

Abstract
Diabetic nephropathy is one of the most common causes of end-stage renal failure, but the factors responsible for the development of diabetic nephropathy have not been fully elucidated. We examined the effect of deletion of the angiotensin-converting enzyme 2 (Ace2) gene on diabetic kidney injury. Ace2−/− mice were crossed with Akita mice (Ins2WT/C96Y), a model of type 1 diabetes mellitus, and four groups of mice were studied at 3 months of age: Ace2+/yIns2WT/WT, Ace2−/yIns2WT/WT, Ace2+/y Ins2WT/C96Y, and Ace2−/yIns2WT/C96Y. Ace2−/y Ins2WT/C96Y mice exhibited a twofold increase in the urinary albumin excretion rate compared with Ace2+/yIns2WT/C96Y mice despite similar blood glucose levels. Ace2−/yIns2WT/C96Y mice were the only group to exhibit increased mesangial matrix scores and glomerular basement membrane thicknesses compared with Ace2+/yIns2WT/WT mice, accompanied by increased fibronectin and α-smooth muscle actin immunostaining in the glomeruli of Ace2−/y Ins2WT/C96Y mice. There were no differences in blood pressure or heart function to account for the exacerbation of kidney injury. Although kidney levels of angiotensin (Ang) II were not increased in the diabetic mice, treatment with an Ang II receptor blocker reduced urinary albumin excretion rate in Ace2−/yIns2WT/C96Y mice, suggesting that acceleration of kidney injury in these mice is Ang II-mediated. We conclude that ACE2 plays a protective role in the diabetic kidney, and ACE2 is an important determinant of diabetic nephropathy.
Activation of the renin-angiotensin system (RAS) and the generation of angiotensin (Ang) II play an important role in the pathogenesis of diabetic nephropathy and blockade of the RAS in both experimental and clinical diabetes mellitus attenuates the development of diabetic kidney injury.1,2,3 The recent discovery of a homologue of classical angiotensin-converting enzyme (ACE), angiotensin- converting enzyme 2 (ACE2), has revealed that Ang peptide processing is more complicated than previously thought.4 Like ACE, ACE2 is membrane-bound, but it is a monocarboxypeptidase that generates Ang (1-9) from the decapeptide Ang I and Ang (1-7) from Ang II.4 ACE2 serves the dual function of degrading the vasoconstrictor Ang II and producing the vasodilator Ang (1-7).4 Moreover, Su et al5 have reported that Ang (1-7) antagonizes Ang II-mediated cell signaling and limits Ang II-induced expression of transforming growth factor-β1. However, the role of ACE2 in mediating diabetic kidney injury has not been fully elucidated.
In diabetic rodent models, early increases in ACE2 mRNA levels, protein expression, and activity have been reported,6,7 whereas ACE2 mRNA and protein levels have been found to decrease in older streptozotocin-induced diabetic rats.8 Taken together, these studies suggest that ACE2 may play an early protective role in the development of diabetic nephropathy. To test this hypothesis directly, we used Akita mice (Ins2WT/C96Y), a model of type I diabetes mellitus, and mice with a deletion of the Ace2 gene (Ace2−/y).9 The Akita mouse harbors a missense mutation of the insulin 2 gene that leads to misfolding of native insulin in the endoplasmic reticulum of pancreatic β cells, activation of the unfolded protein response, and apoptosis, which eventually renders the mice insulin-deficient.10 The Ace2+/yIns2WT/C96Y mouse is a preferred murine model for the study of diabetic nephropathy because the mouse develops sustained hyperglycemia, renal hypertrophy, increased urinary albumin excretion rates (AERs), and mesangial matrix expansion, features that are similar to human diabetic nephropathy.9 We crossed male Ace2+/y-Ins2WT/C96Y mice with female Ace2−/−Ins2WT/WT mice to determine whether the deletion of the AceII gene would exacerbate the functional and structural changes of diabetic nephropathy in the Akita mouse. We then treated Ace2+/yIns2WT/C96Y mice and Ace2−/yIns2WT/C96Y mice with an Ang II type 1 (AT1) receptor blocker and measured kidney Ang peptide levels to determine whether the effect of deletion of the AceII gene in the diabetic mice was Ang II-dependent.
Materials and Methods
Experimental Animals and Protocol
C57BL/6J and diabetic heterozygous Akita (Ins2WT/C96Y) mice were purchased from The Jackson Laboratory (Bar Harbor, ME) and bred in our animal facility. Ace2−/y mice were outbred on the C57BL/6J background for at least seven generations, and littermate control mice, Ace2+/yIns2WT/WT, were used in the current experiments. To generate the Ace2−/yIns2WT/C96Y mice, female Ace2−/− mice were crossed with male Ins2WT/C96Y mice (Figure 1) because the AceII gene is on the X chromosome and the Akita mutation (Ins2WT/C96Y) is a dominant allele. Only male mice were used in the final experiments to simplify the breeding strategy. Throughout the period of study, animals were provided with free access to water and standard 18% protein rodent chow (Harlan Teklad, Madison, WI). Ace2+/yIns2WT/C96Y mice and Ace2−/yIns2WT/C96Y mice were also treated from weaning until 3 months of age with the ARB, irbesartan (50 mg/kg) by daily gavage (kindly provided by Dr. J. Froehlich, Bristol-Myers Squibb Inc., Montreal, QC, Canada). Twenty-four-hour urine volumes were collected at the end of 3 months and albumin excretion rate (AER) measured. All experiments were conducted in accordance with the guidelines of the University of Toronto Animal Care Committee.
Figure 1.

Breeding strategy. Female Ace2−/−Ins2WT/WT mice were bred with male Ace2+/yIns2WT/C96Y mice.
Genotyping
DNA was isolated from tail clips using the sodium dodecyl sulfate-potassium acetate method and the mice were genotyped for the Ins2 and AceII genes. The protocol for the Akita genotyping was obtained from The Jackson Laboratory. Briefly, the primers used were 5′-TGCTGATGCCCTGGCCTGCT-3′ and 5′-TGGTCCCACATATGCACATG-3′ (ACGT Corp., Toronto, ON, Canada). The polymerase chain reaction (PCR) cycling conditions were 94°C for 3 minutes, followed by 12 cycles of 94°C for 20 seconds, 64°C for 30 seconds, and 72°C for 35 seconds, then 25 cycles of 94°C for 20 seconds, 58°C for 30 seconds, and 72°C for 35 seconds, and ended with 72°C for 2 minutes. This PCR reaction amplifies a 280-bp DNA fragment from both mutant and wild-type alleles. The PCR products were then incubated at 37°C with FnU4HI restriction enzyme (1 U/0.2 μl; New England Biolabs, Pickering, ON, Canada), which digested wild-type alleles to 140 bp but not mutant alleles, and were resolved on a 2.5% agarose gel.
Blood Glucose Measurements
Body weights and blood glucose levels were obtained monthly. Blood glucose levels were measured monthly between 8:30 AM and 10:30 AM using an Ascensia Breeze glucometer (Bayer Inc., Toronto, ON, Canada). Approximately 3 μl of blood were collected in conscious mice via tail vein puncture. In pilot studies, we measured the blood glucose concentration weekly in diabetic mice for up to 12 weeks. Hyperglycemia was stable and sustained: between 27.3 and 33.1 mmol/L in the Ace2+/y-Ins2WT/C96Y mice and between 30.1 and 33.3 mmol/L in the Ace2−/yIns2WT/C96Y mice.
Urinary AER Measurements
Twenty-four-hour urine collections were obtained from mice before sacrifice by housing them in individual mouse metabolic cages (Nalgene; Nalge Nunc International, Rochester, NY) with free access to water and rodent mash. Urinary albumin concentration was measured using an indirect competitive enzyme-linked immunosorbent assay according to the manufacturer’s instructions (Albuwell M; Exocell, Philadelphia, PA).
