

Abstract
Primary biliary cirrhosis (PBC) is a chronic autoimmune liver disease characterized by inflammation and destruction of intrahepatic biliary epithelial cells, ultimately leading to liver failure. The serological hallmark of PBC is the presence of high-titer antimitochondrial antibodies (AMA) against the inner lipoyl domain of E2 subunits of 2-oxo-acid dehydrogenase complexes, in particular the E2 component of the pyruvate dehydrogenase complex (PDC-E2). The initiating events triggering the autoimmune response are not yet identified but the hypothesis of molecular mimicry is a widely proposed mechanism for the development of autoimmunity in PBC. Several candidates, including bacteria and viruses have been suggested as causative agents, but also environmental factors, such as chemical xenobiotics, have been implicated in the pathogenesis of primary biliary cirrhosis. In this review, we will discuss our current knowledge of the immunoreactivity of xenobiotically modified PDC peptide antigens. In addition, we will provide a working hypothesis how xenobiotic modification of antigens might occur that ultimately leads to the breaking of self-tolerance and the induction of PBC.
Keywords: Autoantibodies, chemicals, environmental factors, immunopathology, tolerance
Introduction
Primary biliary cirrhosis (PBC) [1] is a progressive autoimmune liver disease with female predilection characterized by immune-mediated destruction of intrahepatic small bile ducts, leading to decreased bile secretion, fibrosis, and eventual liver failure. The serologic signature of PBC is the presence of antimitochondrial antibodies (AMA), the most highly specific autoantibody in human immunopathology. The targets of the AMA response are all members of the family of the 2-oxo-acid dehydrogenase complexes (2-OADC-E2) which include the E2 subunits of the pyruvate dehydrogenase complex (PDC-E2), the branched chain 2-oxo-acid dehydrogenase complex (BCOADC-E2), the 2-oxo-glutaric acid dehydrogenase complex (OGDC-E2), and the dihydrolipoamide dehydrogenase binding protein (E3BP) [2]. AMA can be found in approximately 95% of PBC sera samples [3] but AMA can also be detected in the bile, urine and saliva of PBC patients and AMA titers in these fluids correlate with the sera titer [4–6].
The target antigens for AMA are all localized within the inner mitochondrial matrix and catalyze the oxidative decarboxylation of keto acid substrates. The E2 enzymes have a common structure consisting of an N-terminal domain containing a single or multiple lipoyl groups. Previous studies have demonstrated that the dominant epitopes recognized by AMA are all located within the lipoyl domains of these target antigens [3, 7–9]. Interestingly, there are only five proteins in mammals that contain lipoic acid, and four of the five are autoantigens in PBC. Moreover, when the recombinant forms of the mitochondrial proteins are used diagnostically, a positive AMA is virtually diagnostic, or at least suggests that the person is at substantial risk of future development of PBC [10, 11].
Evidence suggests that PBC has a major genetic basis, with a higher relative risk than nearly any other autoimmune disease [12]. The relative risk of a family member of a first-degree relative of a PBC patient is 50–100-fold higher than the general population [13]. However, there is little, if any, association between PBC and MHC class I or class II alleles [14, 15]. Recently our group evaluated the concordance of PBC in a genetically defined population of twin sets [16]. We identified 16 pairs of twins within a 1400-family cohort including 8 sets each of monozygotic and dizygotic twins. In 5 of 8 sets (0.63 concordance) of the monozygotic twins, both individuals had PBC. Among the 8 pairs of the dizygotic twins, none were found to be concordant for PBC. Interestingly, the age at onset of disease was similar in 4 of 5 concordant sets of monozygotic pairs. However, while the concordance rate of PBC in identical twins is among the highest reported for any autoimmune disease, discordant pairs were also identified suggesting that factors other than genetic predisposition contribute to the development of PBC. Epidemiological studies suggest that environmental factors play a role in triggering or exacerbating PBC [17–19]. We conducted a large scale epidemiological study to evaluate risk factors and co-morbidities in PBC that involved a controlled interview-based study of 1032 patients and a similar number of controls matched for sex, age, race, and geographical location [20]. Data from this study indicate that having a first-degree relative with PBC, history of urinary tract infections, past smoking, or use of hormone replacement increased the risk of PBC. Frequent use of nail polish slightly increased the risk of having PBC. Another study examined the prevalence of PBC and PSC near superfund sites and reported significant clusters of PBC surrounding toxic sites [21].
Autoimmune disease, molecular mimicry and xenobiotics
Autoimmunity is a phenomenon characterized by an inappropriate immune response against self antigens and, if persistent, can result in tissue damage. Autoimmune diseases are controlled by genetic predisposition and environmental factors and both influence susceptibility by modulating the reactivity and functionality of the immune system. The mechanisms responsible for the development of autoimmune diseases remain enigmatic; however, significant hypotheses based on epidemiological and scientific facts have been suggested. One thesis is the concept of molecular mimicry, based on the similarity of pathogen- and host antigen-derived epitopes recognized by the immune system [22, 23]. This mechanism has been suggested to be associated with several systemic autoimmune diseases, including multiple sclerosis [24, 25], systemic lupus erythematosus (SLE) [26] and rheumatoid arthritis [27]. However, there is a need for more mechanistical evidence other than the identification of similar epitopes, either homologous peptide sequences cross-recognized by epitope-specific T lymphocytes or conformational epitopes recognized by antibodies. The paradox is that autoantigens are unable to elicit a primary immune response themselves but can be recognized as targets for effector T cells stimulated by a pathogenic cross-reactive epitope. To break self-tolerance to the autoantigen, the epitope mimic or mimeotope needs to induce activation and proliferation rather than anergy of autoreactive T cells; the autoantigen presented by the host cells of a certain tissue must be recognized by reactive epitope-specific T cells to cause autoimmune disease. Therefore, the pathogen derived epitope has to be sufficiently different from the host derived epitope to stimulate an autoimmune response.
