently found that the TRPL channels translocate between the signaling compartment and the cell body in a light dependent manner. This translocation modifies the ion channel composition of the signaling membrane and induces long-term adaptation [46]. However, the molecular mechanism underlying TRPL translocation remains unclear. In his presentation Huber reported that eGFP-tagged TRPL expressed in the photoreceptor cells formed functional ion channels with properties similar to the native channels and TRPL-eGFP translocation could be directly visualized in intact eyes. TRPL-eGFP failed to translocate to the cell body in flies carrying severe mutations in essential phototransduction proteins, including rhodopsin, Gαq, PLC and the TRP ion channel, or in proteins required for TRP function. The data, furthermore, show that the activation of a small fraction of rhodopsin and of residual amounts of the Gαq protein is sufficient to trigger TRPL-eGFP internalization. Elimination of external Ca2+ inhibited the light dependent translocation of TRPL. Altogether, this study revealed that activation of the phototransduction cascade is mandatory for TRPL internalization suggesting a critical role for the light induced conductance increase and the ensuing Ca2+ influx in the translocation process.
2.2. Phospholipase C and DAG regulation of mammalian TRPC channels
Mammalian TRPCs are homologues of the Drosophila TRP and TRPL channels. Like the Drosophila channels, they are activated by mechanisms dependent upon PLC. In some instances, they function as store-operated channels (see below), in others as channels more directly coupled to PLC. Jim Putney (Research Triangle Park, USA) and co-workers have recently examined in detail the activation mechanism of two TRPCs, TRPC7, a representative of the TRPC3/6/7 subgroup activated by DAG [30], and TRPC5, a representative of the TRPC1/4/5 subgroup, activated downstream of PLC by an unknown mechanism [48]. Tissue culture studies indicate that TRPC7 functions as a subunit of a DAG-activated channel that is also dependent in an unknown manner on the presence of IP3 receptors. Interestingly, with high levels of over expression, this dependence on IP3 receptors is lost, and single channel properties of the TRPC7 channels are altered. For TRPC5 they have utilized a combination of enzyme inhibitors with imaging, whole cell current and excised patch single channel measurements to reveal multiple and complex mechanisms of regulation by the membrane lipid, PIP2.
The role of TRPC2 in pheromone sensing was presented by Frank Zufall (Baltimore, USA). TRPC2 is a PLC dependent channel with known physiological function. The mammalian vomeronasal organ (VNO) plays a critical role in the detection and transduction of pheromonal signals in many mammals [49,50]. Vomeronasal sensory neurons (VSNs) express TRPC2 at high density in their dendritic tips where pheromonal molecules are expected to bind to specific receptors [51]. Mice with a targeted deletion in the TRPC2 gene show dramatic defects in a variety of social behaviors, suggesting that TRPC2 occupies a fundamental role in the transduction machinery underlying the detection of pheromone signals by the VNO. Patch clamp recordings from VSN dendrites have identified a Ca2+ permeable cation channel that is activated by the PLC product DAG. Activation of this channel by DAG is severely defective in VSNs from TRPC2-deficient mice, suggesting that TRPC2 functions as a subunit of this channel. This ion channel provides the best evidence, thus far, for the existence of native DAG-gated cation channels in the mammalian nervous system [52]. Unpublished work by the Zufall lab investigated the modulation of TRPC2 by Ca2+/calmodulin. This work suggests that the DAG-activated channel is subject to strong modulation by Ca2+/calmodulin. This effect could offer a powerful mechanism for the feedback regulation of pheromone sensitivity in the VNO.
2.3. The importance of microdomains in regulation of TRPC channels
TRPC channels display distinct properties and interact to form homomeric or heteromeric channels with different modes of regulation as well as physiological function [53]. Although the exact function of most TRPC channels and their regulation has not been established, increasing data suggest that they are localized and regulated within spatially distinct Ca2+ signaling microdomains [54]. Indu Ambudkar (Bethesda, USA) reported on studies of TRPC channels in their native membranes and showed that both TRPC1 and TRPC3 are assembled in multi-protein complexes and interact with key Ca2+ signaling proteins as well as trafficking and scaffolding proteins [55]. Both protein and lipid components are involved in the assembly of TRPC channels. Furthermore, vesicular trafficking mechanisms, reminiscent of the Drosophila TRPL translocation are critical determinants of constitutive and regulated TRPC channel activity. Interaction(s) of TRPC channels with proteins within the signaling complex regulates their activity in both Drosophila TRP channel [56] and mammalian TRP channels [57]. Stimulation of the cells expressing native TRPC1/3 by carbachol induced TRPC3 recruitment in the apical membrane and TRPC1 in the basolateral membrane requiring SNARE proteins [57,58]. Indu Ambudkar also found using proteomic approach, possible TRPC3 interacting proteins. Accordingly, RACK1, the receptor of activated C kinase 1 interacts with the N-terminus of TRPC3. Critical sites for this interaction are E232 and E233. Elimination of RACK1 binding site reduces the TRPC3 activation by carbachol and OAG. RACK1 also interacts with the IP3 binding domain of the IP3 receptor, which binds to TRPC channels [59].
2.4. Properties of TRPC4
Veit Flockerzi (Homburg, Germany) studied the expression TRPC4 in several species and alternative splicing as a mechanism of regulation. In mouse human and rat tissues but not in bovine tissues he found at least two variants of the TRPC4 protein: the “full-length” TRPC4 protein (Gene Bank accession number U50922) and a slightly smaller variant TRPC4delta (U50921) with apparent relative molecular weights of 102,000 and 97,000. The delta variant lacks the amino acid residues 781–864, which are present within the full-length protein but both variants have a common C-terminal sequence (amino acid residues 865–974). Usually the DNAs of both variants are amplified during PCR using polyA+ RNA from various mouse tissues as templates and primers derived from the sequences encoding the very N- and C-terminal sequences of TRPC4. Several antibodies directed against epitopes common to both variants have been prepared. Those antibodies always detect both proteins in microsomal membrane protein fractions from mouse tissues including brain, vascular endothelium and intestinal smooth muscle. In contrast to the TRPV6 protein both TRPC4 proteins do not change their electrophoretic mobility after incubation in the presence of various glycosidases. Both proteins run at the same position during sucrose density centrifugation and both proteins are eluted in the same fractions after affinity chromatography. So far no mouse tissue could be identified which expresses only one of the two variants indicating that both proteins contribute to a common TRPC4 channel in mouse tissues. The role of such channels in intestinal smooth muscle [60] is under extensive investigation.
2.5. Store operated (SOC) and receptor operated (ROC) TRPC channels
TRPC genes encode largely non-selective cation channels that allow passage of both mono- and divalent cations, including Ca2+. These channels can be activated by PIP2 hydrolysis leading to formation of IP3 and DAG. All TRPCs except TRPC6 have been shown to be susceptible to activation by store depletion, independently of IP3 formation. This mode of activation requires that the TRPC under study be expressed at low levels [61]. Lutz Birnbaumer (Research Triangle Park, USA) explored the effects of tyrosine kinase (TK) inhibitors on SOC, and two forms of ROC: one mediated by endogenous channels as expressed in several cell lines and ROC-mediated by transfected TRPCs. A complex picture emerged: TRPC3 was found to be absolutely dependent on phosphorylation by src on Y226 in the N-terminus. The close structural relatives TRPC6 and TRPC7, which bear the cognate tyrosine of TRPC3 Y226, do not depend on phosphorylation. Furthermore, one cell line failed to exhibit any ROC in response to activation of a transfected Gq-coupled GPCR (the muscarinic M5 receptor), but respond to thapsigargin with a robust SOC, which is inhibited by TK inhibitors. Finally, comparing the potency with which the tyrosine kinase inhibitor, genistein, inhibits SOC and endogenous ROC differs in different cells [62].
2.6. STIM1, the elusive messenger of SOC
The mechanism of coupling between store-operated channels (SOCs) and Ca2+ store depletion has been a contentious issue for many years [63]. The role of TRPC channels in this process has also been controversial. While DAG is a potential mediator of TRPC3, TRPC6, and TYRPC7 channels, the activation of TRPC1, TRPC4, and TRPC5 remains unclear. TRPC1 channels remain the most likely of the TRPC family to function as SOCs, yet their channel properties do not coincide with the known properties of SOCs [53,63]. Recent RNAi screening approaches have revealed the single spanning membrane protein, STIM1, as being an essential component in the activation of SOCs [64–66]. Don Gill (Baltimore, USA) reported on recent studies on STIM1, which is present in the ER and has a luminal unpaired EF-hand functions as the Ca2+ sensor to trigger SOC activation. STIM1 is also present in the plasma membrane (PM) and seems to play an important role in that membrane, perhaps by coupling to the store-operated channel itself. siRNA for STIM1 causes almost complete loss of SOC activity, but has no effect on TRPC3 or TRPC5 channel function. It has been proposed that store-depletion causes insertion of the STIM protein into the PM [64–66], however, Gill and co-workers have not observed such translocation. Interestingly, the close structural STIM1 homologue, STIM2, has a profoundly different effect on SOC. Overexpression of STIM2 causes almost complete inhibition of SOCs. In contrast, STIM2 has no effect on the function of TRPC3 channels. The STIM2 protein is expressed only in ER, and not in the PM. Store-depletion induces redistribution of the ER STIM1 into distinct “puncta”, but does not alter distribution of STIM2 expressed alone. In contrast, when coexpressed with STIM1, STIM2 translocates into puncta upon store-depletion. Double-labeling shows exact coincidence of STIM1 and STIM2 within the puncta, and immunoprecipitation reveals direct interactions between STIM1 and STIM2. Independent of store-depletion, STIM2 is co-localized with and blocks the function of the D76AE87A-STIM1 EF-hand mutant that preexists in puncta and is constitutively coupled to activate SOCs. STIM2 appears to act within the puncta to interfere with the coupling between STIM1 and its downstream target in the activation of SOCs. The results support an “interactional” rather than “insertional” model for STIM1 in SOC activation [67]. STIM1 appears to function as an essential “mediator” of SOC activation, while the role of STIM2 is likely as an important endogenous regulator of the function of STIM1. So far the STIM proteins appear to function independently of TRPC channels.