Histopathology and Electron Microscopy
At 3 months of age, kidneys were harvested for pathological examination. The right kidney was removed before the left, and each was weighed and divided crosswise in three: one section was fixed in 10% neutral buffered formalin (Sigma-Aldrich Co., St. Louis, MO) for 24 hours and then transferred to 90% ethanol for light microscopy and immunohistochemistry, one section was fixed for electron microscopy, and the remaining four sections were snap-frozen for RNA extraction. The formalin-fixed tissue was embedded in paraffin and 2-μm sections were stained with periodic acid-Schiff stain. Slides were scanned digitally by the Advanced Optical Microscope Facility (Princess Margaret Hospital, Toronto, ON, Canada), and mean cross-sectional areas were calculated using Aperio ImageScope software (Aperio Technologies Inc., Vista, CA). Glomerular volume (V̄G) was calculated from the mean cross-sectional area (ĀG) of 33 glomerular profiles on each animal using the following equation:
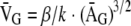 |
where β = 1.38 is the shape coefficient for spheres (the idealized shape of glomeruli) and k = 1.1 is the size distribution coefficient.11 Glomerular mesangial area was scored from 0 to 4, based on the presence or absence of increased mesangial matrix in each glomerular quadrant, on 34 glomerular profiles from a single coronal kidney section, and the mean was entered as the score for the animal. For electron microscopy, tissue was fixed in buffered 1% glutaraldehyde-4% formaldehyde, postfixed in 1% osmium tetroxide, and embedded in epon-araldite. Ultra-thin sections were stained with uranyl acetate and lead citrate and examined with a JEOL 1200 EX-II transmission electron microscope (JEOL, Tokyo, Japan). Glomerular basement membrane (GBM) widths were derived using the orthogonal intercept method and the harmonic mean was calculated on 100 widths for each mouse.
Immunohistochemistry
Formalin-fixed, paraffin-embedded sections were used for all immunohistochemical analysis from mouse kidneys. Heat-induced antigen retrieval was used for all antibodies. Endogenous peroxidase activity was prevented by pretreating all sections with 3% hydrogen peroxide. Primary antibodies and dilutions used were anti-fibronectin (1:1000; BD Transduction, San Diego, CA) and α-smooth muscle actin (α-SMA) (1:200; DAKO, Carpinteria, CA). Fibronectin and α-SMA immunohistochemistry was quantified using Image Pro Plus (Media Cybernetics, Silver Spring, MD) computer image analysis software.
Biochemistry
Mice were injected intraperitoneally 10 to 15 minutes before sacrifice with 0.1 ml of heparin sodium (1000 IU/ml; LEO Pharma Inc., Thornhill, ON, Canada) diluted 1 in 2 with normal saline to prevent blood clotting. Whole blood was collected on ice from the abdominal cavity after the kidneys were harvested. Samples were centrifuged at 3000 rpm at 4°C for 20 minutes, and the plasma was transferred to a new tube and stored at −80°C until analysis. Plasma was used to measure Na+, K+, Cl−, Ca2+, glucose, creatinine, urea, and lactate (Nova Biomedical Stat Profile M7; Centre for Modeling Human Disease, Toronto, ON, Canada, or VITA-TECH, Markham, ON, Canada). Ang I and Ang II concentrations were measured in snap-frozen whole kidney tissue by radioimmunoassay in the Hypertension and Vascular Disease Center Core Laboratory at Wake Forest University School of Medicine, Winston-Salem, NC, as previously described.12
Echocardiographic and Hemodynamic Parameters
Mice were anesthetized with isoflurane/oxygen (1/99%), and measurements were made as previously described.13 Hemodynamic parameters were obtained from the common carotid using a 1.4 French Millar catheter (Millar Inc., Houston, TX) advanced into the proximal aorta and then into the left ventricle. Pressure recordings were filtered at 200 Hz and sampled at 2 kHz (Acq 3.73; BioPac Systems Inc., Goleta, CA). Noninvasive tail-cuff systolic blood pressure (TC-SBP) in the mice was recorded as previously described.13
Real-Time Reverse Transcriptase (RT)-PCR
Renal cortical RNA expression levels for the genes reported in this study were quantified by real-time RT-PCR as described previously.14 In brief, kidney samples from mice were snap-frozen in liquid nitrogen, the cortex was later dissected in an RNA-stabilizing solution (RNAlater; Ambion Inc., Austin, TX), and RNA was extracted using TRIzol reagent (Invitrogen, Carlsbad, CA). One μg of total RNA was reverse-transcribed, and RNA expression levels were quantified by real-time RT-PCR using a sequence detection system (Prism 7700; Applied Biosystems Inc., Foster City, CA) as described previously.15 18S rRNA was used as the endogenous control. Primers and probes for the reported genes are indicated in Table 1.
Table 1.
Primer and Probe Sequences for Real-Time RT-PCR
| mRNA | Primer/probe | Sequence |
|---|---|---|
| ACE | Forward primer | 5′-TGAGAAAAGCACGGAGGTATCC-3′ |
| Reverse primer | 5′-AGAGTTTTGAAAGTTGCTCACATCA-3′ | |
| Probe | 5′-FAM-ACCCTGAAATATGGCACCCGGGC-TAMRA-3′ | |
| ACE2 | Forward primer | 5′-GGATACCTACCCTTCCTACATCAGC-3′ |
| Reverse primer | 5′-CTACCCCACATATCACCAAGCA-3′ | |
| Probe | 5′-FAM-CCACTGGATGCCTCCCTGCCC-TAMRA-3′ | |
| BKB2R | Forward primer | 5′-ATGTTCAACGTCACCACACAAGT-3′ |
| Reverse primer | 5′-GGCAGTTGTCCTTCGAAAGG-3′ | |
| Probe | 5′-FAM-CTCGGGTCTGCTCTTAACGGGA-TAMRA-3′ |
Western Blot Analysis
Kidney tissue was washed in ice-cold phosphate-buffered saline, sonicated in modified RIPA buffer (150 mmol/L sodium chloride, 50 mmol/L Tris-HCl, 1 mmol/L ethylenediaminetetraacetic acid, 1 mmol/L phenylmethyl sulfonyl fluoride, 1% Triton X-100, 1% sodium deoxycholic acid, 0.1% sodium dodecyl sulfate, 5 μg/ml aprotinin, and 5 μg/ml leupeptin) for 10 seconds twice, and then incubated on ice for 25 minutes. After centrifugation at 14,000 rpm for 10 minutes, the protein concentration was determined in the supernate. Proteins were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (10%), transferred to nitrocellulose membranes, and blocked for 1 hour at room temperature with Tris-buffered saline with 0.1% Tween 20 and 5% nonfat dry milk. After blocking, membranes were incubated with a rabbit polyclonal anti-ACE2 antibody, 1:1000, overnight at room temperature (provided by J.M.P.). Immunodetection was performed with chemiluminescence as previously described after incubation with peroxidase-conjugated anti-rabbit IgG.16
Statistical Analysis
Results are expressed as mean ± SEM, unless otherwise specified. Comparisons between multiple groups were performed by one-way analysis of variance. Both parametric and nonparametric tests were used with SPSS software (Chicago, IL), specifically the Fisher’s least significant difference and Tamhane’s T2 methods. P < 0.05 was considered statistically significant.
Results
Whole Animal Studies
Four groups of male mice with a combination of mutant or wild type at the AceII and Ins2 loci were generated: Ace2+/yIns2WT/WT (control C57BL/6J), Ace2−/yIns2WT/WT (ACE2 knockout), Ace2+/yIns2WT/C96Y (single mutant diabetic Akita), and Ace2−/yIns2WT/C96Y (double mutant diabetic ACE2 knockout) mice (Figure 1). Whole animal data, including body, heart, and kidney weights, plasma glucose, and 24-hour albumin excretion, are listed in Table 2. Monthly glucometer blood glucose measurements showed that both the Ace2+/yIns2WT/C96Y mice and Ace2−/yIns2WT/C96Y mice developed early and sustained hyperglycemia (Figure 2b), and weight gains were similar (Figure 2a). Likewise, at 3 months, plasma glucose levels in the diabetic groups were significantly greater than in the nondiabetic groups. There was no significant difference between the plasma glucose concentrations in the Ace2+/yIns2WT/C96Y and Ace2−/yIns2WT/C96Y mice (Table 2). Despite severe hyperglycemia in the Ace2+/yIns2WT/C96Y mice (52.0 ± 5.5 mmol/L) and Ace2−/yIns2WT/C96Y mice (57.8 ± 5.7 mmol/L), body weights remained similar in all four of the groups (Figure 2a and Table 2).
Table 2.
