

Abstract
Adenoviruses are attractive vectors for the delivery of foreign genes into mammalian cells for gene therapy. However, current vectors retain many viral genes that, when expressed at low levels, contribute to the induction of a host immune response against transduced cells. We have developed a helper-dependent packaging system for production of vectors that have large regions of the genome deleted. Helper viruses were constructed with packaging signals flanked by loxP sites so that in 293 cells that stably express the Cre recombinase (293Cre), the packaging signal was efficiently excised, rendering the helper virus genome unpackageable. However, the helper virus DNA was replicated at normal levels and could thus express all of the functions necessary in trans for replication and packaging of a vector genome containing the appropriate cis-acting elements. Serial passage of the vector in helper virus-infected 293Cre cells resulted in an ≈10-fold increase in vector titer per passage. The vector could be partially separated from residual helper virus by cesium chloride buoyant density centrifugation. Large scale preparations of vector yielded semi-purified stocks of ≈1010 transducing virions/ml, with <0.01% contamination by the E1-deleted helper virus. This system should have great utility for the generation of adenovirus-based vectors with increased cloning capacity, increased safety and reduced immunogenicity.
Adenoviruses (Ads) have become popular as vectors for the delivery of foreign genes into mammalian cells for gene therapy, as recombinant viral vaccines, or as general purpose expression vectors for experimental studies. Ads have the advantage that they are well characterized biochemically and genetically, easy to manipulate and can be grown to high titers (1). Viruses rendered replication-defective by deletion of the E1 region can be propagated in 293 cells (2), and deletion of other nonessential regions can increase the cloning capacity further. Ad-based vectors can accommodate ≤105% of the wild-type genome, thus allowing for the insertion of up to 8 kb of foreign DNA in E1/E3 deleted viruses (3). Moreover, Ads infect many different cell types, and can transduce both replicating and nonreplicating cells (4, 5). Finally, Ads have not been associated with any neoplastic disease and usually cause relatively mild, selflimiting infections in immunocompetent individuals.
The potential usefulness of Ad vectors in vivo has been demonstrated in animal models of cancer (6, 7) and inherited disorders such as cystic fibrosis (8, 9, 10, 11). However, results from preclinical studies and preliminary clinical trials of cystic fibrosis using first generation vectors (i.e., E1-deleted viruses), have suggested that virus administration can cause inflammation due to an immune response generated against the virus and the transduced cells (ref. 12 and references cited therein). This is thought to be due, at least in part, to low level expression of viral early and late genes in the transduced cells (9, 13, 14). Immunity to the vector also reduces the effectiveness of repeated administration (14, 15). Second generation vectors have been developed by further attenuating the virus through the deletion or inactivation of other virus genes (E2 or E4; refs. 16, 17, 18, 19, 20, 21, 22). These vectors persist longer in transduced cells and are associated with a decreased inflammatory response (16, 23, 24).
An alternative approach to the design of Ad vectors with improved capacity and safety is the development of helper-dependent vector systems. Indeed, the earliest Ad vectors constructed and studied were of this type (25, 26). However, because working with these systems was technically difficult, this approach was abandoned in favor of helper-independent vectors. Recently, helper-dependent systems have been developed that rely on a complementing virus to provide the necessary proteins in trans for the packaging of an Ad-based vector (27, 28, 29). Although the resulting vectors lack virtually all Ad sequences, except those cis-acting elements required for DNA packaging and replication, use of such vectors thus far has been hampered by the high levels of contaminating helper virus and the low yields of the vector in the final virus preparation. Here we describe a helper-dependent system that produces high titers of vector with very low quantities of contaminating helper virus. The system utilizes a helper virus that has packaging sequences flanked by loxP recognition sites (“floxed”). In 293 cells that express Cre recombinase, the packaging signal is efficiently excised, thus rendering the helper virus DNA unpackagable. The helper virus genome retains the ability to replicate and provide all the functions necessary in trans for the replication and packaging of a vector lacking most Ad sequences (Fig. 1).
Figure 1.
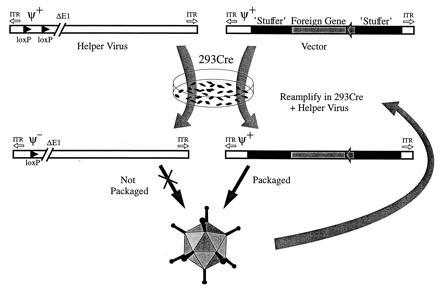
Generation of Ad-based vectors complemented by a helper virus containing a loxP-flanked packaging signal. Infection of Cre-expressing 293 cells with the helper virus results in excision of the viral packaging signal, rendering the helper virus DNA unpackagable. The helper virus will then provide all of the functions necessary in trans for replication and packaging of an Ad vector containing appropriate cis-acting elements [i.e., viral packaging signal (ψ+) and inverted terminal repeats]. Since all functions necessary for virion formation are provided by the helper virus, the majority of the Ad sequences contained in the vector can be replaced by a foreign gene and other non-Ad sequences (stuffer). The titer of the vector can be increased by serial passage in helper virus-infected 293Cre cells.
