

Abstract
Recent cloning efforts have identified thousands of small RNAs including micro RNAs (miRNAs), Piwi-interacting RNAs (piRNAs) and small nucleolar RNAs (snoRNAs). These non-coding small RNAs need to be further validated and characterized by detecting and quantifying their expression in different tissues and during different developmental courses. A simple, accurate and sensitive method for small RNA expression profiling is in high demand. Here, we report such a PCR-based method.
Keywords: micro RNA (miRNA), Piwi-interacting RNA (piRNA), small nucleolar RNAs (snoRNAs), real-time quantitative PCR (Q-PCR), small RNA cDNA (srcDNA)
Numerous small RNAs are expressed by the eukaryotic genome [1,2]. Three classes of non-coding small RNA species have recently been discovered in mammals including micro RNAs (miRNAs) [3], Piwi-interacting RNAs (piRNAs) [4-8] and small nucleolar RNAs (snoRNAs) [9]. Most of the reported and registered small RNAs (∼4,000 miRNAs; ∼172,000 piRNAs and ∼200 snoRNAs as of September 2006) were cloned from different tissues and organisms using various cloning strategies. Expression of most of these small RNAs has not been validated in tissues during different developmental courses. This is not only because the number of small RNAs is increasing on a daily basis, but also due to a lack of a simple and sensitive detection and quantification method. Currently, Northern Blot analysis remains the most commonly used method [3]. Small RNAs are short in length [∼20 nucleotides (nt) for miRNAs, ∼30 nt for piRNAs and 60-200 nt for snoRNA], which makes the conventional detection and quantification methods inapplicable. The low sensitivity of Northern Blot analysis requires a relatively large amount of starting material (10-20 μg of total RNA or >5 μg of small RNA fraction). It is also a labor-intensive procedure involving the use of polyacrylamide gel electrophoresis, electro-transfer, radioisotope-labeled probes and autoradiography. Although two PCR-based quantification methods for miRNAs have been reported [3,10], these approaches require special reagents, such as locked nucleic acid (LNA), or complex modification steps, which hinder their application in conventional molecular biology laboratories. The increasing number of miRNAs and the recent discovery of hundreds of thousands of piRNAs require a high throughput, easy and accurate method to validate all the small RNA candidates. Here we report a PCR-based detection and quantification method for all three classes of small RNAs.
Methods and Materials
Small RNA isolation
Small RNA samples from 15 different mouse tissues [brain, heart, liver, spleen, lung, kidney, stomach, small intestine, colon, ovary, uterus and 4 developing testis samples at post natal day 7 (P7), P14, P21 and adult (8-10 weeks of age)] were isolated using the mirVana™ miRNA isolation kit (Ambion) according to the manufacturer’s instructions. Briefly, 50 to 250 mg of tissue was homogenized in 10 volumes of Lysis/Binding buffer. A 1/10 volume of miRNA Homogenate Additive was added and incubated on ice for 10 min. Total RNA was extracted by adding an equal volume of Acid-Phenol:Chloroform. Small RNA was extracted from the total RNA using a filter cartridge with 100 μl of preheated (95°C) elution solution. The concentration of small RNA was measured using a spectrophotometer NanoDrop, ND-1000 (NanoDrop Technologies).
Construction of small RNA cDNA library and SYBR Green-based real-time quantitative PCR
Small RNA samples isolated from the 15 mouse tissues were polyadenylated at 37°C for 60 min in a 50 μl reaction volume containing 0.5 μg RNA and 1.5 U poly(A) polymerase (Ambion). An equal volume of Acid-Phenol:Chloroform was added, mixed and centrifuged. The aqueous phase was carefully removed to a new tube. The poly(A)-tailed small RNA was purified from the sample using a purification filter cartridge provided in the mirVana Probe & Marker Kit (Ambion). Briefly, Binding/Washing Buffer (12 volumes of the sample) was added to the sample and mixed thoroughly. The mixture was applied onto the purification filter cartridge and centrifuged. The cartridge was then washed with 300 μl of Binding/Washing Buffer. Finally, the poly(A)-tailed small RNA was recovered with two sequential elutions using 25 μl Elution Buffer each time. To generate a small RNA cDNA (srcDNA) library, ∼2 μg of the tailed RNA and 1 μg of RTQ primer (Table 1) were mixed in a 26 μl reaction volume, incubated at 65°C for 10 min, and annealed at 4°C for 20 min. Reverse transcription was carried out with 200 U of SuperScript III reverse-transcriptase (Invitrogen), 1 μl dNTP mix (10 mM each) and 8 μl of 5× Buffer in a final reaction volume of 40 μl at 50°C for 60 min. Finally, the reverse transcriptase was inactivated by incubation at 70°C for 15 min and 1.5U of RNase H (Promega) was added to remove the small RNAs. The sample was purified using the QIAquick spin PCR purification kit (QIAGEN) in a final elution volume of 100 μl. The srcDNA concentration was measured using the NanoDrop spectrophotometer (NanoDrop Technologies). All srcDNA samples were diluted to the same concentration of 25 ng per μl (total about 500 μl). A small RNA-specific primer and a universal reverse primer, RTQ-UNIr, were used for amplification of each of the small RNAs (Table 1). Conventional PCR or real-time quantitative PCR (Q-PCR) was performed using 12.5 μl of AmpliTaq Gold PCR Master Mix or SYBR Green PCR Master Mix (Applied Biosystems), 1 μl (25 ng) of the synthesized srcDNAs and 5 μM of the primers in a 25 μl reaction volume on the 7300 Real-Time PCR System (Applied Biosystems). A 3-step PCR protocol (95°C for 10 min, then 40 cycles of 95°C for 15 sec, 50°C for 30 sec and 60°C for 30 sec) was used. The annealing temperature was adjusted according to the Tm of each of the small RNAs (see Table 1). After PCR, an aliquot of 2 μl of the PCR product was analyzed on a 2% agarose gel.