Although bacteria and viruses are candidates for the induction of autoimmune disease by molecular mimicry and have been discussed elsewhere [28], there are also other environmental factors, xenobiotics or chemical compounds foreign to a living organism. Examples include drugs, pesticides or other organic molecules that have the potential to modify host proteins and render them more immunogenic. There have been a number of chemical xenobiotics associated with several human autoimmune diseases (Table 1). These include SLE, in which factors like UV light and drugs, such as estrogens and aromatic amines are implicated in the development of the disease. Another example is the connective tissue disease scleroderma which can develop after exposure to silica and drugs such as bleomycin and pentazocine [29, 30]. The association of xenobiotics with scleroderma is interesting in that there is an analogy with PBC due to the increased frequency in the coexistence of both autoimmune diseases [31]. Scleroderma-like disease can also be caused by exposure to organic solvents, in particular those that contain vinyl chloride, and epoxy resins [32]. The toxic oil syndrome, caused by ingestion of denaturated rapeseed oil in Spain is another well-known example of connective tissue autoimmune disease induced by chemical exposure. Other chemicals linked to autoimmunity include heavy metals such as mercury [33, 34], iodine [35], canavanine [36] and halothane [37, 38]. Halothane hepatitis is a xenobiotics-induced liver disease that occurs when susceptible individuals develop an immune response against trifluoroacetylated (TFA) protein antigens. Exposure to TFA-conjugated self proteins results in antibody responses against such TFA-self proteins. The lipoylated inner lipoyl domain of human PDC-E2, but not the unlipoylated form, is recognized by anti-TFA [39]. Xenobiotics have the potential to become metabolically activated to intermediates that form covalent adducts with host proteins.
Table 1.
Association of chemical xenobiotics with human autoimmune diseases
| Xenobiotic | Disease | Reference |
|---|---|---|
| Mercury | Immune complex formation, Glomerulonephritis | [33, 34] |
| Iodine | Autoimmune thyroiditis | [35] |
| Vinyl chloride | Scleroderma-like disease | [32] |
| Aromatic amines | Drug-related lupus | [60] |
| Hydrazines | SLE | [61] |
| Silica | Rheumatoid arthritis, SLE, scleroderma | [62] |
| Organic solvents | Systemic sclerosis | [63] |
| Halothane | Hepatitis | [37, 38] |
| Canavanine | SLE-like syndromes | [36] |
| Toxic oil (fatty acid anilids) | “Toxic oil syndrome” (scleroderma-like connective tissue disease) | [64] |
The case for xenobiotics in PBC
The most problematic issue in autoimmunity is the identification of etiologic events that result in immunopathology; these events are conceived only in the general terms of genetic and environmental factors. PBC illustrates this enigma because of its high degree of concordance in identical twins, clinical uniformity and the presence of a highly specific serologic marker, antimitochondrial antibodies (AMA), that are directed at the E2 subunits of 2-oxo-acid dehydrogenase complexes. In addition, the MHC class I and class II-restricted T cell epitopes (peptides 159 – 167 and 163 – 176) of liver-infiltrating, autoreactive T cells appear to localize to the same inner lipoyl domain of PDC-E2 [3, 40] as does the dominant autoreactive B cell epitope [41]. This signature of PBC, the AMA, is a useful probe that enables the dissection of potential environmental triggers and has led to the proposition that the inciting event of PBC is a modification of the major autoantigen, PDC-E2, by xenobiotics.
Many environmental chemicals are metabolized primarily in the liver. During metabolism they may form reactive metabolites, which may modify cellular proteins to form neo-antigens. Although it is not clear how xenobiotics or the modified cellular proteins initiate autoimmunity in PBC, several mechanisms can be proposed. Firstly, the direct toxic effect of the xenobiotics or the neo-antigens my cause abnormal cell death by apoptosis or necrosis, conditions favoring generation of immunogenic autoepitopes. Secondly, the neo-antigen-specific T cells and B cells, once primed, may cross-react with the less immunogenic native autoantigen. Lastly, chemical modification of the native cellular protein such as removal and/or exchange of a hapten on the native peptide have been shown to change processing in APC and lead to the presentation of cryptic peptides [42]. The direct toxic effect of xenobiotics is usually dose-dependent and may manifest in the majority of individuals shortly after drug intake, resulting in conditions such as drug-induced liver injury [38, 43], hence they are relatively easily identified. One such example is halothane hepatitis where a trifluoroacetyl (TFA) metabolite covalently modifies lysine residues of cellular proteins, inducing an antibody response not only against the TFA-modified proteins but also against the lipoyl domain of human PDC-E2, the major autoantigen in PBC [39], implicating not only drug-induced protein modification but also the concept of molecular mimicry as pathogenic mechanisms associated with PBC. On the other hand, the autoimmune effects that follow such drug toxicity may take a prolonged time-period to reach a threshold level that is accompanied by significant toxicity to be clinically manifested [44]. During this time period the autoimmune reactivity may undergo substantial evolution both quantitatively and qualitatively, making the identification of the causative agents a formidable task. Despite this difficulty, progress has been made by using the reactivity of chemically modified PDC-E2 peptides with PBC patient sera as the “footprints” of the original neo-antigens to identify the environmental chemicals responsible for the generation of these immunogenic neo-antigens.
In-vitro evidence for the role of xenobiotics in PBC
Our group has a long-standing interest in defining and dissecting the etiological basis of PBC. Although we have identified the major autoantigens of PBC, the events initiating the recognition of these self-proteins as immunogenic are still under investigation. One of our working hypotheses is that potential modifications of self-proteins by agents such as xenobiotics may alter these self-proteins to cause a breakdown of tolerance perpetuating the immune response by a chronic low-level turnover of the self-protein. To address this hypothesis we initially synthesized a 12-mer peptide containing the inner lipoyl domain of PDC-E2 (amino acids 173 to 184) and replaced the lipoic acid moiety with a panel of 18 synthetic structures designed to mimic a xenobiotically modified lipoyl hapten [45]. We then quantitated the reactivity of sera from PBC patients to these structures by utilizing a microbead ELISA assay. Interestingly, antimitochondrial antibodies (AMAs) from all seropositive patients with PBC, but not controls, reacted against three of these 18 organic modified autoepitopes significantly better than to the native domain. Structural analysis of reactive compounds revealed that features correlating with autoantibody binding include compounds with a halide or methyl halide in the meta- or para-position containing no strong hydrogen bond accepting groups on the phenyl ring. Many chemicals including pharmaceuticals and household detergents have the potential to form such halogenated derivatives as metabolites. Another interesting finding of this study was that modification of the lipoic acid moiety with a trifluoroacetyl (TFA) group resulted in a mimic with higher AMA reactivity than the native ligand lipoic acid, demonstrating a possible mechanism of lipoyl group modification by TFA-containing reagents such as halothane.