MATERIALS AND METHODS
Cell and Virus Culture.
Cell growth, virus propagation, and titration was carried out as previously described (1). Replication competent, E1+ Ad (RCA) was detected by ability to plaque or cause cytopathic effect on monolayers of A549 cells. The 293-derived cell lines that stably express the Cre recombinase, 293Cre1 and 293Cre4 (L.C., M.A., and F.L.G., unpublished results), were propagated in medium supplemented with 0.4 mg/ml G418.
Construction of AdLC8 and AdLC8c Helper Virus.
Plasmids were constructed according to standard protocols (30). The AdLC8 helper virus (Fig. 2) was rescued by cotransfection of 293 cells with pLC8, a plasmid containing a “floxed” packaging signal, and pBHG10, using techniques previously described (1). pLC8 was constructed as follows. A 9.6-kb AscI fragment was removed from pBG18 (31), a plasmid containing the majority of the Ad genome including the Ad packaging signal and the inverted terminal repeats, generating pLC4. A synthetic loxP site with BamHI compatible ends, obtained by annealing two single-stranded oligonucleotides (32), was cloned into pLC4 at a unique BamHI site, located at nucleotide 188 from the left end of the Ad5 genome, generating pLC5. Previously, this region was shown to tolerate similar DNA insertions without affecting virus replication (33). A second loxP site was inserted into pLC5 by first introducing the loxP oligo into the BamHI site of pABS.9 (31), generating pLC7, and subcloning a 1.4-kb XbaI fragment, containing the loxP site and a neo bacterial-expression cassette, into the unique XbaI site of pLC5. Thus, the resulting plasmid, pLC8, contains two loxP sites, separated by 1452 bp, and flanking the Ad packaging signal and neo cassette. The AdLC8cluc helper virus is similar to AdLC8, but lacks the neo expression cassette in E1, and contains the firefly luciferase gene under the regulation of of the human cytomegalovirus (CMV) major immediate-early promoter (−299 to +72 bp relative to the transcription start site) and simian virus 40 polyadenylylation signal inserted in E3 with a “stuffer” sequence. Details of the construction of AdLC8cluc are available on request.
Figure 2.
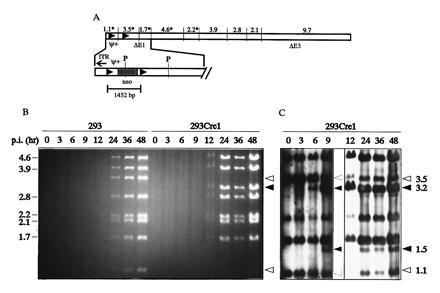
Structure of AdLC8 and analysis of Cre-mediated excision of the loxP flanked packaging signal (ψ) in 293Cre1 cells. (A) AdLC8 contains two loxP sites flanking the viral packaging signal and a neomycin gene (expanded), and was constructed as outlined in the Materials and Methods. The E1 and E3 deletions in AdLC8 remove sequences located between 339-3533 bp and 28,133–30,818 bp of the Ad genome, respectively. A PvuI restriction map of AdLC8, with fragment sizes indicated in kb, is shown above the schematic linear map, and PvuI restriction sites in the expanded regions are indicated (P). Fragments that hybridize to the pLC8-derived probe used during Southern blot analysis are indicated with an asterisk (∗). Cre-mediated recombination between the two loxP sites results in the disappearance of the 1.1- and 3.5-kb PvuI restriction fragments and formation of a novel 3.2-kb fragment. (B) 293 or 293Cre1 cells were infected with AdLC8 at a m.o.i. of 10 and DNA was extracted at times indicated, digested with PvuI, and separated on a 0.8% agarose gel. The 9.7-kb fragment is not shown. (C) DNA from the agarose gel shown in B was transferred to a nylon membrane and hybridized with a probe derived from pLC8 to monitor early Cre-mediated excision events. The 0–9-hr lanes were exposed to x-ray film for 1 hr, and the 12–48-hr lanes for 10 sec. Fragments of unrecombined AdLC8 are indicated by open arrows and recombination products by filled arrows. Molecular sizes (kb), are given on the left and right margins.
Construction of the Helper-Dependent Vector.
pCA35, which contains the Escherichia coli β-galactosidase (lacZ) ORF under the control of the murine CMV immediate-early promoter, was digested with SalI, repaired with the Klenow fragment of DNA polymerase, and recircularized, generating pCA35KS. The murine CMV-lacZ expression cassette was excised from pCA35KS by digestion with XbaI/BglII and inserted into XbaI/BamHI digested pABS.4 (31), generating pABS.4MClacZ, which was then linearized with SalI and ligated into the unique XhoI site of pFG140dx3, a derivative of pFG140 (35), generating pUMA10R. Thus, pUMA10R retains Ad-specific sequences corresponding to 5789 bp of the left end and 6143 bp of the right end of Ad5. The E1-coding region is disrupted by the insertion of pMX2 at the XbaI site (1338 bp). A 1276-kb SwaI fragment from pUMA10R was removed and replaced by a 8270-bp SmaI fragment from bacteriophage lambda DNA (31,617–39,888 bp of the conventional lambda map), generating pRP1001.