Table 1.
Oligonucleotides used in this study
| Name | Sequence (5′ to 3′) | Tm(°C) | Usage |
|---|---|---|---|
| 5′RNA adapter | GACUGGAGCACGAGGACACUGACAUGGACUGAAGGAGUAGAAA | 71 | cDNA library |
| RTQ primer* | CGAATTCTAGAGCTCGAGGCAGGCGACATGGCTGGCTAGT TAAGCTTGGTACCGAGCTCGGATCCACTAGTCC(T)25VN | 75-76 | cDNA library |
| let-7aQ | GAGGTAGTAGGTTGTATAGT | 48 | Q-PCR |
| let-7bQ | GAGGTAGTAGGTTGT T T GT GT |
52 | Q-PCR |
| let-7cQ | GAGGTAGTAGGTTGTAT GT GT |
50 | Q-PCR |
| let-7dQ | GAGGTAGTAGGTTG ATAGT ATAGT |
50 | Q-PCR |
| let-7eQ | GAGGTAG AGGTTGTATAGT AGGTTGTATAGT |
50 | Q-PCR |
| let-7fQ | GAGGTAGTAG TTGTATAGT TTGTATAGT |
46 | Q-PCR |
| let-7gQ | GAGGTAGTAG TTGTA TTGTA AGT AGT |
48 | Q-PCR |
| let-7iQ | GAGGTAGTAG TTGT TTGT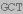 GT GT |
50 | Q-PCR |
| mir-18Q | TAAGGTGCATCTAGTGCAGAT | 50 | Q-PCR |
| mir-19bQ | TGTGCAAATCCATGCAAAACTGA | 52 | Q-PCR |
| piR-t454Q | TATTTTTCTCAGCTCTTATTGGG | 50 | Q-PCR |
| RTQ-UNIr | CGAATTCTAGAGCTCGAGGCAGG | 59 | Q-PCR |
| snoRNA-1 | CCATGATGATAACATAGTTCAGCAGAC | 57 | PCR |
| snoRNA-2 | CTGAAATGATGACCATTTAATTCATGTCTC | 56 | PCR |
| snoRNA-3 | CAAGTGACCCATTGGACTTTTTCTTG | 60 | PCR |
| snoRNA-4 | GAGTCCTGTGTGCATAGTTTTCATC | 56 | PCR |
Among let-7 isoform primers, ones that are unique over the others are highlighted in gray.
In RTQ primer, V is A, G, or C; N is A, G, C, or T.
Results and Discussion
Construction of small RNA cDNA libraries
Small RNAs were isolated from 15 mouse tissues including brain, heart, liver, spleen, lung, kidney, stomach, small intestine, colon, ovary, uterus, and 4 developing testes at P7, P14, P21 and adult. The small RNAs were polyadenylated using a poly(A) polymerase (Fig. 1) and then reverse transcription was performed using the primer RTQ (Table 1) to synthesize the srcDNAs. Primer RTQ is 100 bp in length and consists of 2 variable nucleotides (VN) at its 3′ end, preceded by 25 oligo dTs and a long stuffer sequence (73 bp) at the 5′ end. Mature miRNAs and piRNAs are about 20 and 30 nt in size, respectively. Therefore, the expected srcDNA sizes should range from 120 to 130 bp, which is within the optimal size range for SYBR green-based Q-PCR. Once the srcDNA library is constructed, it can be used up to 500 times for detection and quantification of multiple small RNAs (see below).