2.7. Expression of TRPC proteins in primary human lymphocytes
Many signaling cascades are activated after T-cell receptor (TCR) engagement by antigen-presentation in dendritic cells, macrophages or B-lymphocytes (APCs, antigen presenting cells). Ca2+ entry through calcium release-activated Ca2+ channels (CRAC) is an essential step for T-lymphocyte activation and proliferation [68]. The Ca2+ influx controls the activation of several transcription factors (e.g. NFAT), which regulate the expression of different cytokines such as interleukin (IL)-2, -4, or IFNγ that direct cellular responses. Recent studies discovered CRAC interacting proteins by genome-wide RNA interference (RNAi) screen in Drosophila cells aiming to identify proteins that inhibit SOC. Two interesting proteins were identified: (i) CRAC modulators 1 and 2 (CRACM1 and CRACM2) were identified as modulators of Drosophila CRAC currents and a human ortholog of CRACM1 (encoded by the FLJ14466 gene) was identified [69]. (ii) A modified linkage analysis with single-nucleotide polymorphism arrays, and a Drosophila RNAi screens designed to identify regulators of SOC and NFAT nuclear import, revealed a novel protein designated Orai1, which contains four putative trans-membrane segments. Human with hereditary severe combined immune deficiency (SCID) syndrome show defects in SOC activity. It was found that SCID patients are homozygous for a single missense mutation in ORAI1, and expression of wild-type Orai1 in SCID T cells restores SOC activity [70]. The molecular identity of CRAC channels is still unknown but members of the TRP family appear to be the best candidates to form those channels. To understand the role of TRP channels in Ca2+ signaling, Eva Schwarz, Markus Hoth and co-workers (Homburg, Germany) employed siRNA technology in primary T-lymphocytes. The efficiency of down-regulation was controlled by qRT-PCR. On a single cell level, they analyzed the Ca2+ influx by calcium-imaging experiments where the cells were stimulated with anti-CD3/anti-CD28 coated beads. As a functional assay, they investigated proliferation of siRNA transfected T-lymphocytes in relation to that of control cells. They found a slight reduction in proliferation in TRPC3-siRNA transfected PBLs and hope to get more data and new insights on the role of TRP channels in primary human T-lymphocytes.
3. TRPV channels
3.1. Crystal structures of the N-terminal ankyrin repeat domain of the TRPV1 and TRPV2 ion channels
The TRPV1 and TRPV2 ion channels mediate neuronal responses to many noxious sensory stimuli including heat, low pH, neuropeptides and chemical ligands. All TRPV subfamily members contain an intracellular N-terminal ankyrin repeat domain (ARD)—a prevalent protein-interaction motif [71]. Rachelle Gaudet (Boston, USA) reported on the first crystal structures of the TRPV1- and TRPV2-ARDs. These ARDs, which have six ankyrin repeat structural motifs, reveal several atypical structural features. Repeats one through three display unusually long and flexible fingers with a large number of exposed aromatic residues, whereas repeats five and six have unusually long outer helices. Furthermore, a large counterclockwise twist observed in the stacking of repeats four and five breaks the regularity of the domain, altering the shape of surfaces available for interactions with proteins or other cellular ligands. Both solution studies and crystal packing interactions indicate that the TRPV1- and TRPV2-ARDs do not form homo-oligomers, suggesting that the ARDs may be used for interactions with regulatory factors (see [72]) rather than in promoting tetrameric assembly of the ion channels.
3.2. Phosphoinositide 3-kinase activation underlies NGF-mediated sensitization of TRPV1
Inflammatory hyperalgesia involves sensitization of the pain-transducing ion channel TRPV1 [73]. One proalgesic agent, NGF, signals through the receptor tyrosine kinase TrkA [74]. It is not yet clear, however, whether hyperalgesia is mediated by TrkA activation of phospholipase Cγ (PLCγ), phosphoinositide 3-kinase (PI3K), or both. A sensitization mechanism that utilizes PLCγ predicts that PIP2 inhibits TRPV1 and that hydrolysis of PIP2 will lead to potentiation of TRPV1. Sharona Gordon (Seattle, USA) tested this hypothesis by applying polylysine to sequester acidic lipids in the membrane. Surprisingly, polylysine inhibited TRPV1 instead of potentiating it. Furthermore, direct application of PIP2 to inside-out excised patches dramatically potentiated TRPV1, a result inconsistent with a PLCγ-mediated mechanism of hyperalgesia. Could PI3K activation underlie NGF-mediated hyperalgesia? In support of this idea, Gordon and co-workers found that the p85β subunit of PI3K interacts with the N-terminal region of TRPV1 in yeast two-hybrid experiments and co-immunoprecipitates with TRPV1 from both HEK293 cells and DRG neurons. In addition, TRPV1 from cell lysates interacted with recombinant PI3K-p85 in in vitro pull-down experiments. Using whole-cell voltage clamp, Gordon and co-workers found that wortmannin, an inhibitor of PI3K, completely abolished NGF-mediated sensitization in acutely dissociated DRG neurons. Non-stationary noise analysis indicated that NGF did not alter either unitary conductance or single-channel open probability, but rather acted to increase the number of active TRPV1 channels in the cells. Furthermore, observation of YFP-tagged TRPV1 with total internal reflection fluorescence microscopy showed that the channels translocate to the plasma membrane upon stimulation by NGF. Gordon and co-workers suggest that NGF mediates sensitization of TRPV1 utilizing the PI3K pathway of TrkA by recruiting new channels to the plasma membrane in a mechanism similar to that described for TRPC channels [47].
3.3. Ca2+-dependent regulation of TRPV3
Despite differences in sensitivity, TRPV1, TRPV2 and TRPV3 are all activated by heat and 2-aminoethoxydiphenyl borate (2APB) [75]. A unique feature of the TRPV3 channel is that its response to heat and to 2APB is sensitized upon repetitive stimulations. Michael Zhu (Columbus, USA) reported that the sensitization results from decreases in Ca2+-dependent channel inhibition at both the extracellular and intracellular sides. Extracellular Ca2+ inhibits TRPV3 with two affinity states. It was found that high affinity Ca2+-dependent inhibition of TRPV3 is mediated by Asp641 located at the pore loop. This inhibition is gradually reduced during repeated stimulations. Mutation of Asp641 to Asn abolished the high affinity Ca2+-mediated inhibition and greatly facilitated the activation of TRPV3. The activation and sensitization of TRPV3 are also strongly dependent on the strength of intracellular Ca2+ buffers. During repeated stimulations, the voltage-dependence of TRPV3 activation shifted to less positive voltage, more slowly, with weak Ca2+ buffering than with strong Ca2+ buffering. Zhu and co-workers have identified a calmodulin-binding site at the N-terminus of TRPV3. Interestingly, this falls in a region similar to the calmodulin-binding domain of TRPV1 [72]. Disruption of the calmodulin-binding site eliminated the Ca2+-buffer-dependence of TRPV3 activation as well as the sensitization to repeated stimulations. Thus, Ca2+ inhibits TRPV3 from both the extracellular and intracellular sides. The inhibition is sequentially reduced, appearing as sensitization to repeated stimulations.
3.4. TRPV channels activation by natural agonists and antagonists
Capsaicin and menthol excite and desensitize sensory nerves by acting on two members of the TRP channel superfamily: heat-sensitive TRP vanilloid subtype 1 (TRPV1) and cold-sensitive TRP channel M8, respectively. Camphor has recently been shown to activate TRPV3 [76]. In his keynote lecture David Clapham (Boston, USA) reported that camphor also activates heterologously expressed TRPV1. Activation was enhanced by PLC-coupled receptor stimulation, mimicking inflamed conditions. Similar camphor-activated TRPV1-like currents were observed in isolated rat DRG neurons. Camphor activation of rat TRPV1 was mediated by distinct channel regions from capsaicin, as indicated by camphor activation in the presence of the competitive inhibitor capsazepine, and in a capsaicin-insensitive point mutant. Although camphor activates TRPV1 less effectively, camphor application desensitized TRPV1 more rapidly and completely than capsaicin. Conversely, TRPV3 current sensitized after repeated camphor applications, which is inconsistent with the analgesic role of camphor. Clapham also found that camphor inhibited several other related TRP channels, including ankyrin-repeat TRP 1 (TRPA1). The camphor-induced desensitization of TRPV1 and block of TRPA1 may underlie the analgesic effects of camphor. New results on the actions of carvacrol, eugenol, and thymol were also presented [76].
3.5. Epithelial Ca2+ and Mg2+ channels, TRPV5 and TRPV6
The Ca2+ and Mg2+ balance are tightly maintained by efficient feedback mechanisms involving parathyroid glands, bone, intestine and kidney [77]. Genetic studies as well as molecular cloning strategies recently identified TRPV5 and TRPV6 as the gatekeepers of active Ca2+ and Mg2+ absorption processes [77]. These channels are responsible for the rate-limiting Ca2+ entry in kidney and intestine, respectively, while TRPM6 constitutes the apical entry step in Mg2+ re-absorption. Dis-regulation or malfunction of these influx pathways has been associated with renal wasting and intestinal malabsorption of Ca2+ and Mg2+ [78]. The activity of these channels is controlled at the transcriptional and translational level by hormones and dietary content of divalent cations as studied in various animal models. Besides this long-term control, the epithelial channels can be regulated by trafficking to and from the plasma membrane and by direct activation at the plasma membrane as investigated in several cell models. The recent elucidation of channel-associated proteins has provided new molecular mechanisms underlying these processes [78]. Rene Bindels (Nijmegen, The Netherlands) described several examples, which are important for plasma membrane incorporation and retrieval of TRPV5 [79]. The protein klotho, which is likely involved in aging processes and its absence leads to an increase in serum vitamin D [80], is the main regulator in controlling TRPV5 insertion and presence in the plasma membrane. Hydrolysis of extracellular sugar residues on TRPV5 by klotho keeps the channel in the plasma membrane, which maintains channel activity [79]. Bindels also described the effects of kallikreine (KK), a serum protease, which is involved in blood pressure regulation and Na+ homeostasis and increases TRPV5 activity. It has been shown that KK knock out (ko) mice display hyper-calciuria and the TRPV5 ko mice have increased kallikreine levels [81].