“Rescue” and Amplification of AdRP1001.
Semiconfluent monolayers of 293 cells in 60-mm dishes were transfected (36) with 5 μg of pRP1001 for 4 hr at 37°C, and 18 hr after transfection the cells were infected at a multiplicity of infection (m.o.i.) of 5 plaque-forming units (pfu)/cell with AdLC8 or AdLC8cluc, the medium replaced, and the cells incubated until the monolayers showed complete cytopathic effect (48–72 hr). The cells were scraped into the medium and the virus released by three rounds of freezing and thawing. An aliquot of the resulting crude viral lysate (500 μl) was serially passaged on 60-mm dishes of 293Cre1 or 293Cre4 cells (similar results were obtained with both cell lines). During each round of amplification of the helper-dependent vector, the 293Cre cells were coinfected with AdLC8 or AdLC8cluc at an m.o.i. of 5 for the first two rounds of amplification and at an m.o.i. of 1 for subsequent passages. Amplification of AdRP1001 was monitored by assaying aliquots of crude viral lysate after each passage for lacZ-expressing virus (blue-forming units, bfu) on 293 cells, by counting lacZ-positive cells after 5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside staining as follows: 293 cells were infected with virus inoculum (500 μl), and incubated for 24 hr at 37°C. The infected monolayers were washed once with PBS, fixed with 0.5 ml of 0.2% glutaraldehyde, 2% para-formaldehyde, 2 mM MgCl2 in PBS- for 5 min at 37°C, washed, and stained with 5 mM K4Fe(CN)6, 5 mM K3Fe(CN)6, 2 mM MgCl2, 1 mg/ml 5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside in PBS-. Plates were incubated overnight at room temperature, and bfu determined. Viral lysates were also monitored for helper virus and RCA by plaque assays on 293 and A549 monolayers, respectively. Large-scale virus preparations of AdRP1001 were prepared by infecting 150-mm dishes of 293Cre with 1 ml of crude AdRP1001 stock per 150-mm dish (m.o.i. = ≈1) plus 2 × 107 pfu of helper virus. After complete cytopathic effect, purification of AdRP1001 virions was performed by CsCl buoyant density centrifugation as described (1), and fractions were collected through the viral bands, and assayed for luciferase-expressing virus after 1:1000 dilution and infection of 293 cells. Crude protein extracts were prepared 24 hr postinfection, and assayed for luciferase activity using a commercial kit (Promega) and a Berthold model LB9501 luminometer. The quantity of luciferase present in each sample was calculated by comparison with a standard curve of luminescence vs. luciferase concentration generated from serial dilutions of purified luciferase (Boehringer Mannheim).
Analysis of Viral DNA.
Purification of DNA from infected cells and CsCl purified virions was performed by overnight digestion at 37°C in pronase-SDS buffer (10 mM Tris·HCl, pH 7.4/10 mM EDTA/1% SDS/1 mg/ml pronase), phenol extraction, and ethanol precipitation. Viral DNA was transferred from agarose gels to nylon membranes (Hybond-N, Amersham) by capillary transfer (37), hybridized to digoxigenin-labeled DNA probes, and visualized by chemiluminiscent reaction as specified by the manufacturer (DIG High Prime DNA labeling and detection kit, Boehringer Mannheim).
RESULTS AND DISCUSSION
Efficiency of Excision of the Packaging Signals from AdLC8.
The Cre recombinase, when expressed in 293 cells either transiently from an Ad vector (32) or stably from an integrated copy of the gene (L.C., M.A., and F.L.G., unpublished results), is able to efficiently excise sequences between loxP sites located in the Ad genome. Cloning loxP sites flanking the Ad packaging signal would limit the packaging of a helper virus when Cre is expressed, while allowing it to produce all of the proteins necessary in trans to replicate and package a second vector (Fig. 1). Since the cis-acting elements required for viral DNA replication are independent of those required for packaging of viral DNA into virions, removal of the packaging signal should not impair viral DNA replication, allowing the synthesis of sufficient template for expression of late viral genes to provide helper functions. We first constructed the helper virus AdLC8 (Fig. 2A), that had a “floxed” packaging signal (ψ), and examined the kinetics and efficiency of excision by the Cre recombinase in the 293Cre1 cell line, and the ability of the ψ− viral DNA to replicate. 293Cre1 cells, and 293 cells as controls, were infected with AdLC8 at a m.o.i. of 10. DNA was purified at various times postinfection, digested with PvuI, separated on a 0.8% agarose gel, and the products examined by ethidium bromide staining. Fig. 2B demonstrates that excision of the loxP flanked segment is very efficient as evidenced by the complete elimination in 293Cre1 cells of the 1.1-kb band and almost complete disappearance of the 3.5-kb band, both constituting the left end of the parental virus AdLC8. Instead, a 3.2-kb band was detectable from 12 hr onwards, representing the viral band generated by excision of the floxed cassette. The excision product of 1.5 kb was not detectable on the agarose gel since it remains unreplicated due to the lack of an origin for DNA synthesis. Furthermore, Fig. 2B shows that the recombined virus DNA (AdLC8ψ−) replicates as well in the 293Cre1 line as the parental AdLC8 virus in 293 cells.