Fig. 1.

Schematic illustration of small RNA cDNA (srcDNA) library construction and real-time quantitative PCR (Q-PCR) analysis. Small RNAs were polyadenlyated using a poly(A) polymerase. The poly(A)-tailed RNAs were reverse-transcribed using a primer containing oligo dTs flanked by an adaptor sequence (for sequences see Table 1) to produce srcDNAs. The sample was treated with RNase H to remove the small RNAs from the cDNAs. Conventional PCR or Q-PCR was carried out using a small RNA-specific primer (srSP) and a universal reverse primer, RTQ-UNIr.
Conventional and real-time quantitative PCR analyses of eight let-7 miRNA isoforms
To test the srcDNAs generated, we used let-7 isoforms as a example because these miRNAs were well characterized and identified from many different tissues in the human [11-14], mouse [13,15] and rat [16,17]. Nine let-7 isoforms (a-h) have been reported. Conventional PCR was performed to amplify let-7 miRNAs using the multi-tissue srcDNA libraries as prepared above. We tested all let-7 isoforms except let7-h, which does not exist in either the mouse or human genome. Forty cycles of PCR were carried out and all eight let-7 isoforms were successfully amplified in the 11 mouse tissues and 4 developing testes (Fig. 2A).
Fig. 2.

Characteristics of the real-time quantitative PCR (Q-PCR) assay for let-7 miRNA isforms. (A) PCR amplicons of let-7 miRNA isoforms (a, b, c, d, e, f, g, and i) amplified using srcDNAs prepared from15 mouse tissues. Q-PCRs were performed using SYBR Green with the same amount of srcDNA (25 ng) for each tissue. The primer specific to each let-7 isoform in combination with the universal reverse primer, RTQ-UNIr, were used for amplification. After 40 cycles, products were electrophoresed on 2% agarose gels. NTC stands for a non-template control. A DNA marker was loaded on each side the gel. (B) The dynamic range and sensitivity of the Q-PCR assay. Amplification plot of let-7a miRNA over 10 orders of magnitude generated from a 2-fold (80 ng to 2.5 ng) and a 4-fold (2.5 ng to 0.001 ng) dilution series of the mouse adult testis srcDNAs. The experiments were conducted in triplicate. (C) Standard curve of the let-7a miRNA calculated over the 10 serial dilutions. The slope was -4.3001 and correlation coefficient was 0.9995. (D) Dissociation curves of all the amplicons.
We then performed Q-PCR using 25 ng of each of the 15 srcDNA samples to quantify the expression levels of six of the let-7 isoforms (a, b, c, d, e, and i). Among them, let-7a was the most abundantly expressed isoform (average CT, 17.3) in all tissues tested, whereas let-7i was the least (average CT, 20.3) (Table 2). let-7a also showed the second smallest standard deviation (SD) with a value at 1.06. Although let-7i showed the smallest SD (1.04), its expression levels were the lowest (Table 2). Therefore, we regard let-7a as a better internal control for Q-PCR.
Table 2.
Average threshold cycle (CT) and standard deviation (SD) of let-7 isoforms calculated from Q-PCR.
| miRNA | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 | 13 | 14 | 15 | Average CT | SD |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| let7a | 15.9 | 17.4 | 17.2 | 16.65 | 17.5 | 16.6 | 18.7 | 16.5 | 16.25 | 18.95 | 17.05 | 18.45 | 18.85 | 15.8 | 17.95 | 17.31667 | 1.062791 |
| let7b | 18.25 | 18.15 | 17.8 | 16.9 | 18.2 | 16.85 | 19.7 | 17.55 | 17.05 | 20.8 | 16.9 | 20.25 | 18.8 | 15.85 | 17.5 | 18.03667 | 1.37054 |
| let7c | 16.45 | 17.8 | 17.45 | 17.2 | 18 | 16.95 | 21.45 | 17.75 | 17.15 | 19.8 | 15.7 | 19 | 18.1 | 15.25 | 19.25 | 17.820 | 1.589115 |
| let7d | 16.85 | 18.3 | 17.95 | 18.4 | 17.75 | 17.6 | 19.8 | 17.75 | 17.45 | 19.75 | 17.75 | 20.05 | 19.25 | 16.5 | 18.95 | 18.27333 | 1.079826 |
| let7e | 16.25 | 17.4 | 17.4 | 17.65 | 17.65 | 16.6 | 18.95 | 16.7 | 17.7 | 19.75 | 16.5 | 18.4 | 18.65 | 15.35 | 17.9 | 17.52333 | 1.139841 |
| let7i | 19.5 | 20.35 | 19.65 | 20.95 | 20.2 | 19.65 | 22.05 | 18.75 | 19.4 | 22 | 19.65 | 20.35 | 20.95 | 19.25 | 21.75 | 20.29667 | 1.039826 |
The dynamic range and sensitivity of this method were further tested using let-7a and the SYBR Green PCR system. The adult testis srcDNAs were quantified based on the A260 value using the NanaDrop spectrophotometer and were diluted over 10 orders of magnitude. The Q-PCR assays showed an excellent linear relationship between the log ng of the target srcDNA and CT (Fig. 2B). The slope and intercept were -4.3001 and 23.69, respectively. The CT values correlated well with the srcDNA input (R2=0.9995) (Fig. 2C). Specificity of all the Q-PCR products was analyzed by a dissociation curve (Fig. 2D). The temperature ranged from 80.2 to 80.8°C, suggesting they were from target-specific amplification. These results demonstrate that this Q-PCR assay displays a dynamic range of at least 5 logs and is capable of detecting as little as 0.001ng of the target srcDNA in the Q-PCR assays.