Based on these findings and a quantitative structure-activity relationship (QSAR) analysis of the AMA reactive compounds, we initiated a more comprehensive study using a 15-mer (aa 170-184) inner lipoyl domain PDC-E2 peptide to analyze an extended list of chemical xenobiotics for AMA recognition [46]. We used a refined screening strategy employing a microarray platform where xenobiotic-modified peptide-agarose conjugates are spotted onto glass slides allowing the screening of theoretically thousands of compounds with very small amount of patient sera and reagents (Fig. 1). This list included both aliphatic and aromatic compounds: aliphatic compounds with different chain lengths, halogen substitutions, various degrees of saturation, and aromatic compounds with both halogen and nitro substitutions and heterocyclic compounds (see Table 1 in [46]). Briefly, a total of 107 potential xenobiotic mimics were coupled to the lysine residue of the immunodominant 15 amino acid peptide of the PDC-E2 inner lipoyl domain and spotted onto glass slides. Sera from PBC (n = 47), primary sclerosing cholangitis (n = 15) and healthy volunteers (n = 20) were assayed for Ig reactivity. Of the 107 xenobiotics, 33 (or 64) had significantly higher IgG (or IgM, respectively) reactivity against PBC sera compared to controls. In addition, 9 of 33 (8 out of 64 for IgM) compounds were more reactive than the native lipoylated peptide (Table 2) [46]. Among those compounds were cinnamic acid derivatives carrying hydrophobic halogen or trifluoromethyl substitutions and medium to long chain saturated and unsaturated aliphatic acids. These results let us to hypothesize that the acid moiety attached to the lysine residue of the immunodominant PDC-E2 peptide must preferably be of hydrophobic character and of a certain size to be successfully recognized by the Ag-binding site of AMA. Following extensive ELISA and absorption studies we also concluded that reactivity to 2-octynoic acid was very specific since antibodies recognizing 2-octynoic acid modified PDC peptide did not cross-react with lipoic acid modified peptide; this data allowed further refinement of the optimal requirements of the xenobiotic.
Fig. 1.
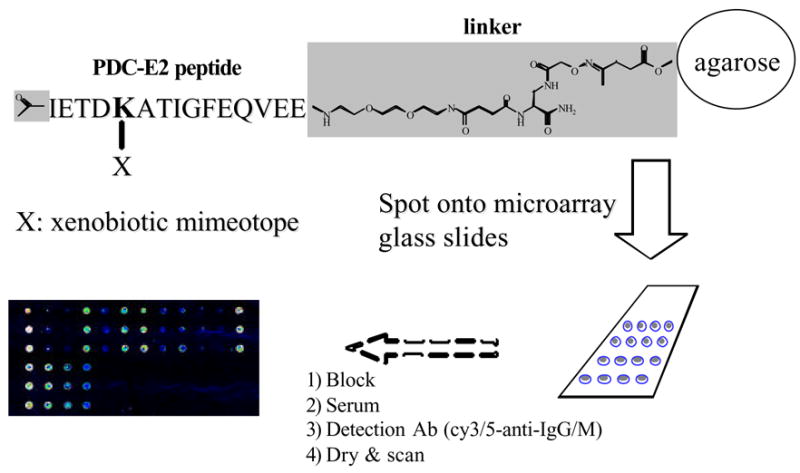
Schematic of microarray based assay utilizing xenobiotic modified peptide-agarose conjugates.
Table 2.
Sera IgG and IgM reactivity of patients with PBC and normal controls against lipoylated PDC-E2 peptide and xenobiotically modified PDC-E2 peptide by microarray.
| Structure | Name | IgG† |
IgM† |
||
|---|---|---|---|---|---|
| PBC | NC | PBC | NC | ||
|
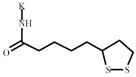
Lipoic acid | 718991* | 378448 | 1030672 ** | 370329 |
|
| |||||
|
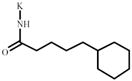
Cyclohexanepentanoic acid | 1339822*** | 404633 | 1657028 *** | 344074 |
|
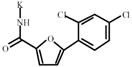
5-(2,4-Dichlorophenyl)-2- furoic acid | 1328219*** | 496957 | 1660965 *** | 473981 |
|
|
11-Bromoundecanoic acid | 1201246*** | 369687 | 1515634 *** | 281940 |
|
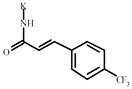
2-(Trifluoromethyl)cinnamic acid | 1004220** | 398648 | 1159470 *** | 311826 |
|
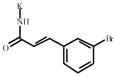
3-Bromocinnamic acid | 846207** | 321628 | 1089700 *** | 281256 |
|
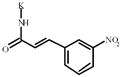
3-Nitrocinnamic acid | 846198** | 373955 | 1027486 *** | 338052 |
|
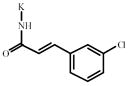
3-Chlorocinnamic acid | 809112** | 405232 | 1037639 *** | 320204 |
|
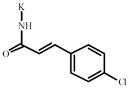
4-Chlorocinnamic acid | 758836** | 345287 | 893271* | 290224 |
|
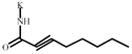
2-Octynoic acid | 757314** | 369752 | 884042** | 354000 |
|
| |||||
|
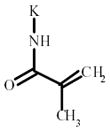
Methacrylic acid | 216631 | 227983 | 201588 | 189025 |
|
| |||||
| PDC peptide only | 253417 | 305013 | 212115 | 219369 | |
p<0.05,
p<0.01,
p<0.001(Student t-test).
Data presented here are mean intensity.
To further define the role of alkynoic acids we then characterized the reactivity of sera with chemical compounds that are structurally similar to 2-octynoic acid but different either in the location of the acetylene bond or in the length of the carbon chain. 2-Octynoic acid-related xenobiotics with varying triple bond location (4-, 5-, 6- or 7-octynoic acid, respectively) or varying chain length (2-butynoic, 2-pentynoic, 2-hexynoic, 2-heptynoic, 2-nonynoin or 2-decynoic acid, respectively) and controls were coupled to the lysine residue of the 15-mer PDC inner lipoyl domain peptide immobilized on agarose beads, spotted on microarray slides and probed with sera from 21 AMA-positive patients and 13 healthy subjects for IgG and IgM reactivity. Acids with the triple bond located at a location other than C-2 (4-, 5-, 6-, or 7-octynoic acid) have significantly lower IgG and IgM reactivity compared to 2-octynoic acid (Fig. 2 A, B). Thence, to define the role of the carbon chain length of 2-alkynamides, we synthesized a panel of 2-alkynamide conjugates of PDC-E2 peptide that varied in their chain length from 4 to 14 carbon atoms. The same panel of sera was used to screen the Ig reactivity of the modified PDC-E2 peptide. There was a significantly higher (p < 0.05) IgG reactivity with PBC sera than with controls for 2-hexyn-, 2-heptyn-, 2-octyn-, 2-nonyn-, 2-decyn-, 2-dodecyn- and 2-tetradecynamides. Significantly greater (p < 0.05) IgM reactivity was seen with all the tested compounds. The highest Ig reactivity with PBC sera was observed for 2-octyn-, 2-nonyn- and 2-decynamides; the 2-nonynamide conjugate reacted significantly better (p < 0.05 for IgG, p < 0.01 for IgM) than the other amides (Fig. 2 C, D). QSAR analysis suggests that the position of the triple bond at position C-2 and a minimum chain length of seven carbon atoms is critical for high reactivity. Among 2-alkynamides with varying carbon chain length, 2-octyn-, 2-nonyn- (particularly) and 2-decynamide exhibited the highest reactivity. Altogether, our data suggests that AMA react specifically with the 2-alkynamide conjugates and emphasizes the important role for reactivity of the conjugated position of the triple bond in alkynoic acids. This reactivity is furthermore mediated by PDC-E2-specific antibodies, since absorption of PBC sera with increasing amounts of recombinant PDC-E2 significantly reduced Ig reactivity in a dose-dependent manner. In summary, our data indicate that alkynamides with a triple bond in the conjugated C-2 position and a chain length of 9 carbon atoms are most favorable for AMA binding. The optimal structure was demonstrated to be derived from 2-nonynoic acid, whose methyl ester is ranked 2324th out of 12945 in terms of occupational exposure with an 80% female prevalence due to its use in cosmetics [47].