To further examine the kinetics of Cre-mediated recombination, Southern blot analysis was performed. DNA from the agarose gel shown in Fig. 2B was transferred to a nylon membrane and probed with a labeled DNA fragment from pLC8. The probe hybridized with fragments of 1123, 1669, 2174, 3489, and 4553 bp of unrecombined AdLC8 (Fig. 2 A and C). Following Cre-mediated excision of ψ (AdLC8ψ−), two additional bands of 1452 bp (representing the excised circle) and 3160 bp were observed. Overexposure of the membrane during autoradiography revealed that no excision occurred prior to 6 hr postinfection in the 293Cre1 line, suggesting that the extreme left end of the virus may not be accessible to Cre during early times after infection, even though Cre is present in 293Cre1 cells at the time of infection. Analysis at later times using a shorter exposure demonstrated that by 12 hr ≈80% of the viral DNA had undergone excision and recombination, and this fraction did not increase at later times. The excised circle represented by a 1.5-kb band was detectable from 9 hr onward by Southern blot, and did not appear to be replicated. No recombination products (3.2-kb and 1.5-kb bands) were detectable after infection of 293 cells with AdLC8 (Fig. 2B and data not shown), indicating that the recombination products observed in 293Cre1 cells derived from Cre-specific events. Since the quantity of unrecombined viral DNA increased with kinetics similar to that of the total viral DNA, we conclude that the unrecombined fraction is from actively replicating virus. It is not known at present why these viral genomes escape Cre-mediated recombination. Nevertheless, the packaging signal was efficiently excised from the majority of the AdLC8 virus in the 293Cre1 cell line, and removal of this sequence did not affect viral DNA replication.
Amplification of Helper-Dependent Vectors with AdLC8.
Removal of the packaging signal from AdLC8 in 293Cre1 cells should allow AdLC8 to provide helper-functions for Ad vectors that lack most, if not all, Ad coding sequences. We examined this by transfecting 293Cre4 cells with pRP1001, a pFG140-derived plasmid with a deletion of Ad5 DNA from 16.1–83.6 mu (Fig. 3A), and infecting the transfected cells with the AdLC8 helper virus 18 hr later. In the presence of AdLC8, pRP1001 is converted to linear molecules that are replicated and packaged into virions, as determined by plating the infected-cell lysates from the transfection/infection on 293 cells and staining for lacZ activity. No blue cells were detected when cells were infected with lysates from control transfection/infections lacking either or both pRP1001 or helper virus. The frequency of AdLC8-mediated conversion of pRP1001 into packageable linear genomes was relatively low (≈80 bfu/μg transfected DNA). It was subsequently determined that there was an increased recovery of AdRP1001 if the initial transfection was performed in 293 cells (≈800 bfu/μg transfected DNA), and later transfections were performed in 293 cells. Serial passage of AdRP1001 through AdLC8-infected 293Cre4 cells resulted in an increase in the titer of helper-dependent vector at each passage (Fig. 3B). After five serial passages, the titer of AdRP1001 was ≈108 bfu/ml. The quantity of AdLC8 that had escaped excision of packaging signals, and formation of RCA (i.e., E1+ virus), were monitored by plaque assays on 293 and A549 cell lines, respectively. The level of AdLC8 present after each passage remained constant at 5 × 105 pfu/ml until passage 5, when infectious titers on 293 cells increased to 5 × 107 pfu/ml (Fig. 3B). RCA were detected after the second amplification through 293Cre4, and RCA titers continued to increase with subsequent passages, accounting for the increase in infectious virus after passage 5. That this was indeed RCA (i.e., E1+, E3− virus) was determined by analysis of the DNA content of several plaque isolates (data not shown). Restriction enzyme analysis of DNA from CsCl-purified virus also showed that the final viral preparation generated restriction patterns consistent with the presence of three distinct viral populations: AdRP1001, AdLC8, and E1+/E3− RCA (data not shown). Formation of E1+ virus from E1− vectors by recombination with the Ad5 sequences in 293 cells has been reported previously (38). Thus, although AdLC8 was capable of providing all the functions necessary to package a helper-dependent vector, its propensity to generate replication-competent virus limited its usefulness.
Figure 3.
