

Abstract
Background
Cytoadherence of Plasmodium falciparum-infected erythrocytes to host endothelium has been associated with pathology in severe malaria, but, despite extensive information on the primary processes involved in the adhesive interactions, the mechanisms underlying disease are poorly understood.
Methods
We compared parasite lines varying in their binding properties to human endothelial cells for their ability to stimulate signaling activity.
Results
In human umbilical vein endothelial cells (HUVECs), which rely on adhesion to intercellular adhesion molecule (ICAM)-1 for binding, signaling is related to the avidity of the parasite line for ICAM-1 and can be blocked either through the use of anti-ICAM-1 monoclonal antibodies or HUVECs with altered ICAM-1 binding properties (i.e., ICAM-1Kilifi). Human dermal microvascular endothelial cells (HDMECs), which can bind infected erythrocytes via ICAM-1 and CD36, have a more complex pattern of signaling behavior, but this is also dependent on adhesive interactions rather than merely contact between cells.
Conclusions
Signaling via apposition of P. falciparum-infected erythrocytes with host endothelium is dependent, at least in part, on the cytoadherence characteristics of the invading isolate. An understanding of the postadhesive processes produced by cytoadherence may help us to understand the variable pathologies seen in malaria disease.
Plasmodium falciparum infection is a major cause of death worldwide, mostly because of the development of severe complications—namely, severe anemia, respiratory acidosis, multiorgan failure, and cerebral malaria (CM). The pathogenesis of cerebral and severe malaria is not fully understood, but organ-specific pathogenesis probably requires sequestration of infected erythrocytes (IEs) on endothelial cells (ECs). In addition, several immune and inflammatory mediators have been heavily implicated in the process leading to severe malaria [1].
A consistent histological finding in CM in both children and adults is the presence of infected and noninfected erythrocytes packed within cerebral vessels [2, 3]. This cytoadhesion process of IEs is mediated by the variable and diverse var gene products encoding P. falciparum erythrocyte membrane protein (PfEMP)-1 that are displayed on the surface of IEs and can bind to many host cellular proteins. PfEMP-1 varies within and between genetically identical parasite clones, which results in changes in their avidity for EC receptors. In most major studies [4, 5], patient samples have varied binding phenotypes but most commonly bind to CD36 and intercellular adhesion molecule (ICAM)-1. In one study [4], ICAM-1 adhesion was higher in isolates from persons with CM; although this did not reach significance, there was a statistically significant association with increased ICAM-1 binding and clinical disease.
The cerebral endothelium performs a critical function with the blood-brain barrier (BBB), forming a tight barrier to maintain homeostasis for adjacent neuronal cells. Several lines of evidence suggest that BBB function is impaired during CM (reviewed in [6]). One mechanism for this may be the intracellular signals produced by IE cytoadherence to EC receptors. Thus sequestration results in microcirculatory changes accumulating IEs, inflammatory cytokines, mononuclear cells, and platelets adjacent to the BBB causing significant local disruption to neuronal function. This may largely explain the coma that can reverse quickly in many treated patients and without apparent residual brain damage.
The mitogen-activated protein kinases (MAPKs) [7] are a family of 3-tier signaling cascades implicated in the cellular responses to a broad range of extracellular stimuli. This is an evolutionarily conserved class of proline-directed serine/threonine kinase pathway found in all eukaryotes, with 3 main members: extracellular signal-regulated kinase (ERK)-1/2, p38, and JNK. The control cascade consists of MAPK kinases and ERKs. MAPK pathways do not function as simple on/off switch signal transducers, so that the physiological outcomes of extracellular stimuli depend on the magnitude, duration, and localization of MAPK activity. In ECs, they exert a major influence on barrier permeability and cell survival [8].