Fig. 2.
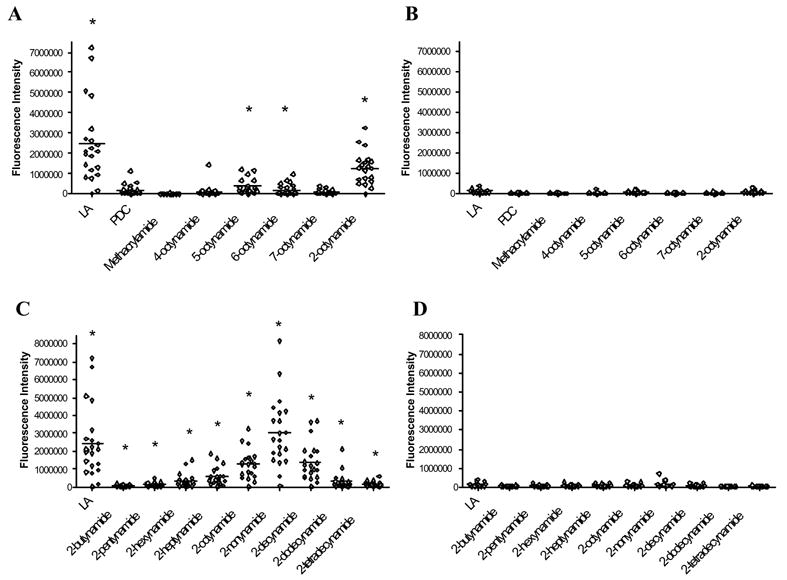
IgM reactivity of n-octynamide- or 2-alkynanide-modified PDC-E2 peptide with PBC sera. PBC patient sera (A, C) and healthy control sera (B, D) were analyzed by microarray for IgM reactivity against PDC-E2 peptide that has been modified with n-octynoic (A, B) or 2-alkynoic acids (C, D). Reactivity of individual samples and mean reactivity are shown. Asterisk indicates a significant difference (p < 0.05, unpaired t test) in IgM reactivity between PBC and control sera.
Conclusion: A model of what causes PBC
The data derived from our studies have led to the assembly of a series of facts that serve as a foundation upon which we have formulated a working model for the pathogenesis of human PBC. These facts include:
A genetic predisposition to develop PBC.
The incredible focused response of autoantibody, CD4+ and CD8+ T cell responses
The higher autoreactive PDC-E2-specific CD4+ T cell precursor frequency in liver and regional lymph node compared to blood in PBC.
The higher autoreactive PDC-E2-specific CD8+ T cell precursor frequency in liver compared to blood in PBC. The number of PDC-E2 specific tetramer positive CD8+ cells is highest in stage I/II.
The finding that a chemically synthesized lipoate mimic in the form of 2-nonynoic acid reacts with a higher affinity than the parent lipoate in PBC.
The ability of our xenobiotics coupled to BSA to break tolerance in experimental animals
Small bile ducts, are not innocent victims; their biology determines their targeting.
In this context, we should also discuss that with the use of recombinant antigens and in our studies of many thousands of PBC sera, there are no false positives. While a small percentage of PBC patients do not have AMA seropositivity, we have “chipped” away at that total with Luminex assays; it is now less than 4% [48]. In the overlap syndrome we believe it is either de novo PBC in a patient with AIH or a dual disease. The frequency of autoreactive PDC-E2 specific CD4+ T cells is approximately the same in AMA negative as AMA positive PBC patients (S. Shimoda, personal communication). PBC reoccurs following liver transplantation [49–51] and there is no convincing evidence that the multi-lineage anti-mitochondrial response changes significantly. Biliary cells also play a role in our model. We do not believe there are any PBC specific biliary cell phenotypes since PBC can reoccur after transplant. Rather, PBC is localized to bile duct cells (and occasionally salivary cells) on the basis of the unique apoptotic properties, the presence of poly Ig and the intense mucosal responses in bile duct epithelial cells [4, 52–54]. The susceptibility genes in PBC remain undefined and will likely be multigenic. Herein we have chosen to focus on the environmental arm of our hypothesis. Thus we postulate that the breaking of tolerance to PDC-E2 takes place as outlined in Fig. 3.
Fig. 3.
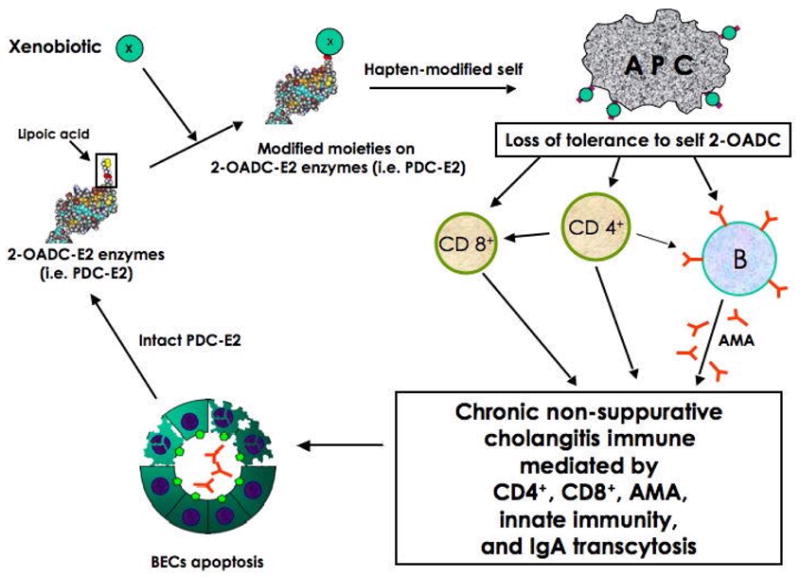
Proposed model of breaking of tolerance in PBC.