Whole Animal Data
| Parameter | Ace2+/yIns2WT/WT | Ace2−/yIns2WT/WT | Ace2+/yIns2WT/C96Y | Ace2−/yIns2WT/C96Y |
|---|---|---|---|---|
| BW (g) | 25.0 ± 0.4 (n = 16) | 24.5 ± 0.6 (n = 19) | 23.1 ± 0.4*† (n = 26) | 23.4 ± 0.5*† (n = 23) |
| KW (g) | 0.185 ± 0.006 (n = 17) | 0.162 ± 0.008 (n = 20) | 0.304 ± 0.011*† (n = 26) | 0.291 ± 0.017*† (n = 23) |
| KW/BW | 0.0072 ± 0.00015 (n = 26) | 0.0063 ± 0.00018 (n = 31) | 0.0134 ± 0.00043*† (n = 43) | 0.0134 ± 0.00049*† (n = 37) |
| HW (g) | 0.141 ± 0.005 (n = 16) | 0.154 ± 0.006 (n = 19) | 0.141 ± 0.005 (n = 26) | 0.146 ± 0.005 (n = 23) |
| Plasma glucose (mmol/L) | 14.2 ± 1.8 (n = 6) | 14.8 ± 0.9 (n = 6) | 52.0 ± 5.5*† (n = 6) | 57.8 ± 5.7*† (n = 7) |
| UV (ml) | 2.87 ± 0.15 (n = 20) | 3.29 ± 0.25 (n = 20) | 19.02 ± 0.88*† (n = 33) | 39.3 ± 1.5*†‡ (n = 28) |
| Urinary albumin (μg/day) | 31.79 ± 2.52 (n = 20) | 37.92 ± 3.71 (n = 20) | 178.77 ± 11.91*† (n = 26) | 398.78 ± 25.60*†‡ (n = 28) |
Body weight (BW), kidney weight (KW), kidney-weight-to-body-weight ratio (KW/BW), heart weight (HW), plasma glucose, 24-hour urine volume (UV), and 24-hour urinary albumin excretion rate were measured upon sacrifice at 3 months of age for all four groups of mice. Data are presented as mean ± SEM with sample sizes indicated in parentheses beside each value.
P < 0.05 compared with Ace2+/yIns2WT/WT.
P < 0.05 compared with Ace2−/yIns2WT/WT.
P < 0.05 compared with Ace2+/yIns2WT/C96Y.
Figure 2.

Body weight and blood glucose measurements. a: Body weights were measured monthly and the values were similar in the four groups of mice. b: Blood glucose was also measured monthly in each group of mice using an Ascensia Breeze glucometer. Both diabetic groups developed early and sustained hyperglycemia compared with the nondiabetic groups. Data are presented as mean ± SEM. *P < 0.05 compared with Ace2+/yIns2WT/WT mice, †P < 0.05 compared with Ace2−/yIns2WT/WT mice, and **P < 0.05 compared with Ace2+/yIns2WT/C96Y mice.
Kidney weights in both diabetic groups were almost twofold greater than in the Ace2+/yIns2WT/WT or Ace2−/yIns2WT/WT groups (Table 2). As expected, plasma creatinine values were similar in the four groups (Table 3), although there was a trend toward lower values in the Ace2−/yIns2WT/C96Y mice (P = 0.095). The urinary AER increased in the diabetic Ace2+/yIns2WT/C96Y mice compared with the nondiabetic Ace2+/yIns2WT/WT mice, and there was a dramatic additive effect of the deletion of the AceII gene on the urinary AER rates in the Ace2−/yIns2WT/C96Y mice (Figure 3). Despite similar blood glucose levels, the Ace2−/yIns2WT/C96Y mice exhibited a near doubling of the urinary AER compared with the Ace2+/yIns2WT/C96Y mice. There were no differences in the measures of plasma electrolytes (Table 3).
Table 3.
Plasma Electrolytes and Renal Function
| Electrolyte | Ace2+/yIns2WT/WT | Ace2−/yIns2WT/WT | Ace2+/yIns2WT/C96Y | Ace2−/yIns2WT/C96Y |
|---|---|---|---|---|
| Na+ (mmol/L) | 142.8 ± 0.9 (n = 6) | 145.2 ± 1.5 (n = 6) | 141.3 ± 2.1 (n = 6) | 137.4 ± 4.9 (n = 8) |
| Cl− (mmol/L) | 110.3 ± 0.9 (n = 6) | 113.0 ± 1.8 (n = 6) | 109.5 ± 1.7 (n = 6) | 105.6 ± 2.7† (n = 8) |
| Ca2+ (mmol/L) | 0.84 ± 0.08 (n = 4) | 1.12 ± 0.02* (n = 3) | 0.84 ± 0.04† (n = 4) | 0.88 ± 0.06† (n = 4) |
| Creatinine (μmol/L) | 42.7 ± 5.7 (n = 6) | 36.7 ± 8.4 (n = 6) | 45.3 ± 5.1 (n = 6) | 31.9 ± 1.9 (n = 7) |
| Lactate (mmol/L) | 6.12 ± 1.00 (n = 4) | 6.07 ± 1.33 (n = 3) | 7.33 ± 1.87 (n = 4) | 6.00 ± 1.24 (n = 4) |
| Urea (mmol/L) | 10.5 ± 0.6 (n = 6) | 11.3 ± 1.1 (n = 6) | 15.0 ± 1.6 (n = 6) | 16.3 ± 2.1*† (n = 8) |
Na+, Cl−, Ca2+, creatinine, lactate, and urea concentrations were measured by CMHD or VITA-TECH from plasma collected from the abdominal cavity after sacrifice at 3 months of age. Data are presented as mean ± SEM with sample sizes indicated in parentheses beside each value.
P < 0.05 compared with Ace2+/yIns2WT/WT mice.
P < 0.05 compared with Ace2−/yIns2WT/WT mice.
Figure 3.

Development of increased urinary AERs in the diabetic mice. The urinary AER was measured with an enzyme-linked immunosorbent assay in 24-hour urine samples collected at 3 months. The AER rate increased in both groups of diabetic mice but the AER was twofold greater in the Ace2−/yIns2WT/C96Y mice compared with the Ace2+/yIns2WT/C96Y mice at 3 months of age. Data are presented as mean ± SEM. *P < 0.05 compared with Ace2+/yIns2WT/WT mice, †P < 0.05 compared with Ace2−/yIns2WT/WT mice, and **P < 0.05 compared with Ace2+/yIns2WT/C96Y mice.
Histomorphometric Studies of the Kidney
Given the marked effect of deletion of the AceII gene on the urinary AER, we further sought to determine whether there were differences in kidney structure (Figure 4 and Table 4). Morphometric assessment showed that mean values for the glomerular volume tended to increase in both diabetic groups, but only the Ace2−/yIns2WT/C96Y mice exhibited a significant increase in glomerular volume compared with the control Ace2+/yIns2WT/WT mice (Table 4). Mean values for the mesangial matrix score also tended to increase in both diabetic groups. The Ace2−/yIns2WT/C96Y mice developed a significant increase in the mesangial matrix score compared with the control Ace2+/yIns2WT/WT mice (Figure 4). In accord with the light microscopy, the GBM thickness was greater in the Ace2−/yIns2WT/C96Y mice than in the control Ace2+/yIns2WT/WT mice (Table 4). Moreover, the GBM thickness was also significantly greater in the Ace2−/yIns2WT/C96Y diabetic mice than in the Ace2+/yIns2WT/C96Y diabetic mice (Table 4).
Figure 4.

Analysis of the mesangial matrix scores in glomeruli. a: Representative light micrographs of PAS-stained kidney sections from each group of mice. b: Mesangial matrix scores were generated on a quadrant scale (0 to 4) based on the amount of PAS-positive material in 34 glomerular profiles per animal. The Ace2−/yIns2WT/C96Y mice were the only diabetic group to develop a significant increase in the mesangial matrix score at 3 months of age compared with the control Ace2+/yIns2WT/WT mice. Data are presented as mean ± SEM on n = 6 per group. *P < 0.05 compared with Ace2+/yIns2WT/WT mice, and †P < 0.05 compared with Ace2−/yIns2WT/WT mice. Original magnifications, ×600.
Table 4.