3.6. Calbindin-D28K dynamically controls TRPV5-mediated Ca2+ transport
TRPV5 and TRPV6 are consistently co-expressed with the Ca2+-binding proteins calbindin–D9K and/or –D28K. Cal-bindins are thought to be involved in facilitated diffusion of Ca2+ from the point of entry to the extrusion site, while simultaneously preventing cytotoxic high Ca2+ levels. J. Hoenderop and co-workers (Nijmegen, The Netherlands) presented a study (in the poster session) in which the role of calbindin–D28K in transepithelial Ca2+ transport and their influence on the activity of TRPV5 was investigated. It was found that TRPV5 associates with calbindin–D28K only when the intracellular Ca2+ concentration is low. Furthermore, in the absence of Ca2+ increased calbindin–D28K abundance was detected in isolated plasma membranes. TIRFM revealed that calbindin–D28K translocates towards the plasma membrane when cells are treated with BAPTA-AM. 45Ca2+-uptake in TRPV5-expressing MDCK cells was increased in the presence of calbindin–D28K but not in the presence of a Ca2+-insensitive calbindin–D28K mutant emphasizing the importance of the Ca2+-binding EF-hand structures. The Ca2+-dependent inhibition of the TRPV5 current was similar in the presence of wild-type or the Ca2+-insensitive calbindin–D28K. Ca2+-insensitive calbindin–D28K that still associates with TRPV5, had a dominant negative effect on transepithelial Ca2+ transport in primary rabbit distal convoluted and connecting tubule cultures. This study thus proposes a novel model in which translocation of calbindin–D28K towards the plasma membrane occurs at low intracellular Ca2+ concentration allowing association with TRPV5 to regulate the channel activity by buffering the local influx of Ca2+.
4. TRPM channels
4.1. Gating mechanisms of TRPM2 channel and their functional consequences in neutrophil granulocytes
TRPM2 is a cation channel with little, but functionally important, permeability for Ca2+. Endogenously expressing TRPM2 is likely to form homomultimers. The intracellular C-terminal tail contains a Nudix box, a common motif of pyrophosphatases. The Nudix box is part of the NUDT9 region homologous with the human NUDT9-H pyrophosphatases. This region governs one main activation mechanism of TRPM2, gating by intracellular ADP-ribose (ADPR). ADPR acts in cooperation with cytosolic Ca2+, which is a co-factor of activation but does not activate TRPM2 by its own [82,83]. To assess the physiological relevance of ADPR in neutrophil granulocytes, Andreas Lückhoff and co-workers, (Aachen, Germany) determined intracellular ADPR concentrations with HPLC of acidic cell extracts. ADPR was present at about 3 μM. At this concentration, ADPR had little effects on TRPM2 in the absence of intracellular Ca2+ but induced a maximal activation in the presence of 10 μM Ca2+. Ca2+ alone did not gate TRPM2. Lückhoff and co-workers also determined cyclic ADPR (cADPR) with a cyclase assay. ADPR and cADPR have been found to act synergistically [84]. However, Lückhoff and co-workers found that cADPR concentrations below 0.3 μM did not affect TRPM2. ADPR and cADPR concentrations were not increased by stimulation with the chemoattractant N-formyl-methionyl-leucyl-phenylalanine (fMLP). They conclude that ADPR at basal concentrations enables regulation by Ca2+ of TRPM2, thereby significantly contributing to the induction of Ca2+ influx and cell activation, particularly chemotaxis.
4.2. TRPM2 activation by cyclic ADP-ribose at body temperature is involved in insulin secretion
There are seven thermosensitive TRP channels in mammals, and there might be other TRP channels sensitive to temperature stimuli [85]. Makoto Tominaga (Okazaki, Japan) and co-workers found that TRPM2, which is phylogenetically close to cold-sensitive TRPM8, can be activated by exposure to warm temperatures (>35 °C) apparently via direct heat-evoked channel gating. β-NAD+- or ADPR-evoked TRPM2 activity was robustly potentiated at elevated temperatures. Tominaga and co-workers also found that, even though cADPR does not activate TRPM2 at 25 °C, co-application of heat and intracellular cADPR dramatically potentiates TRPM2 activity. Heat-activated TRPM2 currents showed the electrophysiological properties almost identical to those observed in the ligand-evoked responses, suggesting that two different stimuli share the common mechanism for TRPM2 activation. Heat and cADPR evoked similar responses in rat insulinoma RIN-5F cells, which express TRPM2 endogenously. In pancreatic islets, TRPM2 was co-expressed with insulin but not with glucagon, and mild heating (around body temperature) of these cells evoked increases in both cytosolic Ca2+ and insulin release, which is KATP channel-independent and cAMP-mediated. Heat-evoked responses in both RIN-5F cells and pancreatic islets were significantly diminished by treatment with TRPM2-specific but not with control siRNA. These results identify TRPM2 as a potential molecular target for cADPR, and suggest that TRPM2 regulates Ca2+ entry into pancreatic β-cells at body temperature depending on production of cADPR-related molecules, thereby regulating insulin secretion [86].
4.3. TRPM4 controls insulin secretion in pancreatic β-cells
TRPM4 is a widely expressed calcium-activated non-selective cation (CAN) channel that conducts mainly Na+ and K+ without appreciable permeation to Ca2+ [87,88]. It is directly activated by [Ca2+]i. In non-excitable cells such as T-lymphocytes, the TRPM4-mediated depolarization reduces the driving force for Ca2+ entry through CRAC with significant impact on Ca2+ oscillations and cytokine production [89]. TRPM4 is also expressed in electrically excitable cells, suggesting that it may critically regulate Ca2+ entry mechanisms in these cells as well [90]. In pancreatic β-cells, changes in membrane potential during glucose stimulation are crucial for determining the shape and frequency of Ca2+ oscillations that triggers insulin secretion. Reinhold Penner (Hawaii, USA) reported that on the basis of its activation mechanism and its functional properties, TRPM4 is an ideal candidate for controlling voltage dependent Ca2+ channels (VDCC) activation. He, therefore, hypothesized that TRPM4 is a key regulatory component that controls membrane potential and provides the basis for Ca2+ entry through VDCCs. Indeed, pancreatic β-cells express TRPM4 proteins and generate large depolarizing membrane currents in response to increased [Ca2+]i. These currents exhibit the characteristics of TRPM4 and can be suppressed by expressing a dominant negative TRPM4 construct. The molecular suppression of TRPM4 significantly decreases insulin secretion in response to a glucose stimulus with profound impact on the amplitude of insulin oscillations. A significant reduction in insulin secretion is also observed with arginine vasopressin treatment, a Gq-coupled receptor agonist in β-cells. Moreover, TRPM4 currents increase during exocytosis in a correlated fashion, suggesting that translocation of TRPM4-containing vesicles via Ca2+-dependent exocytosis may represent a mechanism by which these cells regulate the pool of TRPM4 channels in the plasma membrane.
4.4. Biophysical and pharmacological characterization of TRPM3 splice variants
As far as we know, the TRPM3 gene gives rise to a larger number of different proteins than any other TRP gene. Johannes Oberwinkler and co-workers (Homburg, Germany) reported on one splice event, which generates two proteins (designated TRPM3α1 and TRPM3α2). These proteins differ in their putative pore region (between S5 and S6) by 12 amino acids in length and an additional alanine to proline substitution, but are otherwise identical. Oberwinkler and co-workers found that the biophysical characteristics of these two channel proteins differ profoundly, supporting the notion that the splice event directly affects the properties of the ion-conducting pore. TRPM3α2 channels, possessing the shorter pore, have a significantly higher divalent to monovalent permeability ratio than TRPM3α1 channels. Also, at high extracellular divalent concentration, TRPM3α2 channels carry large divalent inward currents, which cannot be detected in TRPM3α1 channels. Another important difference between TRPM3α2 and TRPM3α1 channels is that the former, but not the latter, has the highly unusual property of being blocked by monovalent cations, especially Na+. Despite these differences, both TRPM3 splice variants share a number of other properties: block by internal Mg2+, outwardly rectifying I/V relationship and constitutive activity.
The significant permeability of TRPM3α2 channels to Ca2+ can be exploited in Ca2+ imaging experiments. Removal of extracellular monovalent cations leads to a much larger Ca2+ influx in TRPM3α2 expressing cells than in non-transfected control cells. Using TRPM3α2 expressing cells, Oberwinkler and co-workers screened for substances that influence TRPM3α2 mediated Ca2+ entry. Although they did not find substances that specifically block TRPM3α2 activity, they were able to identify substances that induce a strong Ca2+ influx in TRPM3α2 expressing cells even in physiological, monovalent-containing, extracellular solution. In electrophysiological experiments, these substances induced a large current in TRPM3α2 expressing cells that retained the biophysical characteristics of the TRPM3α2 mediated current after monovalent removal. Importantly, TRPM3α1 mediated currents were reversibly suppressed by some of the substances that activate TRPM3α2 encoded channels. Altogether these studies demonstrate that the biophysical and pharmacological properties of TRPM3α2 channels provide highly useful tools for the identification of endogenously expressed ionic conductances in cell lines and native tissues that share the unusual characteristics of recombinant TRPM3α2 channels.