Our model does not require the presence of each and every one of these events. However, the common denominator will be an immune response to the lipoyl containing mitochondrial antigens. Our mouse models [55–57], we predict, will develop PBC by a similar mechanism, namely a multi-lineage loss of tolerance to PDC-E2 in a promiscuous “facilitator” host [58, 59]. For decades, and because there is no correlation of AMA titer and disease severity, the AMA was often considered an epiphenomena. Our work over the past two decades argues otherwise and the role of the mitochondrial response must be viewed in the broad context of this multi-lineage response. Our model only requires the initial loss of tolerance and while the initial event and environmental triggers may vary between individuals, the final immune pathways and the immunopathology will be the same.
As previously stated, our proposition is that the inciting event of PBC is a modification of the major autoantigen, PDC-E2, by xenobiotic conjugation. Six distinct “modifications” involving xenobiotic conjugation can be envisioned: (i) xenobiotic modification of the lipoyl moiety (i.e., PDC-E2—LA*); (ii) modification wherein a xenobiotic replaces the lipoic acid (i.e., PDC-E2—X*); (iii) xenobiotic modification of PDC-E2 proximal to the lipoic acid (i.e., PDC-E2*—LA); (iv) concomitant xenobiotic modification of PDC-E2 and the lipoic acid (i.e., PDC-E2*—LA*); (v) concomitant xenobiotic modification of PDC-E2 and replacement of the lipoic acid (i.e., PDC-E2*—X*) (Fig. 4 A); and (vi) carrier protein (CP) modification by a xenobiotic (i.e., CP—X*) (Fig. 4B). The studies detailed herein build on our detailed QSAR work, which has led to the identification of several chemical mimics – i.e., peptide—small molecule conjugates – that react with PBC sera as well as or better than the native autoantigen, and are designed to differentiate these various scenarios. Hence, we believe that the cause of PBC is autoimmunity, reactivity to a self-protein, with subsequent pathology on susceptible biliary epithelial cells.
Fig. 4.
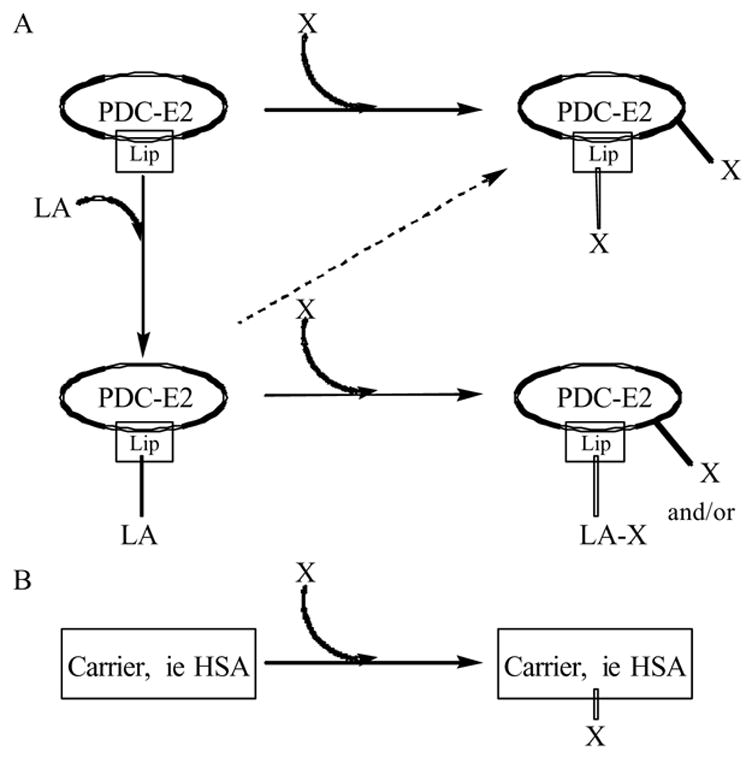
Schematic of possible xenobiotic modifications of PDC-E2 or carrier proteins. Xenobiotics may alter PDC-E2 by either replacing the lipoic acid moiety or by chemically modifying lipoic acid itself and/or modifying PDC-E2 proximal to the lipoyl domain creating an immunological mimic (A). In addition, xenobiotics might be able to modify carrier proteins, such as albumin, which elicits an immune response against the xenobiotic cross-reacting against native PDC-E2 (B). LA, lipoic acid; X, xenobiotic; Lip, lipoyl domain.