Although the sequences of the let-7 isoforms are conserved, primers for each isoform are specific and would not cross-amplify. It is essential for PCR amplification that the 3′end of a primer should match completely to the template. It has been shown that a single base mismatch at or near the 3′ end of a primer dramatically decreases the efficiency of PCR amplification [26-28]. The let-7 isoforms, b, c, g, and i are unique at their 3′ ends, allowing for specific amplification of each isoform (Table 1). let7-a, e and f are the same at the 3′ end, but one nucleotide is different in the middle (Table 1). The SYBR PCR Taq polymerase mix uses a blend of a proof-reading and a regular Taq polymerase. Proof-reading DNA polymerases are known to correct single mismatches on primers that bind to unintended targets [29,30]. The sequencing analyses confirmed that unintended isoforms were not amplified in our PCR assays (data not shown). In addition, a mismatch would lower the annealing temperature and reduce the chance for annealing, thus causing less unintended amplification to occur.
Mature miRNAs are derived from either the 5′-strand, the 3′-strand, or both strands of a duplex in the stem-loop structure of a precursor miRNA (pre-miRNA) [18] [19]. All primers used for amplification of the let-7 isoforms in this study (Table 1) were derived from the 5′-strand and capable of detecting both mature miRNAs (∼22 nt) and pre-miRNAs (∼70 nt). If the pre-miRNAs were amplified, the size of the PCR products would have been ∼170 bp. However, the PCR products were ∼120 bp (Fig. 2A), suggesting that they were derived from mature let-7 miRNAs. Sequencing analyses of the PCR products demonstrated that no pre-miRNAs were detected by our method. These data suggested that the pre-miRNAs were eliminated during the srcDNA synthesis and that the srcDNAs contained only the mature miRNAs. In fact, all pre-miRNAs form a hairpin-loop in their secondary structure and the cDNA synthesis can be prematurely stopped or skipped on these hairpin loops during the reverse transcription (RT) reaction [20-23]. To overcome this problem, RT reactions should be performed at higher temperatures (60-70°C) using a high-temperature-stable Reverse Transcriptase [20,22,23]. Indeed, a recent report on quantitative analyses of pre-miRNA expression in cancer cell lines employed a high-temperature-stable Thermoscript to synthesize cDNAs at 60°C [24]. In our method reported here, however, a regular reverse transcriptase SuperScript III was used and the RT reaction was performed at 50°C. This temperature might not be high enough to melt the secondary structure of the pre-miRNAs, resulting in the lack of srcDNAs from pre-miRNAs during cDNA synthesis. In addition, a stem-loop RT-PCR study using a TaqMan miRNA assay showed that mature synthetic miRNAs were predominantly amplified (99.95%) over the same amount of synthetic pre-miRNAs [25]. Taken together, our PCR method preferentially amplifies mature miRNAs at the conditions described here. It is possible for this method to amplify pre-miRNAs if the RT reaction is performed at a higher temperature using a high-temperature-stable reverse transcriptase during the srcDNA synthesis.