4.5. Regulation of TRPM4 and TRPM5 by Ca2+, voltage and PIP2
TRPM4 and TRPM5 are Ca2+-activated non-selective cation channels that participate in a number of physiological processes including taste transduction and cytokine secretion by immune cells. Both channels can be activated by Ca2+ or by receptor signaling that leads to release of Ca2+ from intracellular stores suggesting that Ca2+ is both sufficient and necessary for channel activation [88]. Emily Liman (Los Angeles, USA) described a second mechanism by which TRPM4 and TRPM5 channels are regulated [91]. Liman showed that the PI(4,5)P2 is a key regulator of the gating of both channels. She presented data that supports a model whereby PIP2 is bound to channels at rest and hydrolysis of PIP2 underlies desensitization of the channels following Ca2+ activation. Her analysis shows that PIP2 enhances the open probability of TRPM4 channels by locking the channels in the voltage-activated state [92]. Studies are underway to identify structural determinants for regulation of TRPM4 and TRPM5 by PIP2. An understanding of the process of desensitization of these channels will contribute to our understanding of their roles in taste sensory transduction and other types of cell signaling.
Bernd Nilius and co-workers (Leuven, Belgium) have recently reported that some TRP channels are voltage dependent. These channels show dramatic shifts of their voltage dependence towards physiological membrane potentials following various physical stimuli such as temperature (TRPV1/M8) or the binding of ligands (TRPV1/V3/M8/M4) [45,93] and Ca2+ (TRPM4/5) [94]. Modifications in a putative S4 voltage sensor and in the C-terminus induce shifts in the voltage dependence. Nilius and co-workers have also shown that small changes in Gibb’s free energy induce large shifts in TRP channel’s voltage dependence which is enhanced by their small gating charge, z (~0.7 for TRPs, 4–13 for classical voltage dependent channels). The small charge of TRP channels might be an important evolutionary structural perquisite for the gating diversity of TRP channels [95]. Nilius emphasized the fact that TRPM4 is a Ca2+-impermeable but Ca2+-activated cation channel, which undergoes a fast desensitization by intracellular Ca2+ ([Ca2+]i). He reported on regulation of the channel by the membrane lipid PIP2 [96]. Application of PIP2 counteracted rundown of TRPM4 currents in whole-cell patch-clamp experiments and attenuated desensitization to [Ca2+]i in inside-out patches. PIP2 shifted the voltage dependence of TRPM4 activation towards negative potentials and increased the Ca2+ sensitivity by approximately 100 times. Similar effects were observed after application of the PLC blocker U73122. Conversely, depletion of cellular PIP2 potently inhibited currents through TRPM4. Both Liman and Nilius groups showed that the TRP-box and the adjacent TRP-domain, which are conserved in most TRP subfamilies are probably not the PIP2 binding domains as previously suggested for TRPM5 and TRPM8 [97]. Nilius showed that mutation of conserved positive residues in the C-terminal TRP box and TRP domain did not affect the stimulatory action of PIP2. However, neutralization of positive residues in a C-terminal pleckstrin homology (PH) domain accelerated current desensitization and abolished the effect of PIP2. In conclusion, PIP2 is a strong positive modulator of TRPM4 and the C-terminal PH domain is strongly implicated in PIP2 action [98].
4.6. Coding of sweet, bitter and umami tastes
TRP channels mediate sensation of temperature taste, pheromones and mechanosensation. In his keynote lecture Charles Zuker (San Diego, USA) gave a general overview about sweet, bitter and umami (the taste of monosodium glutamate) taste. T1R are candidate mammalian taste receptors that combine to assemble two heterotrimeric G-protein coupled receptor complexes: T1R1 + 3, an umami sensor, and T1R2 + 3, a sweet receptor. Using T1R1, T1R2 and T1R3 ko mice, Zuker and co-workers demonstrated that sweet and umami taste are strictly dependent on T1R receptors, and showed that selective elimination of T1R-subunits differentially abolished detection and perception of these two taste modalities [99]. In additional study, knockouts of TRPM5 or PLCβ2, the PLC selectively expressed in taste tissue, abolished sweet, umami and bitter taste reception, but not impact sour or salt tastes. Therefore, despite relying on different receptors, sweet, umami and bitter transduction converge on common signaling molecules (PLC and TRPM5). Zuker and co-workers further found that bitter is encoded independently of sweet and umami, and taste receptor cells are not broadly tuned across these modalities [100].
5. TRPA1 channels
5.1. TRPA1 in audiovestibular and nociceptive cells
Mechanosensory cells, specialized in detecting displacements, are thought to have channels that open in response to mechanical forces [101]. Jaime García-Añoveros (Chicago, USA) reported on his search for potential mechanotransducers. One of them, ankyrin-repeat TRP 1 (TRPA1) is expressed in all of the sensory epithelia of the inner ear. TRPA1 is expressed in support cells and, weakly, in mechanosensory hair cells, where it localizes apically to the cuticular plates and the stereocilia, the mechanotransducing organelles. In dorsal root, trigeminal and nodose ganglia TRPA1 is expressed in most of the smaller, nociceptive neurons, and it localizes to their peripheral nerve processes, were sensory transduction occurs. Exposure of hair cells to agents (siRNA and morpholinos) aimed at reducing TRPA1 levels correlates with diminished mechanotransduction currents, consistent with a role for TRPA1 in hair cell transduction. Heterologously expressed TRPA1 channels can be opened with pungent chemicals like those found in wasabi, mustard, cinnamon, garlic, and mouthwash, consistent with a role of TRPA1 in nociceptive transduction. TRPA1 channels in heterologous cells are blocked by non-specific antagonists such as ruthenium red, gentamicin, gadolinium and amiloride, all known blockers of the hair cell transducer and other mechanosensory channels. Other comparisons between heterologously expressed TRPA1 and the better characterized mechanosensory channel of hair cells, reveal many similarities and a few differences. The channels have a range of many conductance levels that are reduced to 54% by Ca2+. Calcium entering the channel binds to it and causes a brief potentiation followed by closure, and depolarization reopens the Ca2+-closed channels. All these phenomena also occur to the hair cell mechanotransducer, although 1000× faster. These results suggest that TRPA1 channels may be mechanosensory. In hair cells either TRPA1 or a very similar channel protein forms the transduction channel [101,102] and in small neurons of sensory ganglia TRPA1 may mediate mechanonociception [103]. Voltage dependence of the Ca2+ induced channel closure is a property, which is particularly well suited for this channel occurring at hyperpolarization but not at depolarization. Thus, TRPA1 would inactivate to subthreshold (i.e. innocuous) stimulation, and not to supprathreshold (i.e. painful) stimulation, accounting for the lack of desensitization that characterizes pain. The existence of a TRPA1 ortholog in C. elegans and its expression in presumed mechanosensory and nociceptive neurons of the worm suggest that TRPA1 mediates an ancestral form of mechanotransduction.
5.2. Somatosensation and mechanosensation
David Julius (San Francisco, USA) pioneered the studies on the molecular basis of somatosensation—the process whereby we experience touch and temperature. These studies are based on identifying molecules that detect noxious (pain-producing) stimuli. He is also interested in understanding how somatosensation is altered in response to tissue or nerve injury. The approach of his lab has been to identify molecular targets for natural products that mimic the psychophysical effects of commonly encountered somatosensory stimuli, such as heat or cold, and to then ask how these molecules are activated or modulated by noxious stimuli or injury [104–106]. In his presentation Julius focused on members of the TRP channel family (TRPV1, TRPA1) that are expressed by subpopulations of primary afferent sensory neurons and which have been implicated in the detection of thermal stimuli and/or inflammatory agents. Genetic studies support the idea that the capsaicin receptor, TRPV1, functions as an integrator of thermal and chemical stimuli, thereby contributing to sensitization of primary afferent fibers, particularly in the context of peripheral tissue injury and inflammation [106]. Recent data examining a role for this channel in models of pain hypersensitivity, such as that associated with bone cancer were presented [107]. Julius elaborated on generation of pain in bone sarcomas. Osteoclast activation induces bone re-absorption and acidosis, thereby activating TRPV1 in bone sensory fibers, while TRPV1 antagonists decreased pain-related behavior in mouse model of bone sarcoma [107].
Recent work from his group, and others, has shown that TRPA1 is a receptor for mustard oil and related pungent compounds of the isothiocyanate family. Recent data showed that in addition to TRPV1, TRPA1 mediates selective aspects of inflammatory hyperalgesia. This channel is present in a subset of peptinergic neurons. TRPA1 activating compounds affect a subset of sensory fibers and also induce a release of GCRP from sensory nerve endings. TRPA1 has been suggested to function as a transduction channel in a number of sensory modalities, including thermal (cold) nociception, hearing [108], and inflammatory pain [109,110]. Julius studies supports a role for this channel in the latter process by virtue of its ability to function as a “receptor-operated” channel, depolarizing primary afferent fibers in response to endogenous proalgesic agents, such as bradykinin, that activate PLC signaling systems. In this respect, TRPA1 is a receptor operated channel which is sensitized by BK via the PLCβ cascade resulting in IP3 mediated Ca2+ release. Ca2+ is a co-factor for TRPA1 activation and helps gating the channel. Importantly, TRPV1 and TRPA1 co-operate and are both main players in inflammatory and thermal hyperalgesia. All activating effects of MO and other TRPA1 agonists are absent in TRPA1 knock out mice. Strikingly, in TRPA1 ko mice, hearing reflexes were normal, suggesting that TRPA1 is not essential for hair-cell transduction [110] (see also [111]) in contrast to previous report [108]. Data obtained form the TRPA1 ko mouse also suggest that this channel is not cold activated (but see [111]).