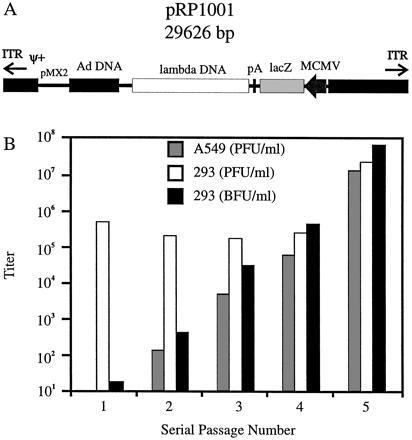
Structure of pRP1001 and vector amplification in AdLC8-infected 293Cre4 cells. (A) pRP1001 (shown in linear form), a derivative of pFG140 (35), contains sequences corresponding to the left end (3–5789 bp interrupted by the insertion of a bacterial plasmid [pMX2] at 1338 bp), and right end (29,792–35,924 bp) of the Ad genome, including the Ad packaging signal (ψ) and inverted terminal repeats. pRP1001 also contains the E. coli β-galactosidase gene (lacZ) between the murine CMV immediate early promoter and the simian virus 40 polyadenylyation signal, and an 8.3-kb insert of lambda DNA. In the presence of AdLC8 in 293 cells, pRP1001 is converted into linear molecules, replicated, and packaged into infectious virions (AdRP1001). (B) Serial passage of AdRP1001 (▪) in AdLC8-infected 293Cre4 cells. The titers on 293 cells (□) and A549 cells (░⃞) are also shown for each passage.
Amplification of AdRP1001 with AdLC8cluc.
Due to the high levels of RCA in helper-dependent vector preparations utilizing AdLC8, we designed a second helper virus, AdLC8cluc, with a luciferase expression cassette inserted in the E3 region (Fig. 4A). This helper virus has three advantages over AdLC8. First, recombination between the virus and Ad5 sequences in the 293 or 293Cre cell lines, resulting in the transfer of E1 sequences to the virus, would result in a virus of ≈38.4 kb. This exceeds the packaging limit for Ads (3), thus reducing or eliminating the potential for generation of RCA. Secondly, the larger genome size of AdLC8cluc should improve separation from the vector during CsCl centrifugation (27). Finally, inclusion of the luciferase cassette provides an easy, rapid, and sensitive assay for contaminating helper virus. Using the same strategy as before, we were able to amplify AdRP1001 to high titers (Fig. 4B). The frequency of AdLC8cluc-mediated conversion of pRP1001 into packagable linear genomes was similar to that of AdLC8 (≈1400 bfu/μg of transfected DNA in AdLC8cluc-infected 293 cells). Serial passage of AdRP1001 through AdLC8cluc-infected 293Cre1 cells resulted in an ≈10-fold increase in the titer of AdRP1001 per passage. Similar kinetics of amplification were observed for other vectors ranging in size from 27.7–33.6 kb (data not shown). Furthermore, the quantity of AdLC8cluc present in stocks remained low, and during the seventh passage, constituted <1% of the total virus. Finally, no RCA were detected. It appeared that more rounds of serial passage were required to obtain high titers of AdRP1001 in AdLC8cluc-infected cells when compared with AdLC8. This may suggest that the presence of RCA in AdLC8-passages also contributed to the replication and packaging of AdRP1001. Nevertheless, the data presented in Fig. 4B clearly illustrate that AdLC8cluc could serve as a helper virus to produce high titer stocks of AdRP1001.
Figure 4.
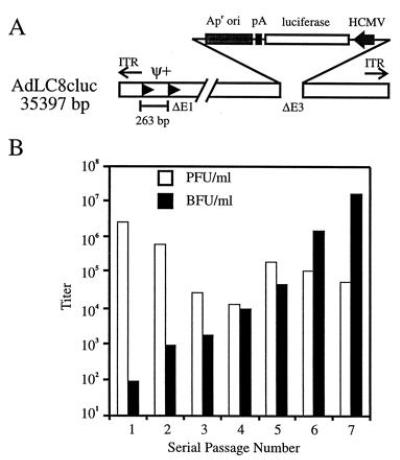
Structure of AdLC8cluc and amplification of AdRP1001 in AdLC8cluc-infected 293Cre1 cells. (A) AdLC8cluc is similar in structure to AdLC8 but lacks the neomycin gene and contains the firefly luciferase gene under regulation of the human CMV promoter and simian virus 40 polyadenylylation signal. (B) Serial passage of AdRP1001 (▪) in AdLC8cluc-infected 293Cre1 cells. The titer of AdLC8cluc at each passage is also shown (□).
Large-Scale Preparation and DNA Analysis of AdRP1001.