Quantification of three testis-expressed small RNAs
The miRNA mir-18 has been reported to be expressed in the adult mouse testis [31]. We also cloned mir-18, mir-19b and a new piRNA piR-t454 from the adult mouse testis and the testis at P21 (our unpublished data). The expression profiles of the two miRNAs and one piRNA relative to the let-7a levels were determined in 15 different mouse tissues. PCR was performed for 30 cycles and PCR products were analyzed on 2% agarose gels (Fig. 3A). let-7a was ubiquitously abundant in all the 15 tissues, but mir-18, mir-19b and piR-t454 showed differential expression patterns. Q-PCRs were also performed on the same samples. The standard curve of let-7 (Fig. 3B) was used to calculate the log input amount of the three small RNAs [(CT -23.567)/-4.2147]. The srcDNA input ranged from1.25-40 ng. The slopes were similar (-3.9 to -4.2) and the CT values were correlated with the srcDNA input (R2 > 0.997) over 6 orders of magnitude (Fig. 3B), indicating that each small RNA was amplified specifically by its primer. The expression levels were normalized by the calculated amount of the control let-7a. Fig. 3C shows the quantification results of the expression levels of the three small RNAs which were similar to the PCR results. mir-18 was abundantly expressed in testis, spleen, small intestine, and colon. Interestingly, expression of mir-18 gradually increased during the 4 stages of testicular development. Generally, mir-19b was expressed more abundantly than mir-18 in all the tissues tested. The expression pattern of mir-19b was similar to that of mir-18 except that higher levels of mir-19 expression were detected in heart, liver, spleen, kidney and stomach as compared with mir-18. Several reports using Northern blot analyses have shown that piRNAs are exclusively expressed in the testis [4-8]. Interestingly, although piR-t454 was detected preferentially in the testis, lower levels of piR-t454 were also detectable in other tissues including the heart, spleen, kidney, and colon. This discrepancy is likely due to the low sensitivity of Northern blot analyses used in those studies [4-8], which failed to detect lower levels of expression in other tissues.
Fig. 3.

Quantitative analyses of three small RNAs (2 miRNAs mir-18, mir-19b, and a piRNA piR-t454) using Q-PCR. (A) PCR amplicons of let-7a, mir-18, mir-19b and piR-t454 RNAs amplified using srcDNAs from 15 mouse tissues. After 30 cycles of PCR, products were electrophoresed on 2% agarose gels. NTC stands for a non-template control. DNA size markers are shown on each side of the gel. (B) Correlation of srcDNA input with CT values for four small RNAs (let-7a, mir-18, mir-19b and piR-t454). Q-PCRs were performed over six orders of magnitude generated from a two-fold dilution series (40 ng to 1.25 ng) of mouse adult testis srcDNAs in triplicate over 40 cycles. (C) Expression levels of mir-18, mir-19b and piR-t454 in 15 mouse tissues. Q-PCRs were performed using the SYBR Green chemistry and srcDNAs prepared from the 15 mouse tissues (n=3). The expression level of each of the 3 small RNAs was normalized against that of let-7a.
Detection of snoRNAs
Four snoRNAs were also analyzed using our method (Fig. 4). We identified all 4 snoRNAs in our cloning efforts (our unpublished data) and three out of the four tested here were cloned previously [9]. snoRNA 1 (GenBank accession No. AJ224029) is 72-nt-long, snoRNA 2 (a novel mouse snoRNA similar to human SNORD111, NR_003079) is 80-nt-long, snoRNA 3 (AF357423) is 119-nt-long, and snoRNA 4 (AF357383) is 184-nt-long. All four snoRNAs were successfully amplified and confirmed by sequencing analyses of the PCR products. snoRNA 1 was ubiquitously expressed in all 15 tissues examined while the other three displayed preferential expression patterns. A previous report showed that snoRNA 3 is brain-specific based upon Northern dot blot analysis. Our PCR assay revealed that snoRNA 3 is preferentially expressed in brain and lung. Several other tissues including the developing testes, stomach, colon and ovary also expressed this snoRNA. Again, the discrepancy is likely a reflection of the low sensitivity of the Northern blot analysis employed in that study [9].
Fig. 4.

PCR amplicons of snoRNAs 1-4 amplified from srcDNAs of 15 mouse tissues. PCRs were performed using SYBR Green with the same amount of srcDNA (25 ng) for each tissue. A primer specific to each snoRNA was used in combination with the universal reverse primer, RTQ-UNIr for PCR. After 40 cycles, PCR products were electrophoresed on 2% agarose gels. NTC was a non-template control. Expected PCR product sizes of each snoRNA with the 100 bp stuffer sequence were indicated. A DNA ladder on each side indicates the size of the products.
Although we observed that the let-7a is ubiquitously expressed in the 15 tissues analyzed, it might not be expressed in other tissues untested or in the same tissues at different developmental stages. Levels of some miRNAs including let-7 miRNAs were reported to be regulated during the development of Caenorhabdits elegans [2,32], Drosophila melanogaster [33,34], mouse embryos [35], and some cancers [36-38]. In these cases, one should use other small RNAs such as 5S ribosomal RNA (GenBank accession No. M31319) or other ubiquitously expressed small RNAs as endogenous controls for Q-PCR.