6. TRPML channels
6.1. Mucolipin 1: endocytosis and cation channel, implications for lysosomal storage disorder
Gideon Bach (Jerusalem, Israel) presented his work on mucolipidosis type IV (ML4). ML4 is a neurodegenerative lysosomal storage disorder characterized by psychomotor retardation and ophthalmological abnormalities, including corneal opacities, retinal degeneration and strabismus. Severely affected as well as milder patients have been described. Over 80% of the ML4 patients are Ashkenazi Jews, with heterozygotes frequency of 1/100. The disease is classified as a mucolipidosis due to the simultaneous lysosomal storage of lipids together with water-soluble substances. A broad spectrum of lipids and acid mucopolysaccharides were identified as the storage substances. Kinetic studies in cells from ML4 patients demonstrated that this heterogeneous storage stems from an abnormal endocytosis process of membrane components, from late endosomes to the lysosomes, and/or delayed efflux to the Golgi apparatus [112]. The ML4 gene was mapped to chromosome 19p13.2–13.3 where a novel gene—MCOLN1, with MLIV-causing mutations, was identified [113]. Two mutations were found among 95% of the Ashkenazi ML4 alleles, including an in acceptor splice site mutation in 72% of the alleles and a partial gene deletion in 23%. Each of these mutations was associated with a defined haplotype in this chromosomal region. Other mutations were mostly identified in single, Ashkenazi and non-Ashkanazi patients, including, missense, nonsense nucleotide deletions and insertions.
MCOLN1 encodes a 580 aa protein—mucolipin 1, which is a member of a new protein family of unknown function at present, the mucolipins. Mucolipin 1 is a glycosylated membrane protein with six trans-membrane domains, a serine lipase and nuclear localization signal motives. Transfection assays indicate that the protein is primarily lysosomal and appears as oligomers. The protein belongs to the TRPML subfamily, with the highest resemblance to polycystins (TRPP). The involvement of this protein in the endocytosis process of membrane components is currently being studied. A population screening program among Ashkenazi population for the detection of heterozygotes is performed in Israel as a prevention program [114].
6.2. TRPML1 is a cathepsin B-cleaved lysosomal H+ channel that regulates lipid hydrolysis and metabolism
Mutations in the gene MCOLN1 coding for the TRP family ion channel TRPML1 lead to the lipid storage disorder mucolipidosis type IV (ML4). The function and role of TRPML1 are not well understood. Shmuel Muallem and co-workers (Dallas, USA) found that TRPML1 is a lysosomal H+ channel that is regulated by cathepsin B (CatB)-mediated cleavage. Both native and recombinant TRPML1 are cleaved resulting in two products. Recombinant TRPML1 is detected as the full length (FL) and as short N- and C-terminal forms, whereas in native cells mainly the cleaved N and C termini are detected. The N-and C-terminal halves of TRPML1 co-immunoprecipitate from cell lysates and co-elute from a Ni2+ column. TRPML1 undergoes proteolytic cleavage that is inhibited by inhibitors of Cathepsin B (CatB) and is altered when TRPML1 is expressed in CatB−/−cells. N-terminal sequencing of the C-terminal fragment of TRPML1 expressed in Sf9 cells indicates a cleavage site at 200R/P201. Consequently, the conserved R200H mutation changes the cleavage pattern of TRPML1. The cleavage inhibits TRPML1 channel activity [115].
ML4 is typified by accumulation of lipids in intracellular organelles, which was hypothesized to be due to altered membrane fusion and fission events. How mutations in TRPML1 lead to aberrant lipolysis is not known. Muallem presented evidence that ML4 is a metabolic disorder that is not associated with aberrant membrane fusion/fission events. Thus, measurement of lysosomal pH revealed that the lysosomes in TRPML1−/−cells obtained from patients with ML4 are over-acidified. TRPML1 functions as a H+ channel and the increased lysosomal acidification in TRPML1−/−cells is likely caused by the loss of TRPML1-mediated H+ leak. Measurement of lipase activity using several substrates revealed a marked reduction in lipid metabolism in TRPML1−/−cells, which was rescued by expression of TRPML1. Furthermore, dissipation of the acidic lysosomal pH of TRPML1−/−cells reversed the lysosomal storage disease phenotype. These finding provide a new mechanism to account for the pathogenesis of ML4 [116].
7. Concluding remarks
Until very recently, studies on TRP channels have been presented and discussed in scientific meetings dedicated to many different fields of biomedicine, reflecting the growing importance of the various TRP channels for understanding fundamental mechanisms common to many cells and tissues, especially where cellular Ca2+ plays an important role. The very successful Minerva-Gentner Symposium in Eilat revealed the need to present and discuss fundamental mechanisms common to all or most TRP channels in specific TRP meetings. These topics included structure–function relationships, physiological and pathological mechanisms of activation, as well as physiological functions of TRP channels. The lack of structural information on TRP channels is an obvious obstacle for future progress of this field. Similarly, the very limited specific pharmacological tools have hindered fast progress in the TRP field.
The symposium emphasized one of the puzzling phenomena related to TRP channels, that has been found in most subfamilies: the ability to activate these channels by unusually large variety of stimuli. These stimuli include large variety of natural compounds (e.g. capsaicin, menthol, camphor, mustard oil, oregano), physical stimuli (e.g. mechanical, temperature, pH, light) specific body-produced chemicals (e.g. pheromones, growth factors, PIP2, DAG, PUFAs) and metabolic stress. Stimuli such as temperature activate channel members having significantly different primary amino acid sequence, and belonging to different subfamilies (e.g. TRPV, TRPM, TRPA). Similarly, metabolic stress also activates members of different subfamilies (Drosophila TRPC channels, TRP/TRPL [40], mammalian TRPM7 [117]). A possible partial answer to this puzzle may arise from the conserved pore region that seems to be susceptible to small changes in the lipid environment of the plasma membrane.
The multiple sites in the protein sequence, typical for protein–protein or protein–lipid interactions of the various TRP channels emphasize the need to study TRP channels in the native cellular environment. The sensitivity of many TRP channel members to the metabolic state, temperature and pH of the cells emphasizes the need to carefully control the conditions of the experiments to avoid conflicting results, which are so far quite common in the TRP field.
The TRP symposium in Eilat provided an enjoyable and an exciting opportunity to learn and discuss the above issues, thus, constituted a milestone in the advancement of the TRP field. Experiments of B. Minke described in this review were supported by the National Institutes of Health (EY 03529).
Acknowledgments
I thank the Minerva Foundation for the generous support, which covered a major fraction of the expenses related to the symposium, Veit Flockerzi for his valuable help in the organization and expenses, the Alomone Labs and the Hebrew University for support. I also thank Esti Ben-Shoan for skilful organization and Ariela Gordon-Shaag and Ben Katz for critical reading of the manuscript. Experiments of B. Minke described in this review were supported by the National Institutes of Health (EY 03529).