Renal Histomorphometry
| Histological parameter | Ace2+/yIns2WT/WT | Ace2−/yIns2WT/WT | Ace2+/yIns2WT/C96Y | Ace2−/yIns2WT/C96Y |
|---|---|---|---|---|
| Glomerular volume (μm3) | 116,396 ± 6011 (n = 6) | 107,523 ± 9270 (n = 6) | 132,908 ± 6675† (n = 6) | 153,949 ± 7866*† (n = 6) |
| Mesangial matrix score | 1.69 ± 0.15 (n = 6) | 1.50 ± 0.13 (n = 7) | 2.06 ± 0.18† (n = 6) | 2.34 ± 0.15*†‡ (n = 6) |
| GBM thickness (nm) | 114.0 ± 0.9 (n = 2) | 107.9 ± 2.6 (n = 3) | 115.9 ± 3.0† (n = 2) | 127.6 ± 1.0*†‡ (n = 3) |
Glomerular volume, mesangial matrix score (0 to 4), and glomerular basement membrane (GBM) thickness were measured at 3 months. Data are presented as mean ± SEM with sample sizes indicated in parentheses beside each value.
P < 0.05 compared with Ace2+/yIns2WT/WT mice.
P < 0.05 compared with Ace2−/yIns2WT/WT mice.
P < 0.05 compared with the Ace2+/yIns2WT/C96Y mice.
To characterize further the mesangial injury, immunohistochemical studies were performed in the four groups of mice (Figures 5 and 6). There was a twofold increase in glomerular fibronectin immunostaining in the Ace2+/yIns2WT/C96Y mice compared with the Ace2+/yIns2WT/WT mice and a threefold increase in fibronectin immunostaining in the Ace2−/yIns2WT/C96Y mice compared with the Ace2+/yIns2WT/WT mice. Fibronectin immunostaining was also greater in the Ace2−/yIns2WT/C96Y mice compared with the Ace2+/yIns2WT/C96Y mice (Figure 5). α-Smooth muscle actin (α-SMA) immunostaining was increased in both of the diabetic groups of mice compared with the Ace2+/yIns2WT/WT mice, but there was also a marked increase in α-SMA in the Ace2−/yIns2WT/C96Y mice compared with the Ace2+/yIns2WT/C96Y mice (Figure 6).
Figure 5.

Histological analysis of fibronectin immunostaining in glomeruli. a: Representative light micrographs of fibronectin immunostaining in kidney sections from each group of mice. b: Fibronectin immunostaining in glomeruli was analyzed by computer image analysis. Fibronectin expression increased in both groups of diabetic mice, but there was a marked increase in fibronectin expression in the glomeruli of the Ace2−/yIns2WT/C96Y mice compared with the Ace2+/yIns2WT/C96Y mice at 3 months of age by measuring brown staining pixel density. Data are presented as mean ± SEM. *P < 0.05 compared with Ace2+/yIns2WT/WT mice, †P < 0.05 compared with Ace2−/yIns2WT/WT mice, and **P < 0.05 compared with the Ace2+/yIns2WT/C96Y mice. Original magnifications, ×600.
Figure 6.

Histological analysis of α-SMA immunostaining in glomeruli. a: Representative light micrographs of α-SMA immunostaining in kidney sections from each group of mice. b: Semiquantitative scoring of α-SMA immuno-staining in glomeruli. α-SMA immunostaining was increased sixfold in the Ace2−/yIns2WT/C96Y mice compared with the Ace2+/yIns2WT/C96Y mice at 3 months of age. Data are presented as mean ± SEM. *P < 0.05 compared with Ace2+/yIns2WT/WT mice, †P < 0.05 compared with Ace2−/yIns2WT/WT mice, and **P < 0.05 compared with the Ace2+/yIns2WT/C96Y mice. Original magnifications, ×600.
Echocardiographic and Hemodynamic Studies
Blood pressure is an important determinant of diabetic kidney injury. Tail-cuff SBP measures revealed no significant differences in SBP among the various groups of mice nor were there any differences in measures of intra-aortic blood pressure (Table 5). In addition, previous studies have shown that deletion of the AceII gene leads to impaired heart function13 and a marked impairment of cardiac function in the double mutant mice might have accounted for the difference in kidney injury. As shown in Table 5, transthoracic echocardiography revealed no significant differences in heart rate, ventricular dimensions, or myocardial contractility in the four groups of mice. The lack of systolic dysfunction was confirmed by hemodynamic measurements that showed no differences in (dP/dt)max or (dP/dt)min in the four groups of mice (Table 5).
Table 5.
Echocardiographic and Hemodynamic Parameters
| Ace2+/yIns2WT/WT | Ace2−/yIns2WT/WT | Ace2+/yIns2WT/C96Y | Ace2−/yIns2WT/C96Y | |
|---|---|---|---|---|
| HR (bpm) | 530 ± 25 (n = 7) | 503 ± 19 (n = 6) | 498 ± 27 (n = 6) | 514 ± 22 (n = 7) |
| LVEDD (mm) | 3.96 ± 0.12 (n = 7) | 4.01 ± 0.16 (n = 6) | 3.76 ± 0.22 (n = 6) | 3.8 ± 0.21 (n = 7) |
| FS (%) | 54.3 ± 3.6 (n = 7) | 51.5 ± 4.1 (n = 6) | 51 ± 4.3 (n = 6) | 50.7 ± 3.9 (n = 7) |
| VCFc (circ/second) | 9.21 ± 0.41 (n = 7) | 9.34 ± 0.52 (n = 6) | 10.11 ± 0.77 (n = 6) | 9.63 ± 0.51 (n = 7) |
| (dP/dt)max (mmHg/second) | 10,621 ± 395 (n = 7) | 9886 ± 523 (n = 9) | 9537 ± 421 (n = 7) | 9282 ± 557 (n = 5) |
| (dP/dt)min (mmHg/second) | −9448 ± 402 (n = 7) | −8780 ± 482 (n = 9) | −8832 ± 403 (n = 7) | −8539 ± 576 (n = 5) |
| TC-SBP (mmHg) | 98 ± 10 (n = 3) | 100 ± 2 (n = 4) | 95 ± 2 (n = 2) | 93 ± 3 (n = 4) |
HR, heart rate; bpm, beats per minute; LVEDD, left ventricular end diastolic dimension; FS, fractional shortening; VCFc, velocity of circumferential fiber shortening; (dP/dt)max/min, positive and negative first derivatives of the left ventricular pressure; TC-SBP, tail-cuff systolic blood pressure. Data are presented as mean ± SEM.
Gene Expression Studies
We analyzed ACE and ACE2 mRNA levels as well as bradykinin B2 receptor mRNA levels in the kidney cortex of the four groups of mice. As expected, ACE2 mRNA levels were not detected in either the Ace2−/yIns2WT/WT mice or the Ace2−/yIns2WT/C96Y mice. ACE mRNA levels fell in the Ace2+/yIns2WT/C96Y mice compared with the Ace2+/yIns2WT/WT mice whereas ACE2 mRNA levels increased. There was no further decline in ACE mRNA levels in the Ace2−/yIns2WT/C96Y mice. Bradykinin B2 receptor mRNA levels increased twofold in both diabetic groups compared with the nondiabetic groups (Figure 7). As expected, Western blot analysis of ACE2 protein levels in the whole kidney homogenates confirmed the measures of ACE2 mRNA levels (Figure 8). There was no ACE2 protein expression in the kidneys of Ace2−/yIns2WT/WT and Ace2−/yIns2WT/C96Y mice. ACE2 protein expression increased in the Ace2+/yIns2WT/C96Y mice compared with the Ace2+/yIns2WT/WT l mice.
Figure 7.

Effect of diabetes mellitus on ACE, ACE2, and the bradykinin B2 receptor expression in the kidney. mRNA levels were measured in the renal cortical tissue by real-time RT-PCR and related to 18S mRNA levels. a: Hyperglycemia was associated with a decrease in ACE expression in the kidney cortex of both groups of diabetic mice. b: ACE2 expression increased in the Ace2+/yIns2WT/C96Y mice compared with the Ace2+/yIns2WT/WT mice and, as expected, was not detected in the mice with deletion of the AceII gene. c: Bradykinin B2 receptor expression increased in both groups of diabetic mice. Data are presented as mean ± SEM. n.d., not detectable. *P < 0.05 compared with Ace2+/yIns2WT/WT mice, and †P < 0.05 compared with both Ace2+/yIns2WT/WT and Ace2−/yIns2WT/WT mice. AU, arbitrary unit.