Acknowledgments
Supported in part from National Institutes of Health grant DK067003.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Kaplan MM, Gershwin ME. Primary biliary cirrhosis. N Engl J Med. 2005;353:1261–73. doi: 10.1056/NEJMra043898. [DOI] [PubMed] [Google Scholar]
- 2.Dubel L, Tanaka A, Leung PS, Van de Water J, Coppel R, Roche T, Johanet C, Motokawa Y, Ansari A, Gershwin ME. Autoepitope mapping and reactivity of autoantibodies to the dihydrolipoamide dehydrogenase-binding protein (E3BP) and the glycine cleavage proteins in primary biliary cirrhosis. Hepatology. 1999;29:1013–8. doi: 10.1002/hep.510290403. [DOI] [PubMed] [Google Scholar]
- 3.Van de Water J, Gershwin ME, Leung P, Ansari A, Coppel RL. The autoepitope of the 74-kD mitochondrial autoantigen of primary biliary cirrhosis corresponds to the functional site of dihydrolipoamide acetyltransferase. J Exp Med. 1988;167:1791–9. doi: 10.1084/jem.167.6.1791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nishio A, Van de Water J, Leung PS, Joplin R, Neuberger JM, Lake J, Bjorkland A, Totterman TH, Peters M, Worman HJ, Ansari AA, Coppel RL, Gershwin ME. Comparative studies of antimitochondrial autoantibodies in sera and bile in primary biliary cirrhosis. Hepatology. 1997;25:1085–9. doi: 10.1002/hep.510250506. [DOI] [PubMed] [Google Scholar]
- 5.Reynoso-Paz S, Leung PS, Van De Water J, Tanaka A, Munoz S, Bass N, Lindor K, Donald PJ, Coppel RL, Ansari AA, Gershwin ME. Evidence for a locally driven mucosal response and the presence of mitochondrial antigens in saliva in primary biliary cirrhosis. Hepatology. 2000;31:24–9. doi: 10.1002/hep.510310106. [DOI] [PubMed] [Google Scholar]
- 6.Tanaka A, Nalbandian G, Leung PS, Benson GD, Munoz S, Findor JA, Branch AD, Coppel RL, Ansari AA, Gershwin ME. Mucosal immunity and primary biliary cirrhosis: presence of antimitochondrial antibodies in urine. Hepatology. 2000;32:910–5. doi: 10.1053/jhep.2000.19254. [DOI] [PubMed] [Google Scholar]
- 7.Leung PS, Chuang DT, Wynn RM, Cha S, Danner DJ, Ansari A, Coppel RL, Gershwin ME. Autoantibodies to BCOADC-E2 in patients with primary biliary cirrhosis recognize a conformational epitope. Hepatology. 1995;22:505–13. [PubMed] [Google Scholar]
- 8.Moteki S, Leung PS, Dickson ER, Van Thiel DH, Galperin C, Buch T, Alarcon-Segovia D, Kershenobich D, Kawano K, Coppel RL, et al. Epitope mapping and reactivity of autoantibodies to the E2 component of 2-oxoglutarate dehydrogenase complex in primary biliary cirrhosis using recombinant 2-oxoglutarate dehydrogenase complex. Hepatology. 1996;23:436–44. doi: 10.1002/hep.510230307. [DOI] [PubMed] [Google Scholar]
- 9.Surh CD, Coppel R, Gershwin ME. Structural requirement for autoreactivity on human pyruvate dehydrogenase-E2, the major autoantigen of primary biliary cirrhosis. Implication for a conformational autoepitope. J Immunol. 1990;144:3367–74. [PubMed] [Google Scholar]
- 10.Mattalia A, Quaranta S, Leung PS, Bauducci M, Van de Water J, Calvo PL, Danielle F, Rizzetto M, Ansari A, Coppel RL, Rosina F, Gershwin ME. Characterization of antimitochondrial antibodies in health adults. Hepatology. 1998;27:656–61. doi: 10.1002/hep.510270303. [DOI] [PubMed] [Google Scholar]
- 11.Metcalf JV, Mitchison HC, Palmer JM, Jones DE, Bassendine MF, James OF. Natural history of early primary biliary cirrhosis. Lancet. 1996;348:1399–402. doi: 10.1016/S0140-6736(96)04410-8. [DOI] [PubMed] [Google Scholar]
- 12.Tsuji K, Watanabe Y, Van De Water J, Nakanishi T, Kajiyama G, Parikh-Patel A, Coppel R, Gershwin ME. Familial primary biliary cirrhosis in Hiroshima. J Autoimmun. 1999;13:171–8. doi: 10.1006/jaut.1999.0299. [DOI] [PubMed] [Google Scholar]
- 13.Parikh-Patel A, Gold E, Mackay IR, Gershwin ME. The geoepidemiology of primary biliary cirrhosis: contrasts and comparisons with the spectrum of autoimmune diseases. Clin Immunol. 1999;91:206–18. doi: 10.1006/clim.1999.4690. [DOI] [PubMed] [Google Scholar]
- 14.Invernizzi P, Battezzati PM, Crosignani A, Perego F, Poli F, Morabito A, De Arias AE, Scalamogna M, Zuin M, Podda M. Peculiar HLA polymorphisms in Italian patients with primary biliary cirrhosis. J Hepatol. 2003;38:401–6. doi: 10.1016/s0168-8278(02)00440-3. [DOI] [PubMed] [Google Scholar]
- 15.Donaldson PT, Baragiotta A, Heneghan MA, Floreani A, Venturi C, Underhill JA, Jones DE, James OF, Bassendine MF. HLA class II alleles, genotypes, haplotypes, and amino acids in primary biliary cirrhosis: a large-scale study. Hepatology. 2006;44:667–74. doi: 10.1002/hep.21316. [DOI] [PubMed] [Google Scholar]
- 16.Selmi C, Mayo MJ, Bach N, Ishibashi H, Invernizzi P, Gish RG, Gordon SC, Wright HI, Zweiban B, Podda M, Gershwin ME. Primary biliary cirrhosis in monozygotic and dizygotic twins: genetics, epigenetics, and environment. Gastroenterology. 2004;127:485–92. doi: 10.1053/j.gastro.2004.05.005. [DOI] [PubMed] [Google Scholar]
- 17.Triger DR. Primary biliary cirrhosis: an epidemiological study. Br Med J. 1980;281:772–5. doi: 10.1136/bmj.281.6243.772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Uibo R, Salupere V. The epidemiology of primary biliary cirrhosis: immunological problems. Hepatogastroenterology. 1999;46:3048–52. [PubMed] [Google Scholar]
- 19.Watson RG, Angus PW, Dewar M, Goss B, Sewell RB, Smallwood RA. Low prevalence of primary biliary cirrhosis in Victoria, Australia. Melbourne Liver Group. Gut. 1995;36:927–30. doi: 10.1136/gut.36.6.927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gershwin ME, Selmi C, Worman HJ, Gold EB, Watnik M, Utts J, Lindor KD, Kaplan MM, Vierling JM. Risk factors and comorbidities in primary biliary cirrhosis: a controlled interview-based study of 1032 patients. Hepatology. 2005;42:1194–202. doi: 10.1002/hep.20907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ala A, Stanca CM, Bu-Ghanim M, Ahmado I, Branch AD, Schiano TD, Odin JA, Bach N. Increased prevalence of primary biliary cirrhosis near Superfund toxic waste sites. Hepatology. 2006;43:525–31. doi: 10.1002/hep.21076. [DOI] [PubMed] [Google Scholar]