References
- 1.Cosens DJ, Manning A. Abnormal electroretinogram from a Drosophila mutant. Nature. 1969;224:285–287. doi: 10.1038/224285a0. [DOI] [PubMed] [Google Scholar]
- 2.Cosens D. Blindness in a Drosophila mutant. J Insect Physiol. 1971;17:285–302. [Google Scholar]
- 3.Minke B, Wu C, Pak WL. Induction of photoreceptor voltage noise in the dark in Drosophila mutant. Nature. 1975;258:84–87. doi: 10.1038/258084a0. [DOI] [PubMed] [Google Scholar]
- 4.Minke B, Selinger Z. Inositol lipid pathway in fly photoreceptors: excitation, calcium mobilization and retinal degeneration. In: Osborne NA, Chader GJ, editors. Progress in Retinal Research. Pergamon Press; Oxford: 1991. pp. 99–124. [Google Scholar]
- 5.Montell C, Rubin GM. Molecular characterization of the Drosophila trp locus: a putative integral membrane protein required for phototransduction. Neuron. 1989;2:1313–1323. doi: 10.1016/0896-6273(89)90069-x. [DOI] [PubMed] [Google Scholar]
- 6.Hardie RC, Minke B. The trp gene is essential for a light-activated Ca2+ channel in Drosophila photoreceptors. Neuron. 1992;8:643–651. doi: 10.1016/0896-6273(92)90086-s. [DOI] [PubMed] [Google Scholar]
- 7.Phillips AM, Bull A, Kelly LE. Identification of a Drosophila gene encoding a calmodulin-binding protein with homology to the trp phototransduction gene. Neuron. 1992;8:631–642. doi: 10.1016/0896-6273(92)90085-r. [DOI] [PubMed] [Google Scholar]
- 8.Niemeyer BA, Suzuki E, Scott K, Jalink K, Zuker CS. The Drosophila light-activated conductance is composed of the two channels TRP and TRPL. Cell. 1996;85:651–659. doi: 10.1016/s0092-8674(00)81232-5. [DOI] [PubMed] [Google Scholar]
- 9.Scott K, Sun Y, Beckingham K, Zuker CS. Calmodulin regulation of Drosophila light-activated channels and receptor function mediates termination of the light response in vivo. Cell. 1997;91:375–383. doi: 10.1016/s0092-8674(00)80421-3. [DOI] [PubMed] [Google Scholar]
- 10.Devary O, Heichal O, Blumenfeld A, et al. Coupling of photoexcited rhodopsin to inositol phospholipid hydrolysis in fly photoreceptors. Proc Natl Acad Sci USA. 1987;84:6939–6943. doi: 10.1073/pnas.84.19.6939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hardie RC, Minke B. Novel Ca2+ channels underlying transduction in Drosophila photoreceptors: implications for phosphoinositide-mediated Ca2+ mobilization. Trends Neurosci. 1993;16:371–376. doi: 10.1016/0166-2236(93)90095-4. [DOI] [PubMed] [Google Scholar]
- 12.Bloomquist BT, Shortridge RD, Schneuwly S, et al. Isolation of a putative phospholipase C gene of Drosophila, norpA, and its role in phototransduction. Cell. 1988;54:723–733. doi: 10.1016/s0092-8674(88)80017-5. [DOI] [PubMed] [Google Scholar]
- 13.Wes PD, Chevesich J, Jeromin A, Rosenberg C, Stetten G, Montell C. TRPC1, a human homolog of a Drosophila store-operated channel. Proc Natl Acad Sci USA. 1995;92:9652–9656. doi: 10.1073/pnas.92.21.9652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhu X, Chu PB, Peyton M, Birnbaumer L. Molecular cloning of a widely expressed human homologue for the Drosophila trp gene. FEBS Lett. 1995;373:193–198. doi: 10.1016/0014-5793(95)01038-g. [DOI] [PubMed] [Google Scholar]
- 15.Zhu X, Jiang MS, Peyton M, et al. trp, a novel mammalian gene family essential for agonist-activated capacitative Ca2+ entry. Cell. 1996;85:661–671. doi: 10.1016/s0092-8674(00)81233-7. [DOI] [PubMed] [Google Scholar]
- 16.Clapham DE. TRP channels as cellular sensors. Nature. 2003;426:517–524. doi: 10.1038/nature02196. [DOI] [PubMed] [Google Scholar]
- 17.Minke B, Cook B. TRP channel proteins and signal transduction. Physiol Rev. 2002;82:429–472. doi: 10.1152/physrev.00001.2002. [DOI] [PubMed] [Google Scholar]
- 18.Montell C. The TRP superfamily of cation channels. Sci STKE. 2005;2005:re3. doi: 10.1126/stke.2722005re3. [DOI] [PubMed] [Google Scholar]
- 19.Nilius B, Voets T. TRP channels: a TR(I)P through a world of multifunctional cation channels. Pflug Arch. 2005;451:1–10. doi: 10.1007/s00424-005-1462-y. [DOI] [PubMed] [Google Scholar]
- 20.Pedersen SF, Owsianik G, Nilius B. TRP channels: an overview. Cell Calcium. 2005;38:233–252. doi: 10.1016/j.ceca.2005.06.028. [DOI] [PubMed] [Google Scholar]
- 21.Voets T, Talavera K, Owsianik G, Nilius B. Sensing with TRP channels. Nat Chem Biol. 2005;1:85–92. doi: 10.1038/nchembio0705-85. [DOI] [PubMed] [Google Scholar]
- 21a.Nilius B, Mahieu F. A road map for TR(I)Ps. Mol Cell. 2006 May 3;22:297–307. doi: 10.1016/j.molcel.2006.04.015. [DOI] [PubMed] [Google Scholar]
- 22.Nilius B, Voets T, Peters J. TRP channels in disease. Sci STKE. 2005;2005:re8. doi: 10.1126/stke.2952005re8. [DOI] [PubMed] [Google Scholar]
- 23.Minke B, Selinger Z. In: Intracellular messengers in invertebrate photoreceptors studied in mutant flies. Boulton A, Baker G, Taylor C, editors. Neuromethods, The Humana Press Inc; Clifton, NJ: 1992. pp. 517–563. [Google Scholar]
- 24.Cook B, Bar YM, Cohen-Ben AH, et al. Phospholipase C and termination of G-protein-mediated signaling in vivo. Nat Cell Biol. 2000;2:296–301. doi: 10.1038/35010571. [DOI] [PubMed] [Google Scholar]
- 25.Hardie RC, Raghu P. Visual transduction in Drosophila. Nature. 2001;413:186–193. doi: 10.1038/35093002. [DOI] [PubMed] [Google Scholar]
- 26.Hardie RC. Regulation of TRP channels via lipid second messengers. Annu Rev Physiol. 2003;65:735–759. doi: 10.1146/annurev.physiol.65.092101.142505. [DOI] [PubMed] [Google Scholar]
- 27.Masai I, Okazaki A, Hosoya T, Hotta Y. Drosophila retinal degeneration A gene encodes an eye-specific diacylglycerol kinase with cysteine-rich zinc-finger motifs and ankyrin repeats. Proc Natl Acad Sci USA. 1993;90:11157–11161. doi: 10.1073/pnas.90.23.11157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Masai I, Suzuki E, Yoon CS, Kohyama A, Hotta Y. Immunolocalization of Drosophila eye-specific diacylgylcerol kinase, rdgA, which is essential for the maintenance of the photoreceptor. J Neurobiol. 1997;32:695–706. doi: 10.1002/(sici)1097-4695(19970620)32:7<695::aid-neu5>3.0.co;2-#. [DOI] [PubMed] [Google Scholar]
- 29.Minke B, Parnas M. Insights on trp channels from in vivo studies in Drosophila. Annu Rev Physiol. 2006;68:649–684. doi: 10.1146/annurev.physiol.68.040204.100939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hofmann T, Obukhov AG, Schaefer M, Harteneck C, Gudermann T, Schultz G. Direct activation of human TRPC6 and TRPC3 channels by diacylglycerol. Nature. 1999;397:259–263. doi: 10.1038/16711. [DOI] [PubMed] [Google Scholar]
- 31.LaLonde MM, Janssens H, Rosenbaum E, et al. Regulation of phototransduction responsiveness and retinal degeneration by a phospholipase D-generated signaling lipid. J Cell Biol. 2005;169:471–479. doi: 10.1083/jcb.200502122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Garcia-Murillas I, Pettitt T, Macdonald E, et al. Lazaro encodes a lipid phosphate phosphohydrolase that regulates phosphatidylinositol turnover during Drosophila phototransduction. Neuron. 2006;49:533–546. doi: 10.1016/j.neuron.2006.02.001. [DOI] [PubMed] [Google Scholar]
- 33.Kwon Y, Montell C. Dependence on the Lazaro phosphatidic acid phosphatase for the maximum light response. Curr Biol. 2006;16:723–729. doi: 10.1016/j.cub.2006.02.057. [DOI] [PubMed] [Google Scholar]
- 34.Elia N, Frechter S, Gedi Y, Minke B, Selinger Z. Excess of Gαe over Gqαe in vivo prevents dark, spontaneous activity of Drosophila photoreceptors. J Cell Biol. 2005;171:517–526. doi: 10.1083/jcb.200506082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cosens D, Perry MM. The fine structure of the eye of a visual mutant. A-type of Drosophila melanogaster. J Insect Physiol. 1972;18:1773–1786. doi: 10.1016/0022-1910(72)90109-6. [DOI] [PubMed] [Google Scholar]
- 36.Wang T, Jiao Y, Montell C. Dissecting independent channel and scaffolding roles of the Drosophila transient receptor potential channel. J Cell Biol. 2005;171:685–694. doi: 10.1083/jcb.200508030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wang T, Xu H, Oberwinkler J, Gu Y, Hardie RC, Montell C. Light activation, adaptation, and cell survival functions of the Na+/Ca2+ exchanger CalX. Neuron. 2005;45:367–378. doi: 10.1016/j.neuron.2004.12.046. [DOI] [PubMed] [Google Scholar]
- 38.Yoon J, Ben-Ami HC, Hong YS, et al. Novel mechanism of massive photoreceptor degeneration caused by mutations in the trp gene of Drosophila. J Neurosci. 2000;20:649–659. doi: 10.1523/JNEUROSCI.20-02-00649.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hong YS, Park S, Geng C, et al. Single amino acid change in the fifth trans-membrane segment of the TRP Ca2+ channel causes massive degeneration of photoreceptors. J Biol Chem. 2002;277:33884–33889. doi: 10.1074/jbc.M204075200. [DOI] [PubMed] [Google Scholar]