The results of Fig. 4B indicate that AdLC8cluc was capable of providing in trans all the functions for replication and amplification of a vector deleted for the majority of Ad sequences. We next determined if this strategy could be scaled up to provide sufficient quantities of AdRP1001 vector for purification and further characterization. Twenty 150-mm dishes (≈4 × 108 cells) of 293Cre1 cells were coinfected with AdRP1001 from passage 7 (Fig. 4B), and AdLC8cluc at an m.o.i. ≈1 for both viruses, and the infected cells harvested when the cells exhibited ≈80% cytopathic effect. The resulting virus was centrifuged to equilibrium in a CsCl density gradient. Two viral bands were visible which differed in their apparent density. The top band (lower buoyant density), containing the AdRP1001 vector with a lower DNA content (see below), was removed and purified on a second CsCl gradient. Only one viral band was visible after the second centrifugation. The lower band (higher buoyant density) and residual AdRP1001 were removed from the first gradient and recentrifuged. After the second centrifugation, two bands were again visible. Fractions (≈40 μl) were collected through these bands, and each fraction was analyzed for the presence of pfu and bfu. As shown in Fig. 5, these two activities could be readily separated by fractionation. The majority of AdLC8cluc was located in fraction 1 whereas the peak of AdRP1001 was in fraction 5. This difference in buoyant density is presumably due to differences in DNA content between AdLC8cluc (≈35.3 kb) and AdRP1001 (≈29.6 kb), as has been noted previously (27, 28). Furthermore, when fractions were assayed for their ability to induce luciferase activity, this activity correlated well (R2 = 0.963) with the relative titer of AdLC8cluc, indicating that luciferase activity is a good indicator of the level of contaminating helper virus. Using the data in Fig. 5, luciferase production by AdLC8cluc was calculated to be ≈2.6 × 105 pg luciferase/pfu on 293 cells.
Figure 5.
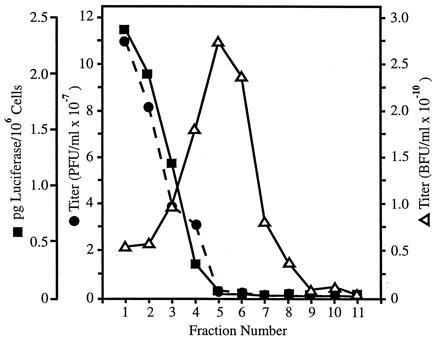
Gradient profile of CsCl-purified virus from large scale preparation of AdRP1001 in AdLC8cluc-infected 293Cre1 cells. Fractions (≈40 μl) were collected through the two visible virus bands from the CsCl gradient, and each fraction was analyzed for the presence of AdRP1001 (bfu) and AdLC8cluc (pfu), as indicated in the Materials and Methods. An aliquot of each fraction was also used to infect 293 cells, and cell extracts prepared at 24 hr postinfection were assayed for luciferase activity. The luciferase values were compared with a standard curve of luciferase activity to determine the amount of luciferase protein produced per 106 infected cells.
We also measured AdRP1001 and helper virus titers isolated from the lower density band after CsCl centrifugation. We obtained a titer of 8.6 × 109 bfu/ml, or a total yield of 6.5 × 109 AdRP1001 transducing units from 20–150-mm dishes of 293Cre cells. The ratio of AdRP1001 to AdLC8cluc in this stock was ≈16,000:1, or a 30-fold enhancement in the purity of AdRP1001 over the crude stock used in the purification. These results contrast with helper-dependent systems developed previously in which the major component of the “purified” vector was helper virus, with between 100- to 1000-fold more helper virus than vector (27, 28). Indeed, our yield of nearly 1010 virions is ≈1000-fold higher than that obtained using these other systems. Furthermore, it has been shown that the vectors in these systems underwent DNA recombination, generating a heterogeneous population of vector consisting of monomer, dimer, and more complex DNA structures. In contrast, restriction analysis of purified AdRP1001 showed that the DNA structure of the vector was identical to that predicted from the sequence of the initial transfecting plasmid pRP1001 (data not shown). When serial dilutions of AdRP1001 were used to infect 293 (permissive cells) or A549 cells (nonpermissive), a linear dependence was observed, as expected for “one-hit” kinetics (data not shown), indicating that infection by a single recombinant gave rise to a blue cell. We conclude that the helper-dependent system described above can produce high-titer vector preparations with very low quantities of E1− helper virus.
The vector utilized in this study retained ≈11.9 kb of Ad5 DNA. Since the sequences required in cis for Ad DNA replication and packaging are predicted to comprise only about 500 bp of DNA (39), the quantity of viral sequences contained in the vector could be further reduced. This would serve two functions. (i) It would eliminate all Ad-specific genes in the transduced cells, removing any potential contribution to immunogenicity caused by low-level expression of viral genes. (ii) It would allow for cloning of larger inserts of foreign DNA (theoretically as much as ≈37 kb) into the vector. It is important to note that the virus from AdLC8cluc-amplified vector stocks that generated plaques on 293 cells was the E1-deleted helper virus, and constituted <0.01% of the total virus present; thus, preparations of helper-dependent vector would be safer than current E1-deleted vectors. An Ad-based vector was recently described that contained the dystrophin gene, under the regulation of the creatine kinase promoter, and a lacZ reporter gene, for a total insert size approximately 28 kb (AdDYSβ-gal, ref. 29). We were able to amplify this vector (kindly provided by S. Kochanek, Baylor College of Medicine) 10- to 20-fold per serial passage in our helper system, demonstrating the feasability of this approach for other vectors. We are currently modifying and refining vectors, by removing additional Ad sequences and incorporating various lambda stuffer sequences, to facilitate the construction and amplification of Ad-based vectors carrying a variety of foreign genes using the AdLC8cluc/293Cre system.