Figure 8.

Western blot analysis of ACE2 expression in the kidney. a: Western blots for ACE2 and β-actin protein levels in the renal cortical tissue in the four groups of mice. b: Densitometry measures of ACE2 protein levels were related to β-actin. Hyperglycemia was associated with an increase in ACE2 expression in the kidney cortex of the Ace2+/yIns2WT/C96Y mice compared with the Ace2+/yIns2WT/WT mice. No ACE2 protein expression was detected in the Ace2−/yIns2WT/WT mice and the Ace2−/yIns2WT/C96Y mice. Data are presented as mean ± SEM. *P < 0.05 compared with Ace2+/yIns2WT/WT mice.
Angiotensin Receptor Blockade
We have reported that Ang II receptor blockade attenuates development of glomerulosclerosis in aging Ace2−/y mice.13 Accordingly, we treated Ace2+/yIns2WT/C96Y mice and Ace2−/yIns2WT/C96Y mice with the Ang II type 1 receptor blocker (ARB), irbesartan, to determine whether an ARB would reduce the urinary AER. As shown in Figure 9, treatment with the ARB reduced the urinary AER in the Ace2−/yIns2WT/C96Y mice to the same levels observed in the Ace2+/yIns2WT/C96Y mice. α-SMA immunostaining was also reduced to normal levels in the glomeruli from the Ace2−/yIns2WT/C96Y mice (α-SMA staining score, 0.01±.005).
Figure 9.

Effect of angiotensin II type 1 receptor blockade on the urinary AER. Four groups of mice were studied: Ace2+/yIns2WT/C96Y mice and Ace2−/yIns2WT/C96Y, either untreated or treated with the angiotensin II type 1 receptor blocker irbesartan (IRB) from weaning until 3 months of age (n = 6 in each group). The urinary AER was measured with an enzyme-linked immunosorbent assay in 24-hour urine samples. The AER was greater in untreated Ace2−/yIns2WT/C96Y mice compared with untreated Ace2+/yIns2WT/C96Y mice but this difference was prevented by treatment with IRB. Data are presented as mean ± SEM. **P < 0.05 compared with Ace2+/yIns2WT/C96Y mice, §P < 0.05 compared with IRB Ace2+/yIns2WT/C96Y mice, and ‡P < 0.05 compared with Ace2−/yIns2WT/C96Y mice.
The effect of irbesartan on blood pressure was determined in 8-week-old Ace2+/yIns2WT/WT and Ace2−/y Ins2WT/WT mice. The mice received 50 mg/kg/day as above, and blood pressure measurements were obtained after 4 weeks of treatment. Blood pressure values tended to be greater in the Ace2−/yIns2WT/WT compared with the Ace2+/yIns2WT/WT mice although the differences did not reach statistical significance (Table 6). Treatment with irbesartan reduced aortic SBP, diastolic blood pressure, and mean arterial blood pressure in both groups of mice.
Table 6.
Blood Pressure in Mice Treated with Irbesartan (50 mg/kg/day) for 4 Weeks
| Ace2+/y Ins2WT/WT | Ace2+/y Ins2WT/WT + IRB | Ace2−/y Ins2WT/WT | Ace2−/y Ins2WT/WT + IRB | |
|---|---|---|---|---|
| Aortic SBP (mmHg) | 114 ± 6 (n = 8) | 101 ± 5 (n = 8)* | 121 ± 6 (n = 8) | 104 ± 5 (n = 8)* |
| Aortic DBP (mmHg) | 78 ± 3 (n = 8) | 70 ± 4 (n = 8) | 81 ± 4 (n = 8) | 69 ± 5 (n = 8)* |
| MABP (mmHg) | 90 ± 4 (n = 8) | 80 ± 4 (n = 8)* | 94 ± 4 (n = 8) | 81 ± 6 (n = 8)* |
Eight-week-old Ace2+/yIns2WT/WT mice and Ace2−/y Ins2WT/WT mice were treated with the angiotensin receptor blocker irbesartan (50 mg/kg/day) for 4 weeks before measurement of blood pressure. Data are presented as mean ± SEM.
P < 0.05 compared with untreated mice.
Previous studies have also suggested that deletion of the AceII gene leads to increased generation of Ang II in tissue.13 To relate the functional studies to Ang peptide levels, we measured the Ang peptides, Ang I and Ang II, in whole kidney samples from the four groups of mice (Table 7). The concentrations of Ang I and Ang II were lower in the Ace2−/yIns2WT/C96Y mice compared with the control Ace2+/yIns2WT/WT mice and Ace2−/yIns2WT/WT mice.
Table 7.
Angiotensin Peptide Measures in Whole Kidney
| Ace2+/yIns2WT/WT | Ace2−/yIns2WT/WT | Ace2+/yIns2WT/C96Y | Ace2−/yIns2WT/C96Y | |
|---|---|---|---|---|
| Ang I (fmol/mg protein) | 7.84 ± 0.77 (n = 5) | 7.61 ± 0.70 (n = 6) | 6.43 ± 0.63 (n = 6) | 5.04 ± 0.91*† (n = 6) |
| Ang II (fmol/mg protein) | 30.5 ± 3.5 (n = 5) | 30.8 ± 4.6 (n = 6) | 22.7 ± 3.6 (n = 6) | 18.5 ± 3.3*† (n = 6) |
Whole kidney Ang peptide levels at 3 months. Ang I and Ang II concentrations were measured in kidney tissue in the four groups of mice at 3 months of age (n = 5 in each group). Data are presented as mean ± SEM.
P < 0.05 compared with Ace2+/yIns2WT/WT.
P < 0.05 compared with Ace2−/yIns2WT/WT mice.
Discussion
The rationale for this study was twofold: first, activation of the RAS plays an important role in the development of experimental and clinical diabetic nephropathy,1,2,3 and second, the recent discovery of an ACE homologue, ACE2, has revised our understanding of Ang peptide processing.4 Tikellis and colleagues8 were the first to report that ACE2 expression was reduced in the kidneys of rats with longstanding diabetes mellitus, whereas more recently, Ye and colleagues6 and Wysocki and colleagues7 reported that there was an early increase in ACE2 expression and activity in the kidneys of the diabetic db/db mouse. Taken together, these studies suggested that ACE2 might play an early protective role in the development of diabetic nephropathy and that a decline in expression of ACE2 might contribute to the development of glomerular injury. However, definitive data regarding the role of ACE2 in the pathogenesis of diabetic nephropathy has been lacking.
The development of microalbuminuria is the first functional abnormality that characterizes the natural history of nephropathy in patients with type 1 diabetes mellitus, and the AER is a primary outcome in many clinical studies of diabetic nephropathy.1,2,17,18 In addition, treatment-induced declines in albuminuria often predict a reduction in the rate of progression of diabetic nephropathy.18,19 Accordingly, the first goal of the current study was to determine whether deletion of the AceII gene would affect the urinary AER in the Ace2+/yIns2WT/C96Y mouse.20,21,22,23 A major finding was that the diabetic Ace2−/yIns2WT/C96Y mice exhibited a twofold increase in urinary albumin excretion compared with the diabetic Ace2+/yIns2WT/C96Y mice. In a recent study, Ye and colleagues24 have reported that pharmacological blockade of ACE2 increases the AER in diabetic db/db mice, a model of type 2 diabetes mellitus. Taken together with the current study, this work supports the hypothesis that ACE2 is an important determinant of albuminuria in the diabetic mouse.
Mauer and colleagues25 have reported structure-function correlations in studies of nephropathy in patients with type 1 diabetes mellitus and therefore a second goal of this study was to relate the exacerbation of urinary AER in the Ace2−/yIns2WT/C96Y mice to structural changes in the kidneys. Our findings of kidney hypertrophy in the two groups of diabetic mice confirm previous studies.22,23 The mean kidney weight was numerically greater in the Ace2−/yIns2WT/C96Y mice than in the Ace2+/yIns2WT/C96Y mice, although the difference did not reach statistical significance. At the light microscopic level, the Ace2−/yIns2WT/C96Y mice were the only group to exhibit a significant increase in glomerular volume compared with the control Ace2+/yIns2WT/WT mice at 3 months of age. Glomerular hypertrophy and mesangial matrix expansion are early features of human diabetic nephropathy. Mean values for the glomerular volume and mesangial matrix scores tended to increase in both diabetic groups;the increases were statistically significant in the Ace2−/yIns2WT/C96Y mice. In addition to these light microscopic changes in the glomerular mesangium, an increase in the deposition of extracellular matrix proteins in the glomeruli of the Ace2−/yIns2WT/C96Y mice was also reflected in the measures of GBM thickness. Exacerbation of AER in the Ace2−/yIns2WT/C96Y mice compared with the Ace2+/yIns2WT/C96Y mice was also associated with a significantly greater mean value for the GBM thickness, and the Ace2−/yIns2WT/C96Y mice were the only diabetic mice to exhibit a greater mean value for GBM thickness than the Ace2+/yIns2WT/WT mice and Ace2−/yIns2WT/WT mice.