With an increasing number of novel non-coding small RNAs being identified, a simple and accurate expression profiling method is highly demanded. The method that we report here can be used for validation of all cloned and predicted small RNAs, and for identification of tissue- or disease-specific small RNA species, which might be physiologically and clinically critical.
Acknowledgements
The authors would like to thank David Young and Myra Godfrey for editing the text. The Nevada Genomic Center is acknowledged for sequencing assays. This work was supported by a grant from the National Institute of Health (HD048855), and in part, by the start-up funds from the University of Nevada, Reno, to W.Y.
Footnotes
This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errorsmaybe discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Reference List
- [1].Hopper AK. Cellular dynamics of small RNAs. Critical Reviews in Biochemistry and Molecular Biology. 2006;41:3–19. doi: 10.1080/10409230500405237. [DOI] [PubMed] [Google Scholar]
- [2].Soares LMM, Valcarcel J. The expanding transcriptome: the genome as the ‘Book of Sand’. Embo Journal. 2006;25:923–931. doi: 10.1038/sj.emboj.7601023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Berezikov E, Cuppen E, Plasterk RH. Approaches to microRNA discovery. Nat.Genet.38 Suppl. 2006:S2–S7. doi: 10.1038/ng1794. [DOI] [PubMed] [Google Scholar]
- [4].Aravin A, Gaidatzis D, Pfeffer S, Lagos-Quintana M, Landgraf P, Iovino N, Morris P, Brownstein MJ, Kuramochi-Miyagawa S, Nakano T, Chien MC, Russo JJ, Ju JY, Sheridan R, Sander C, Zavolan M, Tuschl T. A novel class of small RNAs bind to MILI protein in mouse testes. Nature. 2006;442:203–207. doi: 10.1038/nature04916. [DOI] [PubMed] [Google Scholar]
- [5].Girard A, Sachidanandam R, Hannon GJ, Carmell MA. A germline-specific class of small RNAs binds mammalian Piwi proteins. Nature. 2006;442:199–202. doi: 10.1038/nature04917. [DOI] [PubMed] [Google Scholar]
- [6].Grivna ST, Beyret E, Wang Z, Lin HF. A novel class of small RNAs in mouse spermatogenic cells. Genes & Development. 2006;20:1709–1714. doi: 10.1101/gad.1434406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Lau NC, Seto AG, Kim J, Kuramochi-Miyagawa S, Nakano T, Bartel DP, Kingston RE. Characterization of the piRNA complex from rat testes. Science. 2006;313:363–367. doi: 10.1126/science.1130164. [DOI] [PubMed] [Google Scholar]
- [8].Watanabe T, Takeda A, Tsukiyama T, Mise K, Okuno T, Sasaki H, Minami N, Imai H. Identification and characterization of two novel classes of small RNAs in the mouse germline: retrotransposon-derived siRNAs in oocytes and germline small RNAs in testes. Genes & Development. 2006;20:1732–1743. doi: 10.1101/gad.1425706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Huttenhofer A, Kiefmann M, Meierw-Ewert S, O’Brien J, Lehrach H, Bachellerie JP, Brosius J. RNomics: an experimental approach that identifies 201 candidates for novel, small, non-messenger RNAs in mouse. Embo Journal. 2001;20:2943–2953. doi: 10.1093/emboj/20.11.2943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Chen CF, Ridzon DA, Broomer AJ, Zhou ZH, Lee DH, Nguyen JT, Barbisin M, Xu NL, Mahuvakar VR, Andersen MR, Lao KQ, Livak KJ, Guegler KJ. Real-time quantification of microRNAs by stem-loop RT-PCR. Nucleic Acids Research. 2005;33:e179. doi: 10.1093/nar/gni178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Dostie J, Mourelatos Z, Yang M, Sharma A, Dreyfuss G. Numerous microRNPs in neuronal cells containing novel microRNAs (vol 9, pg 180, 2003) Rna-A Publication of the Rna Society. 2003;9:631–632. doi: 10.1261/rna.2141503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Kasashima K, Nakamura Y, Kozu T. Altered expression profiles of microRNAs during TPA-induced differentiation of HL-60 cells. Biochemical and Biophysical Research Communications. 2004;322:403–410. doi: 10.1016/j.bbrc.2004.07.130. [DOI] [PubMed] [Google Scholar]