Acknowledgments
We thank C. Evelegh and J. Rudy for excellent technical assistance, S. Kochanek for providing AdDYSβ-gal, and S. Bacchetti and J. Bramson for critically reading the manuscript. This work was supported by grants from the National Institutes of Health, Natural Sciences and Engineering Research Council, Medical Research Council, and the National Cancer Institute of Canada (NCIC). R.J.P. is supported by a Natural Sciences and Engineering Research Council Postdoctoral Fellowship and L.C. by an Ontario Graduate Scholarship. M.A.R. is a Research Scientist of the NCIC and F.L.G. is a Terry Fox Research Scientist of the NCIC.
Footnotes
The publication costs of this article were defrayed in part by page charge payment. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. §1734 solely to indicate this fact.
Abbreviations: Ad, adenovirus; RCA, replication competent E1+ Ad; bfu, blue-forming units; pfu, plaque-forming units; CMV, cytomegalovirus; m.o.i., multiplicity of infection.
References
- 1.Hitt M, Bett A J, Addison C L, Prevec L, Graham F L. In: Methods in Molecular Genetics. Adolph K W, editor. Vol. 7. San Diego: Academic; 1995. pp. 13–30. [Google Scholar]
- 2.Graham F L, Smiley J, Russell W C, Nairn R. J Gen Virol. 1977;36:59–72. doi: 10.1099/0022-1317-36-1-59. [DOI] [PubMed] [Google Scholar]
- 3.Bett A J, Prevec L, Graham F L. J Virol. 1993;67:5911–5921. doi: 10.1128/jvi.67.10.5911-5921.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Brody S L, Crystal R G. Ann NY Acad Sci. 1994;716:90–101. doi: 10.1111/j.1749-6632.1994.tb21705.x. [DOI] [PubMed] [Google Scholar]
- 5.Bramson J L, Graham F L, Gauldie J. Curr Opin Biotechnol. 1995;6:590–595. doi: 10.1016/0958-1669(95)80097-2. [DOI] [PubMed] [Google Scholar]
- 6.Addison C L, Braciak T, Ralston R, Muller W J, Gauldie J, Graham F L. Proc Natl Acad Sci USA. 1995;92:8522–8526. doi: 10.1073/pnas.92.18.8522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Addison C L, Gauldie J, Muller W J, Graham F L. Int J Oncol. 1995;7:1253–1260. doi: 10.3892/ijo.7.6.1253. [DOI] [PubMed] [Google Scholar]
- 8.Rosenfeld M A, Yoshimura K, Trapnell B C, Yoneyama K, Rosenthal E R, Dalemans W, Fukayama M, Bargon J, Steir L E, Stratford-Perricaudet L, Perricaudet M, Guggino W B, Pavirani A, Lecocq J-P, Crystal R G. Cell. 1992;68:143–155. doi: 10.1016/0092-8674(92)90213-v. [DOI] [PubMed] [Google Scholar]
- 9.Engelhardt J F, Simon R H, Yang Y, Zepeda M, Weber-Pendleton S, Doranz B, Grossman M, Wilson J M. Hum Gene Ther. 1993;4:759–769. doi: 10.1089/hum.1993.4.6-759. [DOI] [PubMed] [Google Scholar]
- 10.Zabner J, Coutre L A, Gregory R J, Graham S M, Smith A E, Welsh M J. Cell. 1993;75:207–216. doi: 10.1016/0092-8674(93)80063-k. [DOI] [PubMed] [Google Scholar]
- 11.Crystal R G, McElvaney N G, Rosenfeld M A, Chu C, Mastrangeli A, Hay J G, Brody S L, Jaffe H A, Eissa N T, Danel C. Nat Genet. 1994;8:42–51. doi: 10.1038/ng0994-42. [DOI] [PubMed] [Google Scholar]
- 12.Wilmott R W, Amin R S, Perez C R, Wert S E M, Keller G, Boivin G P, Hirsch R, De Inocencio J, Lu P, Reising S F, Yei S, Whitsett J A, Trapnell B C. Hum Gene Ther. 1996;6:301–318. doi: 10.1089/hum.1996.7.3-301. [DOI] [PubMed] [Google Scholar]
- 13.Yang Y, Nunes F A, Berencsi K, Furth E E, Gonczol E, Wilson J M. Proc Natl Acad Sci USA. 1994;91:4407–4411. doi: 10.1073/pnas.91.10.4407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dong J-Y, Wang D, Van Ginkel F W, Pascual D W, Frizzell R A. Hum Gene Ther. 1996;7:319–331. doi: 10.1089/hum.1996.7.3-319. [DOI] [PubMed] [Google Scholar]
- 15.Yei S, Mittereder N, Wert S, Whitsett J A, Wilmott R W, Trapnell B C. Hum Gene Ther. 1994;5:731–744. doi: 10.1089/hum.1994.5.6-731. [DOI] [PubMed] [Google Scholar]