Immunohistochemical studies of the kidneys were also performed in the four groups of mice. Both groups of diabetic mice exhibited greater fibronectin immunostaining in glomeruli compared with the Ace2+/yIns2WT/WT mice and the Ace2−/yIns2WT/WT mice, but fibronectin expression was most pronounced in the Ace2−/yIns2WT/C96Y mice. These observations are in accord with previous in vitro studies that have related fibronectin expression to glucose-induced activation of protein kinase C in glomerular mesangial cells, and with in vivo studies of experimental models of diabetic nephropathy.26,27 Pharmacological blockade of ACE2 also increases fibronectin expression in the glomeruli of the diabetic db/db mouse.24 We have previously reported14 that deletion of the AceII gene is associated with increased glomerular α-SMA staining in 12-month-old Ace2−/y mice, and the rationale for examining α-SMA in the current study is that de novo expression of α-SMA reflects activation of the mesangial cell and the development of a myofibroblast-like phenotype that has been linked to Ang II and glomerular injury.28 Deletion of the AceII gene was not associated with any increased glomerular α-SMA immunostaining nor was diabetes mellitus, per se, associated with marked increases in glomerular α-SMA expression in the Ace2+/yIns2WT/C96Y mice at 3 months of age. However, there was a significant increase in glomerular expression of α-SMA in the Ace2−/yIns2WT/C96Y mice compared with all of the other groups of mice.
A further goal of the current study was to determine the mechanism(s) responsible for the acceleration of the functional and structural changes of diabetic nephropathy in the Ace2−/yIns2WT/C96Y mice. Numerous clinical and experimental studies have shown that glycemic control and blood pressure are critical determinants of the rate of progression of diabetic nephropathy.1,2,18,29 Although the plasma glucose concentrations at 3 months in the diabetic groups of mice were greater than those reported by Gurley and colleagues23 (24.8 ± 3.6 mmol/L) and Kakoki and colleagues22 (38.8 ± 1.9 mmol/L), we observed early and sustained increases in the blood glucose concentrations. At the time of sacrifice, the blood glucose levels were also similar in both groups of diabetic mice, supporting our conclusion that differences in glycemic control were not responsible for the accelerated diabetic kidney injury in the Ace2−/yIns2WT/C96Y mice.
It was also important to evaluate heart function in the Ace2−/yIns2WT/C96Y mice because it has been reported that AceII-null mice exhibit decreased cardiac contractility,13 although recent studies suggest that the phenotype is variable.30 It was also possible that sustained hyperglycemia might have further exacerbated impairment in cardiac function in the Ace2−/yIns2WT/C96Y mice. Mean values for fractional shortening and velocity of circumferential fiber shortening, measures of heart function, and tail-cuff SBP were similar in the four groups of mice. It is surprising that we did not see a significant cardiac phenotype in the AceII-null mice and this may be attributable to the C57BL/6J background. In addition, the failure to detect differences in blood pressure between our groups may have been a function of the number of animals that we studied; blood pressure values did tend to be higher in the Ace2−/yIns2WT/WT mice than in the Ace2+/yIns2WT/WT mice although the differences did not reach statistical significance. Importantly, tail-cuff SBP was not increased in the Ace2−/yIns2WT/C96Y mice compared with the Ace2+/yIns2WT/C96Y mice, so we conclude that neither differences in blood pressure nor differences in heart function were responsible for the accelerated kidney injury in the Ace2−/yIns2WT/C96Y mice.
Activation of the RAS plays a key role in the progression of clinical and experimental diabetic nephropathy and blockade of the RAS has been shown to have important effects on the rate of progression of diabetic kidney disease.2,3 ACE2 plays an important role in the processing of angiotensin peptides in the kidney, as suggested in recent studies by Tikellis and colleagues8 and Wysocki and colleagues.7 Indeed, we observed that ACE2 mRNA levels and protein levels increased and ACE mRNA levels decreased in the kidneys of the Ace2+/yIns2WT/C96Y mice in accord with the findings of Wysocki and colleagues7 in diabetic db/db mice. High glucose concentrations activate the RAS at the level of renin31,32 and/or angiotensinogen,31,33,34 and these effects would be expected to increase activity of the RAS in the kidney.33 Therefore, we predicted that the accelerated kidney injury in Ace2−/yIns2WT/C96Y mice might be Ang II-dependent.
To test the hypothesis that exacerbation of diabetic kidney injury in the Ace2−/yIns2WT/C96Y mice was Ang II-dependent, we treated diabetic mice with 50 mg/kg of the Ang II receptor antagonist irbesartan from weaning to 3 months of age. We have previously reported that this dose of irbesartan reduces glomerular injury in aged Ace2−/y mice.14 Another major finding of the current study was that angiotensin receptor blockade with irbesartan reduced the urinary AER in Ace2−/yIns2WT/C96Y mice to values similar to those of the Ace2+/yIns2WT/C96Y mice. This finding is consistent with the hypothesis that acceleration of diabetic kidney injury in Ace2−/y-Ins2WT/C96Y mice is Ang II-dependent. We did not measure blood pressure in the irbesartan-treated diabetic mice although we found that 50 mg/kg/day of irbesartan was sufficient to lower blood pressure in both Ace2+/yIns2WT/WT mice and Ace2−/yIns2WT/WT mice.
To relate our findings further to activation of the RAS, we measured kidney angiotensin peptide levels in the four groups of mice. We predicted that the Ang II levels would be greatly increased in the Ace2−/yIns2WT/C96Y mice.30,35 Surprisingly, we found that Ang I levels and Ang II levels were not increased in the kidneys of Ace2+/yIns2WT/C96Y mice compared with Ace2+/yIns2WT/WT mice and that deletion of the ace2 gene did not lead to an increase in Ang II concentrations in samples of whole kidney from the Ace2−/yIns2WT/C96Y mice. Moreover, we could not confirm that Ang I and Ang II levels were increased in the Ace2−/yIns2WT/WT kidneys compared with the control Ace2+/yIns2WT/WT kidneys, as previously reported.13 This may be attributable to the C57BL/6J background, as recently suggested by Gurley and colleagues.30
It is possible that whole kidney Ang peptide levels did not accurately reflect the local microenvironment of glomeruli or efferent arterioles in our mice. For example, kinetic models of Ang II production and distribution in the renal cortex predict that AT1 receptors in the glomerulus respond primarily to arterially delivered Ang II, whereas peritubular AT1 receptors bind mostly to locally generated Ang II, in a paracrine manner.36,37 Moreover, the normalization of glomerular α-SMA expression by irebsartan that we observed further supports the hypothesis that there was increased Ang II activity in the glomeruli of the in Ace2−/yIns2WT/C96Y mice.
Two additional mechanisms of glomerular injury have been reported recently in studies of the Ins2WT/C96Y mouse. Kakoki and colleagues22 found that deletion of the bradykinin B2 receptor (BKB2R) also accelerated glomerular injury in the Ins2WT/C96Y mouse. We therefore measured BKB2R mRNA levels in the kidneys of our Ace2+/yIns2WT/C96Y mice and Ace2−/yIns2WT/C96Y mice to determine whether deletion of the AceII gene had an unexpected effect on BKB2R expression in the kidney. We observed a near doubling in BKB2R mRNA levels in the Ace2+/yIns2WT/C96Y mice, in accord with Kakoki and colleagues,22 whereas deletion of the AceII gene did not affect BKB2R mRNA levels. A recent study by Susztak and colleagues38 reported that podocyte apoptosis occurred with the onset of diabetes in Ace2+/yIns2WT/C96Y mice and preceded the development of albuminuria, whereas Liebau and colleagues39 found that Ang II increases apoptosis in human podocytes. Taken together, an increase in Ang II coupled with hyperglycemia in the Ace2−/yIns2WT/C96Y mice might induce greater levels of podocyte apoptosis, accounting for the accelerated nephropathy seen in this group. In this regard, ACE2 is expressed on glomerular podocytes as well as mesangial cells.24 Further studies will be necessary to determine whether deletion of the AceII gene leads to more podocyte apoptosis in Ace2−/yIns2WT/C96Y mice.