- 22.Oldstone MB. Molecular mimicry and autoimmune disease. Cell. 1987;50:819–20. doi: 10.1016/0092-8674(87)90507-1. [DOI] [PubMed] [Google Scholar]
- 23.von Herrath MG, Oldstone MB. Virus-induced autoimmune disease. Curr Opin Immunol. 1996;8:878–85. doi: 10.1016/S0952-7915(96)80019-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Talbot PJ, Paquette JS, Ciurli C, Antel JP, Ouellet F. Myelin basic protein and human coronavirus 229E cross-reactive T cells in multiple sclerosis. Ann Neurol. 1996;39:233–40. doi: 10.1002/ana.410390213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wucherpfennig KW, Strominger JL. Molecular mimicry in T cell-mediated autoimmunity: viral peptides activate human T cell clones specific for myelin basic protein. Cell. 1995;80:695–705. doi: 10.1016/0092-8674(95)90348-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Poole BD, Scofield RH, Harley JB, James JA. Epstein-Barr virus and molecular mimicry in systemic lupus erythematosus. Autoimmunity. 2006;39:63–70. doi: 10.1080/08916930500484849. [DOI] [PubMed] [Google Scholar]
- 27.van Eden W, Hogervorst EJ, Hensen EJ, van der Zee R, van Embden JD, Cohen IR. A cartilage-mimicking T-cell epitope on a 65K mycobacterial heat-shock protein: adjuvant arthritis as a model for human rheumatoid arthritis. Curr Top Microbiol Immunol. 1989;145:27–43. doi: 10.1007/978-3-642-74594-2_3. [DOI] [PubMed] [Google Scholar]
- 28.Bach JF. The effect of infections on susceptibility to autoimmune and allergic diseases. N Engl J Med. 2002;347:911–20. doi: 10.1056/NEJMra020100. [DOI] [PubMed] [Google Scholar]
- 29.Shanklin DR, Smalley DL. The immunopathology of siliconosis. History, clinical presentation, and relation to silicosis and the chemistry of silicon and silicone. Immunol Res. 1998;18:125–73. doi: 10.1007/BF02788777. [DOI] [PubMed] [Google Scholar]
- 30.Yoshida SH, Swan S, Teuber SS, Gershwin ME. Silicone breast implants: immunotoxic and epidemiologic issues. Life Sci. 1995;56:1299–310. doi: 10.1016/0024-3205(95)00081-x. [DOI] [PubMed] [Google Scholar]
- 31.Gershwin ME, Ansari AA, Mackay IR, Nakanuma Y, Nishio A, Rowley MJ, Coppel RL. Primary biliary cirrhosis: an orchestrated immune response against epithelial cells. Immunol Rev. 2000;174:210–25. doi: 10.1034/j.1600-0528.2002.017402.x. [DOI] [PubMed] [Google Scholar]
- 32.D'Cruz D. Autoimmune diseases associated with drugs, chemicals and environmental factors. Toxicol Lett. 2000;112–113:421–32. doi: 10.1016/s0378-4274(99)00220-9. [DOI] [PubMed] [Google Scholar]
- 33.Havarinasab S, Hultman P. Organic mercury compounds and autoimmunity. Autoimmun Rev. 2005;4:270–5. doi: 10.1016/j.autrev.2004.12.001. [DOI] [PubMed] [Google Scholar]
- 34.Rowley B, Monestier M. Mechanisms of heavy metal-induced autoimmunity. Mol Immunol. 2005;42:833–8. doi: 10.1016/j.molimm.2004.07.050. [DOI] [PubMed] [Google Scholar]
- 35.Rose NR, Bonita R, Burek CL. Iodine: an environmental trigger of thyroiditis. Autoimmun Rev. 2002;1:97–103. doi: 10.1016/s1568-9972(01)00016-7. [DOI] [PubMed] [Google Scholar]
- 36.Montanaro A, Bardana EJ., Jr Dietary amino acid-induced systemic lupus erythematosus. Rheum Dis Clin North Am. 1991;17:323–32. [PubMed] [Google Scholar]
- 37.Gut J. Molecular basis of halothane hepatitis. Arch Toxicol Suppl. 1998;20:3–17. doi: 10.1007/978-3-642-46856-8_1. [DOI] [PubMed] [Google Scholar]
- 38.Liu ZX, Kaplowitz N. Immune-mediated drug-induced liver disease. Clin Liver Dis. 2002;6:755–74. doi: 10.1016/s1089-3261(02)00025-9. [DOI] [PubMed] [Google Scholar]
- 39.Christen U, Quinn J, Yeaman SJ, Kenna JG, Clarke JB, Gandolfi AJ, Gut J. Identification of the dihydrolipoamide acetyltransferase subunit of the human pyruvate dehydrogenase complex as an autoantigen in halothane hepatitis. Molecular mimicry of trifluoroacetyl-lysine by lipoic acid. Eur J Biochem. 1994;223:1035–47. doi: 10.1111/j.1432-1033.1994.tb19082.x. [DOI] [PubMed] [Google Scholar]
- 40.Kita H, Matsumura S, He XS, Ansari AA, Lian ZX, Van De Water J, Coppel RL, Kaplan MM, Gershwin ME. Quantitative and functional analysis of PDC-E2-specific autoreactive cytotoxic T lymphocytes in primary biliary cirrhosis. J Clin Invest. 2002;109:1231–1240. doi: 10.1172/JCI14698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shimoda S, Van de Water J, Ansari A, Nakamura M, Ishibashi H, Coppel RL, Lake J, Keeffe EB, Roche TE, Gershwin ME. Identification and precursor frequency analysis of a common T cell epitope motif in mitochondrial autoantigens in primary biliary cirrhosis. J Clin Invest. 1998;102:1831–40. doi: 10.1172/JCI4213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Griem P, Wulferink M, Sachs B, Gonzalez JB, Gleichmann E. Allergic and autoimmune reactions to xenobiotics: how do they arise? Immunol Today. 1998;19:133–41. doi: 10.1016/s0167-5699(97)01219-x. [DOI] [PubMed] [Google Scholar]
- 43.Gluud C. Acute, serious drug-induced liver injury. J Hepatol. 2002;37:675–7. doi: 10.1016/s0168-8278(02)00312-4. [DOI] [PubMed] [Google Scholar]
- 44.Ju C. Immunological mechanisms of drug-induced liver injury. Curr Opin Drug Discov Devel. 2005;8:38–43. [PubMed] [Google Scholar]
- 45.Long SA, Quan C, Van de Water J, Nantz MH, Kurth MJ, Barsky D, Colvin ME, Lam KS, Coppel RL, Ansari A, Gershwin ME. Immunoreactivity of organic mimeotopes of the E2 component of pyruvate dehydrogenase: connecting xenobiotics with primary biliary cirrhosis. J Immunol. 2001;167:2956–63. doi: 10.4049/jimmunol.167.5.2956. [DOI] [PubMed] [Google Scholar]
- 46.Amano K, Leung PS, Rieger R, Quan C, Wang X, Marik J, Suen YF, Kurth MJ, Nantz MH, Ansari AA, Lam KS, Zeniya M, Matsuura E, Coppel RL, Gershwin ME. Chemical xenobiotics and mitochondrial autoantigens in primary biliary cirrhosis: identification of antibodies against a common environmental, cosmetic, and food additive, 2-octynoic acid. J Immunol. 2005;174:5874–83. doi: 10.4049/jimmunol.174.9.5874. [DOI] [PubMed] [Google Scholar]
- 47.NIOSH. National Occupational Exposure Survey 1981–1983 1988–1990; Pub. Nos. 88-106, 89-103. Health N.I.f.O.S.a., Cincinnati, OH.