- 16.Engelhardt J F, Ye X, Doranz B, Wilson J M. Proc Natl Acad Sci USA. 1994;91:6196–6200. doi: 10.1073/pnas.91.13.6196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Armentano D, Sookdeo C C, Hehir K M, Gregory R J, St. George J A, Prince G A, Wadsworth S C, Smith A E. Hum Gene Ther. 1995;6:1343–1353. doi: 10.1089/hum.1995.6.10-1343. [DOI] [PubMed] [Google Scholar]
- 18.Shaack J, Guo X, Ho W, Y-W, Karlok M, Chen C, Ornelles D. J Virol. 1995;69:4079–4085. doi: 10.1128/jvi.69.7.4079-4085.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Amalfitano A, Begy C R, Chamberlain J S. Proc Natl Acad Sci USA. 1996;93:3352–3356. doi: 10.1073/pnas.93.8.3352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gorziglia M I, Kadan M J, Yei S, Lim J, Lee G M, Luthra R, Trapnell B C. J Virol. 1996;70:4173–4178. doi: 10.1128/jvi.70.6.4173-4178.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Krougliak V, Graham F L. Hum Gene Ther. 1995;6:1575–1586. doi: 10.1089/hum.1995.6.12-1575. [DOI] [PubMed] [Google Scholar]
- 22.Yeh P, Dedieu J-F, Orsini C, Vigne E, Denefle P, Perricaudet M. J Virol. 1996;70:559–565. doi: 10.1128/jvi.70.1.559-565.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yang Y, Nunes F A, Berencsi K, Gonczol E, Engelhardt J F, Wilson J M. Nat Genet. 1994;7:362–369. doi: 10.1038/ng0794-362. [DOI] [PubMed] [Google Scholar]
- 24.Goldman M J, Litzky L A, Engelhardt J F, Wilson J M. Hum Gene Ther. 1995;6:839–851. doi: 10.1089/hum.1995.6.7-839. [DOI] [PubMed] [Google Scholar]
- 25.Solnick D. Cell. 1981;24:135–143. doi: 10.1016/0092-8674(81)90509-2. [DOI] [PubMed] [Google Scholar]
- 26.Thummel C, Tijan R, Grodzicker T. Cell. 1981;23:825–836. doi: 10.1016/0092-8674(81)90447-5. [DOI] [PubMed] [Google Scholar]
- 27.Mitani K, Graham F L, Caskey C T, Kochanek S. Proc Natl Acad Sci USA. 1995;92:3854–3858. doi: 10.1073/pnas.92.9.3854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fisher K J, Choi H, Burda J, Chen S-J, Wilson J M. Virology. 1996;217:11–22. doi: 10.1006/viro.1996.0088. [DOI] [PubMed] [Google Scholar]
- 29.Kochanek S, Clemens P R, Mitani K, Chan S, Caskey C T. Proc Natl Acad Sci USA. 1996;93:5731–5736. doi: 10.1073/pnas.93.12.5731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sambrook J, Fritch E F, Maniatis T. Molecular Cloning: A Laboratory Manual. 2nd Ed. Plainview, NY: Cold Spring Harbor Lab. Press; 1989. [Google Scholar]
- 31.Bett A J. Ph.D. thesis. Hamilton, ON Canada: McMaster Univ.; 1995. [Google Scholar]
- 32.Anton M, Graham F L. J Virol. 1995;69:4600–4606. doi: 10.1128/jvi.69.8.4600-4606.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bautista D S, Hitt M, McGrory J, Graham F L. Virology. 1991;182:578–596. doi: 10.1016/0042-6822(91)90599-7. [DOI] [PubMed] [Google Scholar]
- 34.DeWet J, Wood K V, DeLuca M, Helinski D R, Subramani S. Mol Cell Biol. 1987;7:725–737. doi: 10.1128/mcb.7.2.725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Graham F L. EMBO J. 1984;3:2917–2922. doi: 10.1002/j.1460-2075.1984.tb02232.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Graham F L, Van der Eb A J. Virology. 1973;52:456–467. doi: 10.1016/0042-6822(73)90341-3. [DOI] [PubMed] [Google Scholar]
- 37.Southern E. J Mol Biol. 1975;98:503–517. doi: 10.1016/s0022-2836(75)80083-0. [DOI] [PubMed] [Google Scholar]
- 38.Lochmuller H, Jani A, Huard J, Prescott S, Simoneau M, Massie B, Karpati G, Acsadi G. Hum Gene Ther. 1994;5:1485–1491. doi: 10.1089/hum.1994.5.12-1485. [DOI] [PubMed] [Google Scholar]
- 39.Grable M, Hearing P. J Virol. 1992;66:723–731. doi: 10.1128/jvi.66.2.723-731.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]