Our understanding of the role of ACE2 in human kidney disease is incomplete. In a recent study from Japan, Konoshita and colleagues40 compared ACE2 mRNA levels in the kidneys of a group of diabetic patients with a mixed group of patients with primary glomerular disease, nephrosclerosis, and lupus nephritis. They reported that ACE2 mRNA levels were similar in kidneys from these two disease groups, but they did not compare the levels of expression to a relatively normal group of kidneys.40 More recently, Lely and colleagues41 reported ACE2 immunostaining was uniformly increased in the glomeruli and peritubular capillaries of the kidney in a variety of renal diseases that included diabetic nephropathy. Because deletion of the AceII gene was associated with accelerated kidney injury in our mice, it is tempting to speculate that ACE2 is renoprotective and that reduced ACE2 expression might contribute to the progression of kidney disease.
In summary, we conclude that ACE2 is an important determinant of diabetic kidney injury. This conclusion is based on structure-function studies in the Ace2−/yIns2WT/C96Y mice that show an increased urinary AER, in association with increased glomerular volume, increased mesangial matrix expansion, increased fibronectin and α-SMA expression, and increased GBM thickening compared with the Ace2+/yIns2WT/C96Y mice. Differences in blood glucose levels, blood pressure, and heart function do not account for the accelerated kidney injury. The reduction in urinary AER by Ang II type 1 receptor blockade in the Ace2−/yIns2WT/C96Y mice suggests that Ang II is responsible for exacerbation of kidney injury in this experimental model.
Acknowledgments
We thank Dr. Golam Kabir, Miria Bartolini, Lois Kelsey, and the staff of the Division of Comparative Medicine at the University of Toronto for expert technical assistance.
Footnotes
Address reprint requests to J.W. Scholey M.D., Medical Sciences Building, Room 7326, One King’s College Circle, University of Toronto, Toronto, ON, Canada, M5S 1A8. E-mail: james.scholey@utoronto.ca.
Supported by the Canadian Diabetes Association (operating grant to G.Y.O., A.M.H., and J.W.S.) and the Canadian Institutes of Health Research [New Emerging Team Program (Genes, Gender and Glomerular-Based Diseases) to D.W.W. and A.M.H.], and Kidney Foundation of Canada Krescent program research fellowship award to H.R.
J.W.S. is the recipient of a Canadian Institutes of Health Research-AMGEN Canada Research Chair in Nephrology.
References
- Lewis EJ, Hunsicker LG, Bain RP, Rohde RD, for the Collaborative Study Group The effect of angiotensin-converting enzyme inhibition on diabetic nephropathy. N Engl J Med. 1993;329:1456–1462. doi: 10.1056/NEJM199311113292004. [DOI] [PubMed] [Google Scholar]
- Lewis EJ, Hunsicker LG, Clarke WR, Berl T, Pohl MA, Lewis JB, Ritz E, Atkins RC, Rohde R, Raz I, for the Collaborative Study Group Renoprotective effect of the angiotensin-receptor antagonist irbesartan in patients with nephropathy due to type 2 diabetes. N Engl J Med. 2001;345:851–860. doi: 10.1056/NEJMoa011303. [DOI] [PubMed] [Google Scholar]
- Brenner BM, Cooper ME, de Zeeuw D, Keane WF, Mitch WE, Parving HH, Remuzzi G, Snapinn SM, Zhang Z, Shahinfar S, RENAAL Study Investigators Effects of losartan on renal and cardiovascular outcomes in patients with type 2 diabetes and nephropathy. N Engl J Med. 2001;345:861–869. doi: 10.1056/NEJMoa011161. [DOI] [PubMed] [Google Scholar]
- Burrell LM, Johnston CI, Tikellis C, Cooper ME. ACE2, a new regulator of the renin-angiotensin system. Trends Endocrinol Metab. 2004;15:166–169. doi: 10.1016/j.tem.2004.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su Z, Zimpelmann J, Burns KD. Angiotensin-(1-7) inhibits angiotensin II-stimulated phosphorylation of MAP kinases in proximal tubular cells. Kidney Int. 2006;69:2212–2218. doi: 10.1038/sj.ki.5001509. [DOI] [PubMed] [Google Scholar]
- Ye M, Wysocki J, Naaz P, Salabat MR, LaPointe MS, Batlle D. Increased ACE 2 and decreased ACE protein in renal tubules from diabetic mice: a renoprotective combination? Hypertension. 2004;43:1120–1125. doi: 10.1161/01.HYP.0000126192.27644.76. [DOI] [PubMed] [Google Scholar]
- Wysocki J, Ye M, Soler MJ, Gurley SB, Xiao HD, Bernstein KE, Coffman TM, Chen S, Batlle D. ACE and ACE2 activity in diabetic mice. Diabetes. 2006;55:2132–2139. doi: 10.2337/db06-0033. [DOI] [PubMed] [Google Scholar]
- Tikellis C, Johnston CI, Forbes JM, Burns WC, Burrell LM, Risvanis J, Cooper ME. Characterization of renal angiotensin-converting enzyme 2 in diabetic nephropathy. Hypertension. 2003;41:392–397. doi: 10.1161/01.HYP.0000060689.38912.CB. [DOI] [PubMed] [Google Scholar]
- Breyer MD, Böttinger E, Brosius FC, III, Coffman TM, Harris RC, Heilig CW, Sharma K. Mouse models of diabetic nephropathy. J Am Soc Nephrol. 2005;16:27–45. doi: 10.1681/ASN.2004080648. [DOI] [PubMed] [Google Scholar]
- Oyadomari S, Koizumi A, Takeda K, Gotoh T, Akira S, Araki E, Mori M. Targeted disruption of the Chop gene delays endoplasmic reticulum stress-mediated diabetes. J Clin Invest. 2002;109:525–532. doi: 10.1172/JCI14550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller PL, Scholey JW, Rennke HG, Meyer TW. Glomerular hypertrophy aggravates epithelial cell injury in nephrotic rats. J Clin Invest. 1990;85:1119–1126. doi: 10.1172/JCI114543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allred AJ, Chappell MC, Ferrario CM, Diz DI. Differential actions of renal ischemic injury on the intrarenal angiotensin system. Am J Physiol. 2000;279:F636–F645. doi: 10.1152/ajprenal.2000.279.4.F636. [DOI] [PubMed] [Google Scholar]
- Crackower MA, Sarao R, Oudit GY, Yagil C, Kozieradzki I, Scanga SE, Oliveira-dos-Santos AJ, da Costa J, Zhang L, Pei Y, Scholey J, Ferrario CM, Manoukian AS, Chappell MC, Backx PH, Yagil Y, Penninger JM. Angiotensin-converting enzyme 2 is an essential regulator of heart function. Nature. 2002;417:822–828. doi: 10.1038/nature00786. [DOI] [PubMed] [Google Scholar]
- Oudit GY, Herzenberg AM, Kassiri Z, Wong D, Reich H, Khokha R, Crackower MA, Backx PH, Penninger JM, Scholey JW. Loss of angiotensin-converting enzyme 2 leads to the late development of angiotensin II-dependent glomerulosclerosis. Am J Pathol. 2006;168:1808–1820. doi: 10.2353/ajpath.2006.051091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kassiri Z, Oudit GY, Sanchez O, Dawood F, Mohammed FF, Nuttall RK, Edwards DR, Liu PP, Backx PH, Khokha R. Combination of tumor necrosis factor-alpha ablation and matrix metalloproteinase inhibition prevents heart failure after pressure overload in tissue inhibitor of metalloproteinase-3 knockout mice. Circ Res. 2005;97:380–390. doi: 10.1161/01.RES.0000178789.16929.cf. [DOI] [PubMed] [Google Scholar]