- 40.Agam K, von-Campenhausen M, Levy S, et al. Metabolic stress reversibly activates the Drosophila light-sensitive channels TRP and TRPL in vivo. J Neurosci. 2000;20:5748–5755. doi: 10.1523/JNEUROSCI.20-15-05748.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Minke B, Agam K. TRP gating is linked to the metabolic state and maintenance of the Drosophila photoreceptor cells. Cell Calcium. 2003;33:395–408. doi: 10.1016/s0143-4160(03)00052-6. [DOI] [PubMed] [Google Scholar]
- 42.Gu Y, Oberwinkler J, Postma M, Hardie RC. Mechanisms of light adaptation in Drosophila photoreceptors. Curr Biol. 2005;15:1228–1234. doi: 10.1016/j.cub.2005.05.058. [DOI] [PubMed] [Google Scholar]
- 43.Hardie RC, Raghu P, Moore S, Juusola M, Baines RA, Sweeney ST. Calcium influx via TRP channels is required to maintain PIP2 levels in Drosophila photoreceptors. Neuron. 2001;30:149–159. doi: 10.1016/s0896-6273(01)00269-0. [DOI] [PubMed] [Google Scholar]
- 44.Lisman JE, Brown JE. The effects of intracellular iontophoretic injection of calcium and sodium ions on the light response of Limulus ventral photoreceptors. J Gen Physiol. 1972;59:701–719. doi: 10.1085/jgp.59.6.701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Voets T, Droogmans G, Wissenbach U, Janssens A, Flockerzi V, Nilius B. The principle of temperature-dependent gating in cold-and heat-sensitive TRP channels. Nature. 2004;430:748–754. doi: 10.1038/nature02732. [DOI] [PubMed] [Google Scholar]
- 46.Bahner M, Frechter S, Da Silva N, Minke B, Paulsen R, Huber A. Light-regulated subcellular translocation of Drosophila TRPL channels induces long-term adaptation and modifies the light-induced current. Neuron. 2002;34:83–93. doi: 10.1016/s0896-6273(02)00630-x. [DOI] [PubMed] [Google Scholar]
- 47.Bezzerides VJ, Ramsey IS, Kotecha S, Greka A, Clapham DE. Rapid vesicular translocation and insertion of TRP channels. Nat Cell Biol. 2004;6:709–720. doi: 10.1038/ncb1150. [DOI] [PubMed] [Google Scholar]
- 48.Vazquez G, Wedel BJ, Aziz O, Trebak M, Putney JW., Jr The mammalian TRPC cation channels. Biochim Biophys Acta. 2004;1742:21–36. doi: 10.1016/j.bbamcr.2004.08.015. [DOI] [PubMed] [Google Scholar]
- 49.Leypold BG, Yu CR, Leinders-Zufall T, Kim MM, Zufall F, Axel R. Altered sexual and social behaviors in trp2 mutant mice. Proc Natl Acad Sci USA. 2002;99:6376–6381. doi: 10.1073/pnas.082127599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Stowers L, Holy TE, Meister M, Dulac C, Koentges G. Loss of sex discrimination and male–male aggression in mice deficient for TRP2. Science. 2002;295:1493–1500. doi: 10.1126/science.1069259. [DOI] [PubMed] [Google Scholar]
- 51.Liman ER, Corey DP, Dulac C. TRP2: a candidate transduction channel for mammalian pheromone sensory signaling. Proc Natl Acad Sci USA. 1999;96:5791–5796. doi: 10.1073/pnas.96.10.5791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lucas P, Ukhanov K, Leinders-Zufall T, Zufall F. A diacylglycerolgated cation channel in vomeronasal neuron dendrites is impaired in TRPC2 mutant mice: mechanism of pheromone transduction. Neuron. 2003;40:551–561. doi: 10.1016/s0896-6273(03)00675-5. [DOI] [PubMed] [Google Scholar]
- 53.Putney JW. Physiological mechanisms of TRPC activation. Pflug Arch. 2005;451:29–34. doi: 10.1007/s00424-005-1416-4. [DOI] [PubMed] [Google Scholar]
- 54.Lockwich TP, Liu X, Singh BB, Jadlowiec J, Weiland S, Ambudkar IS. Assembly of Trp1 in a signaling complex associated with caveolin-scaffolding lipid raft domains. J Biol Chem. 2000;275:11934–11942. doi: 10.1074/jbc.275.16.11934. [DOI] [PubMed] [Google Scholar]
- 55.Ambudkar IS. Ca2+ signaling microdomains:platforms for the assembly and regulation of TRPC channels. Trends Pharmacol Sci. 2006;27:25–32. doi: 10.1016/j.tips.2005.11.008. [DOI] [PubMed] [Google Scholar]
- 56.Tsunoda S, Sierralta J, Sun Y, et al. A multivalent PDZ-domain protein assembles signaling complexes in a G-protein-coupled cascade. Nature. 1997;388:243–249. doi: 10.1038/40805. [DOI] [PubMed] [Google Scholar]
- 57.Bandyopadhyay BC, Swaim WD, Liu X, Redman RS, Patterson RL, Ambudkar IS. Apical localization of a functional TRPC3/TRPC6-Ca2+-signaling complex in polarized epithelial cells. Role in apical Ca2+ influx. J Biol Chem. 2005;280:12908–12916. doi: 10.1074/jbc.M410013200. [DOI] [PubMed] [Google Scholar]
- 58.Singh BB, Lockwich TP, Bandyopadhyay BC, et al. VAMP2-dependent exocytosis regulates plasma membrane insertion of TRPC3 channels and contributes to agonist-stimulated Ca2+ influx. Mol Cell. 2004;15:635–646. doi: 10.1016/j.molcel.2004.07.010. [DOI] [PubMed] [Google Scholar]
- 59.Yuan JP, Kiselyov K, Shin DM, et al. Homer binds TRPC family channels and is required for gating of TRPC1 by IP3 receptors. Cell. 2003;114:777–789. doi: 10.1016/s0092-8674(03)00716-5. [DOI] [PubMed] [Google Scholar]
- 60.Freichel M, Philipp S, Cavalie A, Flockerzi V. TRPC4 and TRPC4-deficient mice. Novartis Found Symp. 2004;258:189–199. [PubMed] [Google Scholar]
- 61.Kiselyov K, Muallem S. Fatty acids, diacylglycerol., Ins(1,4,5)P3 receptors and Ca2+ influx. Trends Neurosci. 1999;22:334–337. doi: 10.1016/s0166-2236(99)01426-5. [DOI] [PubMed] [Google Scholar]
- 62.Kawasaki BT, Liao Y, Birnbaumer L. Role of Src in C3 transient receptor potential channel function and evidence for a heterogeneous makeup of receptor- and store-operated Ca2+ entry channels. Proc Natl Acad Sci USA. 2006;103:335–340. doi: 10.1073/pnas.0508030102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Putney JW, Jr, McKay RR. Capacitative calcium entry channels. Bioessays. 1999;21:38–46. doi: 10.1002/(SICI)1521-1878(199901)21:1<38::AID-BIES5>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- 64.Liou J, Kim ML, Heo WD, et al. STIM is a Ca2+ sensor essential for Ca2+-store-depletion-triggered Ca2+ influx. Curr Biol. 2005;15:1235–1241. doi: 10.1016/j.cub.2005.05.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Roos J, DiGregorio PJ, Yeromin AV, et al. STIM1, an essential and conserved component of store-operated Ca2+ channel function. J Cell Biol. 2005;169:435–445. doi: 10.1083/jcb.200502019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Zhang SL, Yu Y, Roos J, et al. STIM1 is a Ca2+ sensor that activates CRAC channels and migrates from the Ca2+ store to the plasma membrane. Nature. 2005;437:902–905. doi: 10.1038/nature04147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Spassova MA, Soboloff J, He LP, Xu W, Dziadek MA, Gill DL. STIM1 has a plasma membrane role in the activation of store-operated Ca2+ channels. Proc Natl Acad Sci USA. 2006;103:4040–4045. doi: 10.1073/pnas.0510050103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Penner R, Fleig A. Store-operated calcium entry: a tough nut to CRAC. Sci STKE. 2004;2004:e38. doi: 10.1126/stke.2432004pe38. [DOI] [PubMed] [Google Scholar]
- 69.Vig M, Peinelt C, Beck A, et al. CRACM1 is a plasma membrane protein essential for store-operated Ca2+ entry. Science. 2006 doi: 10.1126/science.1127883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Feske S, Gwack Y, Prakriya M, et al. A mutation in Orai1 causes immune deficiency by abrogating CRAC channel function. Nature. 2006 doi: 10.1038/nature04702. [DOI] [PubMed] [Google Scholar]
- 71.Clapham DE, Julius D, Montell C, Schultz G. International Union of Pharmacology. XLIX. Nomenclature and structure–function relationships of transient receptor potential channels. Pharmacol Rev. 2005;57:427–450. doi: 10.1124/pr.57.4.6. [DOI] [PubMed] [Google Scholar]
- 72.Rosenbaum T, Gordon-Shaag A, Munari M, Gordon SE. Ca2+/calmodulin modulates TRPV1 activation by capsaicin. J Gen Physiol. 2004;123:53–62. doi: 10.1085/jgp.200308906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Julius D, Basbaum AI. Molecular mechanisms of nociception. Nature. 2001;413:203–210. doi: 10.1038/35093019. [DOI] [PubMed] [Google Scholar]
- 74.Chuang ED, Prescott H, Kong, et al. Bradykinin and nerve growth factor release the capsaicin receptor from PtdIns(4, 5)P2-mediated inhibition. Nature. 2001;411:957–962. doi: 10.1038/35082088. [DOI] [PubMed] [Google Scholar]
- 75.Hu HZ, Gu Q, Wang C, et al. 2-Aminoethoxydiphenyl borate is a common activator of TRPV1, TRPV2, and TRPV3. J Biol Chem. 2004;279:35741–35748. doi: 10.1074/jbc.M404164200. [DOI] [PubMed] [Google Scholar]
- 76.Xu H, Delling M, Jun JC, Clapham DE. Oregano, thyme and clove-derived flavors and skin sensitizers activate specific TRP channels. Nat Neurosci. 2006;9:628–635. doi: 10.1038/nn1692. [DOI] [PubMed] [Google Scholar]
- 77.Hoenderop JG, Nilius B, Bindels RJ. Calcium absorption across epithelia. Physiol Rev. 2005;85:373–422. doi: 10.1152/physrev.00003.2004. [DOI] [PubMed] [Google Scholar]