- [13].Lagos-Quintana M, Rauhut R, Yalcin A, Meyer J, Lendeckel W, Tuschl T. Identification of tissue-specific microRNAs from mouse. Current Biology. 2002;12:735–739. doi: 10.1016/s0960-9822(02)00809-6. [DOI] [PubMed] [Google Scholar]
- [14].Suh MR, Lee Y, Kim JY, Kim SK, Moon SH, Lee JY, Cha KY, Chung HM, Yoon HS, Moon SY, Kim VN, Kim KS. Human embryonic stem cells express a unique set of microRNAs. Developmental Biology. 2004;270:488–498. doi: 10.1016/j.ydbio.2004.02.019. [DOI] [PubMed] [Google Scholar]
- [15].Poy MN, Eliasson L, Krutzfeldt J, Kuwajima S, Ma XS, MacDonald PE, Pfeffer B, Tuschl T, Rajewsky N, Rorsman P, Stoffel M. A pancreatic islet-specific microRNA regulates insulin secretion. Nature. 2004;432:226–230. doi: 10.1038/nature03076. [DOI] [PubMed] [Google Scholar]
- [16].Kim J, Krichevsky A, Grad Y, Hayes GD, Kosik KS, Church GM, Ruvkun G. Identification of many microRNAs that copurify with polyribosomes in mammalian neurons. Proceedings of the National Academy of Sciences of the United States of America. 2004;101:360–365. doi: 10.1073/pnas.2333854100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Miska EA, Alvarez-Saavedra E, Townsend M, Yoshii A, Sestan N, Rakic P, Constantine-Paton M, Horvitz HR. Microarray analysis of microRNA expression in the developing mammalian brain. Genome Biology. 2004;5 doi: 10.1186/gb-2004-5-9-r68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Lee Y, Ahn C, Han JJ, Choi H, Kim J, Yim J, Lee J, Provost P, Radmark O, Kim S, Kim VN. The nuclear RNase III Drosha initiates microRNA processing. Nature. 2003;425:415–419. doi: 10.1038/nature01957. [DOI] [PubMed] [Google Scholar]
- [19].Bartel DP. MicroRNAs: Genomics, biogenesis, mechanism, and function. Cell. 2004;116:281–297. doi: 10.1016/s0092-8674(04)00045-5. [DOI] [PubMed] [Google Scholar]
- [20].Geiszt M, Lekstrom K, Leto TL. Analysis of mRNA transcripts from the NAD(P)H oxidase 1 (Nox1) gene - Evidence against production of the NADPH oxidase homolog-1 short (NOH-1S) transcript variant. Journal of Biological Chemistry. 2004;279:51661–51668. doi: 10.1074/jbc.M409325200. [DOI] [PubMed] [Google Scholar]
- [21].Mader RM, Schmidt WM, Sedivy R, Rizovski B, Braun J, Kalipciyan M, Exner M, Steger GG, Mueller MW. Reverse transcriptase template switching during reverse transcriptase-polymerase chain reaction: Artificial generation of deletions in ribonucleotide reductase mRNA. Journal of Laboratory and Clinical Medicine. 2001;137:422–428. doi: 10.1067/mlc.2001.115452. [DOI] [PubMed] [Google Scholar]
- [22].Ro S, Kang SH, Farrelly AM, Ordog T, Partain R, Fleming N, Sanders KM, Kenyon JL, Keef KD. Template switching within exons 3 and 4 of K(V)11.1 (HERG) gives rise to a 5 ′ truncated cDNA. Biochemical and Biophysical Research Communications. 2006;345:1342–1349. doi: 10.1016/j.bbrc.2006.05.032. [DOI] [PubMed] [Google Scholar]
- [23].Zhang YJ, Pan HY, Gao SJ. Reverse transcription slippage over the mRNA secondary structure of the LIP1 gene. Biotechniques. 2001;31:1286–+. doi: 10.2144/01316st02. [DOI] [PubMed] [Google Scholar]