- Reich H, Tritchler D, Herzenberg AM, Kassiri Z, Zhou X, Gao W, Scholey JW. Albumin activates ERK via EGF receptor in human renal epithelial cells. J Am Soc Nephrol. 2005;16:1266–1278. doi: 10.1681/ASN.2004030222. [DOI] [PubMed] [Google Scholar]
- The Diabetes Control and Complications Trial Research Group The effect of intensive treatment of diabetes on the development and progression of long-term complications in insulin-dependent diabetes mellitus. N Engl J Med. 1993;329:977–986. doi: 10.1056/NEJM199309303291401. [DOI] [PubMed] [Google Scholar]
- UKPDS 10, UK Prospective Diabetes Study Group Urinary albumin excretion over 3 years in diet-treated type 2 (non-insulin-dependent) diabetic patients, and association with hypertension, hyperglycaemia, and hypertriglyceridaemia. Diabetologia. 1993;36:1021–1029. [PubMed] [Google Scholar]
- Mogensen CE, Christensen CK. Predicting diabetic nephropathy in insulin-dependent patients. N Engl J Med. 1984;311:89–93. doi: 10.1056/NEJM198407123110204. [DOI] [PubMed] [Google Scholar]
- Fujita H, Haseyama T, Kayo T, Nozaki J, Wada Y, Ito S, Koizumi A. Increased expression of glutathione S-transferase in renal proximal tubules in the early stages of diabetes: a study of type-2 diabetes in the Akita mouse model. Exp Nephrol. 2001;9:380–386. doi: 10.1159/000052636. [DOI] [PubMed] [Google Scholar]
- Qi Z, Fujita H, Jin J, Davis LS, Wang Y, Fogo AB, Breyer MD. Characterization of susceptibility of inbred mouse strains to diabetic nephropathy. Diabetes. 2005;54:2628–2637. doi: 10.2337/diabetes.54.9.2628. [DOI] [PubMed] [Google Scholar]
- Kakoki M, Takahashi N, Jennette JC, Smithies O. Diabetic nephropathy is markedly enhanced in mice lacking the bradykinin B2 receptor. Proc Natl Acad Sci USA. 2004;101:13302–13305. doi: 10.1073/pnas.0405449101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurley SB, Clare SE, Snow KP, Hu A, Meyer TW, Coffman TM. Impact of genetic background on nephropathy in diabetic mice. Am J Physiol. 2006;290:F214–F222. doi: 10.1152/ajprenal.00204.2005. [DOI] [PubMed] [Google Scholar]
- Ye M, Wysocki J, William J, Soler MJ, Cokic I, Batlle D. Glomerular localization and expression of angiotensin-converting enzyme 2 and angiotensin-converting enzyme: implications for albuminuria in diabetes. J Am Soc Nephrol. 2006;17:3067–3075. doi: 10.1681/ASN.2006050423. [DOI] [PubMed] [Google Scholar]
- Mauer SM, Steffes MW, Ellis EN, Sutherland DE, Brown DM, Goetz FC. Structural-functional relationships in diabetic nephropathy. J Clin Invest. 1984;74:1143–1155. doi: 10.1172/JCI111523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koya D, Jirousek MR, Lin YW, Ishii H, Kuboki K, King GL. Characterization of protein kinase C beta isoform activation on the gene expression of transforming growth factor-beta, extracellular matrix components, and prostanoids in the glomeruli of diabetic rats. J Clin Invest. 1997;100:115–126. doi: 10.1172/JCI119503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma K, Jin Y, Guo J, Ziyadeh FN. Neutralization of TGF-beta by anti-TGF-beta antibody attenuates kidney hypertrophy and the enhanced extracellular matrix gene expression in STZ-induced diabetic mice. Diabetes. 1996;45:522–530. doi: 10.2337/diab.45.4.522. [DOI] [PubMed] [Google Scholar]
- Johnson RJ, Alpers CE, Yoshimura A, Lombardi D, Prtizl P, Floege J, Schwartz SM. Renal injury from angiotensin II-mediated hypertension. Hypertension. 1992;19:464–474. doi: 10.1161/01.hyp.19.5.464. [DOI] [PubMed] [Google Scholar]
- UKPDS 33, UK Prospective Diabetes Study Group Intensive blood-glucose control with sulphonylureas or insulin compared with conventional treatment and risk of complications in patients with type 2 diabetes. Lancet. 1998;352:837–853. [PubMed] [Google Scholar]
- Gurley SB, Allred A, Le TH, Griffiths R, Mao L, Philip N, Haystead TA, Donoghue M, Breitbart RE, Acton SL, Rockman HA, Coffman TM. Altered blood pressure responses and normal cardiac phenotype in ACE2-null mice. J Clin Invest. 2006;116:2218–2225. doi: 10.1172/JCI16980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsieh TJ, Fustier P, Zhang SL, Filep JG, Tang SS, Ingelfinger JR, Fantus IG, Hamet P, Chan JS. High glucose stimulates angiotensinogen gene expression and cell hypertrophy via activation of the hexosamine biosynthesis pathway in rat kidney proximal tubular cells. Endocrinology. 2003;144:4338–4349. doi: 10.1210/en.2003-0220. [DOI] [PubMed] [Google Scholar]
- Vidotti DB, Casarini DE, Cristovam PC, Leite CA, Schor N, Boim MA. High glucose concentration stimulates intracellular renin activity and angiotensin II generation in rat mesangial cells. Am J Physiol. 2004;286:F1039–F1045. doi: 10.1152/ajprenal.00371.2003. [DOI] [PubMed] [Google Scholar]
- Singh R, Singh AK, Alavi N, Leehey DJ. Mechanism of increased angiotensin II levels in glomerular mesangial cells cultured in high glucose. J Am Soc Nephrol. 2003;14:873–880. doi: 10.1097/01.asn.0000060804.40201.6e. [DOI] [PubMed] [Google Scholar]
- Zhang SL, Filep JG, Hohman TC, Tang SS, Ingelfinger JR, Chan JSD. Molecular mechanisms of glucose action on angiotensinogen gene expression in rat proximal tubular cells. Kidney Int. 1999;55:454–464. doi: 10.1046/j.1523-1755.1999.00271.x. [DOI] [PubMed] [Google Scholar]
- Li N, Zimpelmann J, Cheng K, Wilkins JA, Burns KD. The role of angiotensin converting enzyme 2 in the generation of angiotensin 1-7 by rat proximal tubules. Am J Physiol. 2005;288:F353–F362. doi: 10.1152/ajprenal.00144.2004. [DOI] [PubMed] [Google Scholar]
- Schalekamp MA, Danser AH. Angiotensin II production and distribution in the kidney—II. Model-based analysis of experimental data. Kidney Int. 2006;69:1553–1557. doi: 10.1038/sj.ki.5000305. [DOI] [PubMed] [Google Scholar]
- Schalekamp MA, Danser AH. Angiotensin II production and distribution in the kidney: I. A kinetic model. Kidney Int. 2006;69:1543–1552. doi: 10.1038/sj.ki.5000303. [DOI] [PubMed] [Google Scholar]
- Susztak K, Raff AC, Schiffer M, Böttinger EP. Glucose-induced reactive oxygen species cause apoptosis of podocytes and podocyte depletion at the onset of diabetic nephropathy. Diabetes. 2006;55:225–233. [PubMed] [Google Scholar]
- Liebau MC, Lang D, Bohm J, Endlich N, Bek MJ, Witherden I, Mathieson PW, Saleem MA, Pavenstadt H, Fischer KG. Functional expression of the renin-angiotensin system in human podocytes. Am J Physiol. 2006;290:F710–F719. doi: 10.1152/ajprenal.00475.2004. [DOI] [PubMed] [Google Scholar]
- Konoshita T, Wakahara S, Mizuno S, Motomura M, Aoyama C, Makino Y, Kawai Y, Kato N, Koni I, Miyamori I, Mabuchi H. Tissue gene expression of renin-angiotensin system in human type 2 diabetic nephropathy. Diabetes Care. 2006;29:848–852. doi: 10.2337/diacare.29.04.06.dc05-1873. [DOI] [PubMed] [Google Scholar]
- Lely AT, Hamming I, van Goor H, Navis GJ. Renal ACE2 expression in human kidney disease. J Pathol. 2004;204:587–593. doi: 10.1002/path.1670. [DOI] [PubMed] [Google Scholar]