- 48.Oertelt S, Rieger R, Selmi C, Invernizzi P, Ansari AA, Coppel RL, Podda M, Gershwin ME. A highly sensitive bead assay for antimitochondrial antibodies: chipping away at AMA negative PBC. Hepatology. 2006 doi: 10.1002/hep.21583. In press. [DOI] [PubMed] [Google Scholar]
- 49.Kotlyar DS, Campbell MS, Reddy KR. Recurrence of diseases following orthotopic liver transplantation. Am J Gastroenterol. 2006;101:1370–8. doi: 10.1111/j.1572-0241.2006.00586.x. [DOI] [PubMed] [Google Scholar]
- 50.Mattalia A, Luttig B, Rosina F, Leung PS, Van de Water J, Bauducci M, Ciancio A, Boker KH, Worman H, Cooper RL, Manns M, Ansari A, Rizzetto M, Gershwin ME. Persistence of autoantibodies against recombinant mitochondrial and nuclear pore proteins after orthotopic liver transplantation for primary biliary cirrhosis. J Autoimmun. 1997;10:491–7. doi: 10.1006/jaut.1997.0156. [DOI] [PubMed] [Google Scholar]
- 51.Van de Water J, Gerson LB, Ferrell LD, Lake JR, Coppel RL, Batts KP, Wiesner RH, Gershwin ME. Immunohistochemical evidence of disease recurrence after liver transplantation for primary biliary cirrhosis. Hepatology. 1996;24:1079–84. doi: 10.1002/hep.510240517. [DOI] [PubMed] [Google Scholar]
- 52.Malmborg AC, Shultz DB, Luton F, Mostov KE, Richly E, Leung PS, Benson GD, Ansari AA, Coppel RL, Gershwin ME, Van de Water J. Penetration and co-localization in MDCK cell mitochondria of IgA derived from patients with primary biliary cirrhosis. J Autoimmun. 1998;11:573–80. doi: 10.1006/jaut.1998.0220. [DOI] [PubMed] [Google Scholar]
- 53.Surh CD, Roche TE, Danner DJ, Ansari A, Coppel RL, Prindiville T, Dickson ER, Gershwin ME. Antimitochondrial autoantibodies in primary biliary cirrhosis recognize cross-reactive epitope(s) on protein X and dihydrolipoamide acetyltransferase of pyruvate dehydrogenase complex. Hepatology. 1989;10:127–33. doi: 10.1002/hep.1840100202. [DOI] [PubMed] [Google Scholar]
- 54.Worman HJ, Courvalin JC. Antinuclear antibodies specific for primary biliary cirrhosis. Autoimmun Rev. 2003;2:211–7. doi: 10.1016/s1568-9972(03)00013-2. [DOI] [PubMed] [Google Scholar]
- 55.Irie J, Wu Y, Wicker L, Rainbow DB, Nalesnik M, Hirsch R, Peterson L, Leung P, Cheng CM, Mackay IR, Gershwin M, Ridgway W. NOD.c3c4 congenic mice develop autoimmune biliary disease that serologically and pathogenetically models human primary biliary cirrhosis. J Exp Med. 2006 doi: 10.1084/jem.20051911. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Oertelt S, Lian ZX, Cheng CM, Chuang YH, Padgett KA, He XS, Ridgway WM, Ansari AA, Coppel RL, Li MO, Flavell RA, Kronenberg M, Mackay IR, Gershwin ME. Anti-mitochondrial antibodies and primary biliary cirrhosis in TGF-beta receptor II dominant-negative mice. J Immunol. 2006;177:1655–60. doi: 10.4049/jimmunol.177.3.1655. [DOI] [PubMed] [Google Scholar]
- 57.Wakabayashi K, Lian ZX, Moritoki Y, Lan RY, Tsuneyama K, Chuang YH, Yang GX, Ridgway W, Ueno Y, Ansari AA, Coppel RL, Mackay IR, Gershwin ME. IL-2 receptor alpha−/− mice and the development of primary biliary cirrhosis. Hepatology. 2006;44:1240–1249. doi: 10.1002/hep.21385. [DOI] [PubMed] [Google Scholar]
- 58.Aoki CA, Roifman CM, Lian ZX, Bowlus CL, Norman GL, Shoenfeld Y, Mackay IR, Eric Gershwin M. IL-2 receptor alpha deficiency and features of primary biliary cirrhosis. J Autoimmun. 2006;27:50–3. doi: 10.1016/j.jaut.2006.04.005. [DOI] [PubMed] [Google Scholar]
- 59.Lan RY, Ansari AA, Lian ZX, Gershwin ME. Regulatory T cells: development, function and role in autoimmunity. Autoimmun Rev. 2005;4:351–63. doi: 10.1016/j.autrev.2005.01.007. [DOI] [PubMed] [Google Scholar]
- 60.Reidenberg MM. Aromatic amines and the pathogenesis of lupus erythematosus. Am J Med. 1983;75:1037–42. doi: 10.1016/0002-9343(83)90885-9. [DOI] [PubMed] [Google Scholar]
- 61.Yung RL, Richardson BC. Drug-induced lupus. Rheum Dis Clin North Am. 1994;20:61–86. [PubMed] [Google Scholar]
- 62.Steenland K, Goldsmith DF. Silica exposure and autoimmune diseases. Am J Ind Med. 1995;28:603–8. doi: 10.1002/ajim.4700280505. [DOI] [PubMed] [Google Scholar]
- 63.Silman AJ, Newman J. Epidemiology of systemic sclerosis. Curr Opin Rheumatol. 1996;8:585–9. doi: 10.1097/00002281-199611000-00015. [DOI] [PubMed] [Google Scholar]
- 64.Posada de la Paz M, Philen RM, Borda AI. Toxic oil syndrome: the perspective after 20 years. Epidemiol Rev. 2001;23:231–47. doi: 10.1093/oxfordjournals.epirev.a000804. [DOI] [PubMed] [Google Scholar]