- 78.van AM, Hoenderop JG, Bindels RJ. The epithelial calcium channels TRPV5 and TRPV6: regulation and implications for disease. Naunyn Schmiedebergs Arch Pharmacol. 2005;371:295–306. doi: 10.1007/s00210-005-1021-2. [DOI] [PubMed] [Google Scholar]
- 79.Chang Q, Hoefs S, van der Kemp AW, Topala CN, Bindels RJ, Hoenderop JG. The β-glucuronidase klotho hydrolyzes and activates the TRPV5 channel. Science. 2005;310:490–493. doi: 10.1126/science.1114245. [DOI] [PubMed] [Google Scholar]
- 80.Kuro-o M, Matsumura Y, Aizawa H, et al. Mutation of the mouse klotho gene leads to a syndrome resembling ageing. Nature. 1997;390:45–51. doi: 10.1038/36285. [DOI] [PubMed] [Google Scholar]
- 81.Picard N, Van AM, Campone C, et al. Tissue kallikrein-deficient mice display a defect in renal tubular calcium absorption. J Am Soc Nephrol. 2005;16:3602–3610. doi: 10.1681/ASN.2004110923. [DOI] [PubMed] [Google Scholar]
- 82.Perraud AL, Fleig A, Dunn CA, et al. ADP-ribose gating of the calcium-permeable LTRPC2 channel revealed by Nudix motif homology. Nature. 2001;411:595–599. doi: 10.1038/35079100. [DOI] [PubMed] [Google Scholar]
- 83.Kuhn FJ, Luckhoff A. Sites of the NUDT9-H domain critical for ADP-ribose activation of the cation channel TRPM2. J Biol Chem. 2004;279:46431–46437. doi: 10.1074/jbc.M407263200. [DOI] [PubMed] [Google Scholar]
- 84.Kolisek M, Beck A, Fleig A, Penner R. Cyclic ADP-ribose and hydrogen peroxide synergize with ADP-ribose in the activation of TRPM2 channels. Mol Cell. 2005;18:61–69. doi: 10.1016/j.molcel.2005.02.033. [DOI] [PubMed] [Google Scholar]
- 85.Patapoutian A, Peier AM, Story GM, Viswanath V. ThermoTRP channels and beyond: mechanisms of temperature sensation. Nat Rev Neurosci. 2003;4:529–539. doi: 10.1038/nrn1141. [DOI] [PubMed] [Google Scholar]
- 86.Togashi K, Hara Y, Tominaga T, et al. TRPM2 activation by cyclic ADP-ribose at body temperature is involved in insulin secretion. EMBO J. 2006 doi: 10.1038/sj.emboj.7601083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Petersen OH. Cation channels: homing in on the elusive CAN channels. Curr Biol. 2002;12:R520–R522. doi: 10.1016/s0960-9822(02)01027-8. [DOI] [PubMed] [Google Scholar]
- 88.Fleig A, Penner R. The TRPM ion channel subfamily: molecular, biophysical and functional features. Trends Pharmacol Sci. 2004;25:633–639. doi: 10.1016/j.tips.2004.10.004. [DOI] [PubMed] [Google Scholar]
- 89.Launay P, Cheng H, Srivatsan S, Penner R, Fleig A, Kinet JP. TRPM4 regulates calcium oscillations after T cell activation. Science. 2004;306:1374–1377. doi: 10.1126/science.1098845. [DOI] [PubMed] [Google Scholar]
- 90.Launay P, Fleig A, Perraud AL, Scharenberg AM, Penner R, Kinet JP. TRPM4 is a Ca2+-activated non-selective cation channel mediating cell membrane depolarization. Cell. 2002;109:397–407. doi: 10.1016/s0092-8674(02)00719-5. [DOI] [PubMed] [Google Scholar]
- 91.Liu D, Liman ER. Intracellular Ca2+ and the phospholipid PIP2 regulate the taste transduction ion channel TRPM5. Proc Natl Acad Sci USA. 2003;100:15160–15165. doi: 10.1073/pnas.2334159100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Zhang Z, Okawa H, Wang Y, Liman ER. Phosphatidylinositol 4,5-bisphosphate rescues TRPM4 channels from desensitization. J Biol Chem. 2005;280:39185–39192. doi: 10.1074/jbc.M506965200. [DOI] [PubMed] [Google Scholar]
- 93.Nilius B, Talavera K, Owsianik G, Prenen J, Droogmans G, Voets T. Gating of TRP channels: a voltage connection? J Physiol. 2005;567:35–44. doi: 10.1113/jphysiol.2005.088377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Nilius B, Prenen J, Tang J, et al. Regulation of the Ca2+ sensitivity of the non-selective cation channel TRPM4. J Biol Chem. 2005;280:6423–6433. doi: 10.1074/jbc.M411089200. [DOI] [PubMed] [Google Scholar]
- 95.Owsianik G, D’hoedt D, Voets T, Nilius B. Structure–function relationship of the TRP channel superfamily. Rev Physiol Biochem Pharmacol. 2006;156:61–90. [PubMed] [Google Scholar]
- 96.Nilius B, Mahieu F, Prenen J, et al. The Ca2+-activated cation channel TRPM4 is regulated by phosphatidylinositol-4,5-biphosphate. EMBO J. 2006;25:467–478. doi: 10.1038/sj.emboj.7600963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Rohacs T, Lopes CM, Michailidis I, Logothetis DE. PI(4,5)P2 regulates the activation and desensitization of TRPM8 channels through the TRP domain. Nat Neurosci. 2005;8:626–634. doi: 10.1038/nn1451. [DOI] [PubMed] [Google Scholar]
- 98.Nilius B, Mahieu F, Prenen J, et al. The Ca2+-activated cation channel TRPM4 is regulated by phosphatidylinositol-4,5-biphosphate. EMBO J. 2006;25:467–478. doi: 10.1038/sj.emboj.7600963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Zhao GQ, Zhang Y, Hoon MA, et al. The receptors for mammalian sweet and umami taste. Cell. 2003;115:255–266. doi: 10.1016/s0092-8674(03)00844-4. [DOI] [PubMed] [Google Scholar]
- 100.Zhang Y, Hoon MA, Chandrashekar J, et al. Coding of sweet, bitter, and umami tastes: different receptor cells sharing similar signaling pathways. Cell. 2003;112:293–301. doi: 10.1016/s0092-8674(03)00071-0. [DOI] [PubMed] [Google Scholar]
- 101.Sukharev S, Corey DP. Mechanosensitive channels: multiplicity of families and gating paradigms. Sci STKE. 2004;2004:re4. doi: 10.1126/stke.2192004re4. [DOI] [PubMed] [Google Scholar]
- 102.Corey DP, García-Añoveros J, Holt JR, et al. TRPA1 is a candidate for the mechanosensitive transduction channel of vertebrate hair cells. Nature. 2004;432:723–730. doi: 10.1038/nature03066. [DOI] [PubMed] [Google Scholar]
- 103.Nagata K, Duggan A, Kumar G, García-Añoveros J. Nociceptor and hair cell transducer properties of TRPA1, a channel for pain and hearing. J Neurosci. 2005;25:4052–4061. doi: 10.1523/JNEUROSCI.0013-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Caterina MJ, Schumacher MA, Tominaga M, Rosen TA, Levine JD, Julius D. The capsaicin receptor: a heat-activated ion channel in the pain pathway. Nature. 1997;389:816–824. doi: 10.1038/39807. [DOI] [PubMed] [Google Scholar]
- 105.Caterina MJ, Rosen TA, Tominaga M, Brake AJ, Julius D. A capsaicin-receptor homologue with a high threshold for noxious heat. Nature. 1999;398:436–441. doi: 10.1038/18906. [DOI] [PubMed] [Google Scholar]
- 106.Caterina MJ, Leffler A, Malmberg AB, et al. Impaired nociception and pain sensation in mice lacking the capsaicin receptor. Science. 2000;288:306–313. doi: 10.1126/science.288.5464.306. [DOI] [PubMed] [Google Scholar]
- 107.Ghilardi JR, Rohrich H, Lindsay TH, et al. Selective blockade of the capsaicin receptor TRPV1 attenuates bone cancer pain. J Neurosci. 2005;25:3126–3131. doi: 10.1523/JNEUROSCI.3815-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Corey DP, García-Añoveros J, Holt JR, et al. TRPA1 is a canidate for the mechanosensitive transduction channel of vertebrate hair cells. Nature. 2004;432:723–730. doi: 10.1038/nature03066. [DOI] [PubMed] [Google Scholar]
- 109.Bautista DM, Movahed P, Hinman A, et al. Pungent products from garlic activate the sensory ion channel TRPA1. Proc Natl Acad Sci USA. 2005;102:12248–12252. doi: 10.1073/pnas.0505356102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Bautista DM, Jordt SE, Nikai T, et al. TRPA1 mediates the inflammatory actions of environmental irritants and proalgesic agents. Cell. 2006;124:1269–1282. doi: 10.1016/j.cell.2006.02.023. [DOI] [PubMed] [Google Scholar]
- 111.Kwan KY, Allchorne AJ, Vollrath MA, et al. TRPA1 contributes to cold, mechanical, and chemical nociception but is not essential for hair-cell transduction. Neuron. 2006;50:277–289. doi: 10.1016/j.neuron.2006.03.042. [DOI] [PubMed] [Google Scholar]
- 112.Bach G. Mucolipin 1: endocytosis and cation channel—a review. Pflug Arch. 2005;451:313–317. doi: 10.1007/s00424-004-1361-7. [DOI] [PubMed] [Google Scholar]
- 113.Bargal R, Avidan N, Ben-Asher E, et al. Identification of the gene causing mucolipidosis type IV. Nat Genet. 2000;26:118–123. doi: 10.1038/79095. [DOI] [PubMed] [Google Scholar]
- 114.Bach G, Webb MB, Bargal R, Zeigler M, Ekstein J. The frequency of mucolipidosis type IV in the Ashkenazi Jewish population and the identification of 3 novel MCOLN1 mutations. Hum Mutat. 2005;26:591. doi: 10.1002/humu.9385. [DOI] [PubMed] [Google Scholar]
- 115.Kiselyov K, Chen J, Rbaibi Y, et al. TRP-ML1 is a lysosomal monovalent cation channel that undergoes proteolytic cleavage. J Biol Chem. 2005;280:43218–43223. doi: 10.1074/jbc.M508210200. [DOI] [PubMed] [Google Scholar]
- 116.Soyombo AA, Tjon-Kon-Sang S, Rbaibi Y, et al. TRP-ML1 regulates lysosomal pH and acidic lysosomal lipid hydrolytic activity. J Biol Chem. 2006;281:7294–7301. doi: 10.1074/jbc.M508211200. [DOI] [PubMed] [Google Scholar]
- 117.Aarts M, Iihara K, Wei WL, et al. A key role for TRPM7 channels in anoxic neuronal death. Cell. 2003;115:863–877. doi: 10.1016/s0092-8674(03)01017-1. [DOI] [PubMed] [Google Scholar]