- [24].Schmittgen TD, Jiang JM, Liu Q, Yang LQ. A high-throughput method to monitor the expression of microRNA precursors. Nucleic Acids Research. 2004;32 doi: 10.1093/nar/gnh040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Chen C, Ridzon DA, Broomer AJ, Zhou Z, Lee DH, Nguyen JT, Barbisin M, Xu NL, Mahuvakar VR, Andersen MR, Lao KQ, Livak KJ, Guegler KJ. Real-time quantification of microRNAs by stem-loop RT-PCR. Nucleic Acids Res. 2005;33:e179. doi: 10.1093/nar/gni178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Gibbs RA, Nguyen PN, Caskey CT. Detection of Single Dna-Base Differences by Competitive Oligonucleotide Priming. Nucleic Acids Research. 1989;17:2437–2448. doi: 10.1093/nar/17.7.2437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Lindeman R, Hu SP, Volpato F, Trent RJ. Polymerase Chain-Reaction (Pcr) Mutagenesis Enabling Rapid Nonradioactive Detection of Common Beta-Thalassemia Mutations in Mediterraneans. British Journal of Haematology. 1991;78:100–104. doi: 10.1111/j.1365-2141.1991.tb04389.x. [DOI] [PubMed] [Google Scholar]
- [28].Saiki RK, Gelfand DH, Stoffel S, Scharf SJ, Higuchi R, Horn GT, Mullis KB, Erlich HA. Primer-Directed Enzymatic Amplification of Dna with A Thermostable Dna-Polymerase. Science. 1988;239:487–491. doi: 10.1126/science.2448875. [DOI] [PubMed] [Google Scholar]
- [29].Carver TE, Hochstrasser RA, Millar DP. Proofreading Dna - Recognition of Aberrant Dna Termini by the Klenow Fragment of Dna-Polymerase-I. Proceedings of the National Academy of Sciences of the United States of America. 1994;91:10670–10674. doi: 10.1073/pnas.91.22.10670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Gale JM, Tafoya GB. Evaluation of 15 polymerases and phosphorothioate primer modification for detection of UV-induced C: G to T: A mutations by allele-specific PCR. Photochemistry and Photobiology. 2004;79:461–469. doi: 10.1562/2003-11-12-ra.1. [DOI] [PubMed] [Google Scholar]
- [31].Yu ZR, Raabe T, Hecht NB. MicroRNA Mirn122a reduces expression of the posttranscriptionally regulated germ cell transition protein 2 (Tnp2) messenger RNA (mRNA) by mRNA cleavage. Biology of Reproduction. 2005;73:427–433. doi: 10.1095/biolreprod.105.040998. [DOI] [PubMed] [Google Scholar]
- [32].Reinhart BJ, Slack FJ, Basson M, Pasquinelli AE, Bettinger JC, Rougvie AE, Horvitz HR, Ruvkun G. The 21-nucleotide let-7 RNA regulates developmental timing in Caenorhabditis elegans. Nature. 2000;403:901–906. doi: 10.1038/35002607. [DOI] [PubMed] [Google Scholar]
- [33].Pasquinelli AE, Reinhart BJ, Slack F, Martindale MQ, Kuroda MI, Maller B, Hayward DC, Ball EE, Degnan B, Muller P, Spring J, Srinivasan A, Fishman M, Finnerty J, Corbo J, Levine M, Leahy P, Davidson E, Ruvkun G. Conservation of the sequence and temporal expression of let-7 heterochronic regulatory RNA. Nature. 2000;408:86–89. doi: 10.1038/35040556. [DOI] [PubMed] [Google Scholar]
- [34].Sempere LF, Dubrovsky EB, Dubrovskaya VA, Berger EM, Ambros V, Ros A. The expression of the let-7 small regulatory RNA is controlled by ecdysone duriniz metamorphosis in Drosophila melanogaster. Developmental Biology. 2002;244:170–179. doi: 10.1006/dbio.2002.0594. [DOI] [PubMed] [Google Scholar]
- [35].Schulman BRM, Esquela-Kerscher A, Slack FJ. Reciprocal expression of lin-41 and the microRNAs let-7 and mir-125 during mouse embryogenesis. Developmental Dynamics. 2005;234:1046–1054. doi: 10.1002/dvdy.20599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Akao Y, Nakagawa Y, Naoe T. let-7 microRNA functions as a potential growth suppressor in human colon cancer cells. Biological & Pharmaceutical Bulletin. 2006;29:903–906. doi: 10.1248/bpb.29.903. [DOI] [PubMed] [Google Scholar]
- [37].Johnson SM, Grosshans H, Shingara J, Byrom M, Jarvis R, Cheng A, Labourier E, Reinert KL, Brown D, Slack FJ. RAS is regulated by the let-7 MicroRNA family. Cell. 2005;120:635–647. doi: 10.1016/j.cell.2005.01.014. [DOI] [PubMed] [Google Scholar]
- [38].Takamizawa J, Konishi H, Yanagisawa K, Tomida S, Osada H, Endoh H, Harano T, Yatabe Y, Nagino M, Nimura Y, Mitsudomi T, Takahashi T. Reduced expression of the let-7 microRNAs in human lung cancers in association with shortened postoperative survival. Cancer Research. 2004;64:3753–3756. doi: 10.1158/0008-5472.CAN-04-0637. [DOI] [PubMed] [Google Scholar]