EC surface receptors not only mediate firm adhesion but are also involved in signal transduction processes [9-11]. For example, ICAM-1 is a fundamental component in many immune-related processes, where it mediates cell-cell interactions, allowing for signal transduction as well as activating these pathways directly. ICAM-1 ligation at the cell surface can lead to a range of signaling-mediated activities, including p38 MAPK-inducing heat-shock protein 27 phosphorylation in pulmonary microvascular ECs, which in turn modulates cytoskeletal rearrangements and neutrophil migration toward EC junctions [12]; interleukin (IL)-1α, IL-1β, and IL-6 expression mediated through p38 and ERK-1/2 MAPK [13]; tyrosine phosphorylation of cytoskeletal proteins focal adhesion kinase, paxillin, and p130 in cerebral ECs, mediated by Rho activation [14]; and up-regulation of vascular adhesion molecule-1 on endothelial cells mediated by ERK-1 and AP-1 [15].
We propose that cytoadherence contributes to pathogenic processes in severe malaria through activating cell-signaling processes and that the variable pathologic characteristics seen in malaria could be due partly to P. falciparum variant-specific signaling activity. To examine this, we used a coculture, in vitro model where cultured IEs were placed in direct contact with human EC layers. ECs were then assessed for cell signal activity. The signaling proteins chosen for study were JNK, ERK-1/2 (p42/44), and p38, which represent the 3 major MAPK families and have previously been shown to be activated by ICAM-1 stimulation.
MATERIALS AND METHODS
P. falciparum parasite culture
For our experiments, we used IEs derived from the IT line—namely, ItG, A4, and C24. These clones are genetically identical but differ in their binding phenotype through expression of different var genes. ItG and A4 both bind to CD36 and ICAM-1 but differ in their avidity for the latter [16, 17], whereas C24 is unable to bind to ICAM-1 but shows strong adhesion to CD36.
IEs were cultured for up to 5 days under standard laboratory conditions, with regular replacement of growth medium and addition of washed red blood cells (RBCs) as appropriate [18]. The culture was synchronized using sorbitol 3 days before experiments. The levels of mature trophozoites were enriched using Plasmagel flotation [19] before experimental work. Parasitemia was adjusted to 50% and hematocrit to 3% in binding medium, and this was used in coincubation experiments.
Culture of human umbilical vein endothelial cells (HUVECs) and human dermal microvascular endothelial cells (HDMECs)
Primary human cell lines HUVECs and HDMECs were purchased from PromoCell. All experiments were performed on ECs at passage 4.
ICAM-1Kilifi HUVECs were harvested from umbilical cords using a technique modified from that suggested by Jaffe et al. [20] after relevant ethical permission was obtained. Briefly, the umbilical vein was cannulated, then 2% collagenase solution was infused and the cord incubated for 20 min at 37°C. The vein was then flushed with Promocell growth medium into a 50-mL Falcon tube. The cells were washed with growth medium and then placed into tissue-culture flasks.
Determination of the ICAM-1Kilifi genotype
Genomic human DNA was extracted from umbilical cord blood collected at the time of HUVEC harvesting. The ICAM-1Kilifi genotype was determined using an allele-specific polymerase chain reaction method detailed elsewhere [21].
Signaling experiment procedures
ECs were seeded on 75-cm2 culture flasks and allowed to reach confluence over the course of 24-36 h. The confluent cells were treated with 1 ng/mL tumor necrosis factor in M199 medium (Sigma) supplemented with 1% fetal calf serum (FCS) for 8 h, which is known to up-regulate and maintain EC ICAM-1 expression (N.J., data not shown) [22]. After this, the cells were quiesced by overnight incubation in M199/1% FCS (basic medium without growth factors).
These cells were then used in coculture experiments. To block specific adhesion to ICAM-1 and CD36, 10 μg/mL monoclonal antibody (MAb) 15.2 (Serotec) and a 1:1000 dilution of MAb8A6 (gift from J. Barnwell), respectively, were added to the ECs 30 min before the addition of IEs. For IE binding experiments, the medium was removed and an IE culture added. Incubation with identically prepared uninfected erythrocytes (3% hematocrit and 4 mL vol for each flask) was used as a baseline control. After 15 min, the IE culture was aspirated, and the ECs were washed twice with ice-cold PBS. After washing, flasks were checked carefully by microscopy for the presence of any adherent IEs, but none were seen.
Cellular proteins were extracted using lysis buffer (Cell Signaling Technology) supplemented with 1 mmol/L phenylmethylsulfonyl fluoride and sonication. Cell debris was removed by centrifugation at 14,000 rpm for 10 min to remove cell debris; at all steps, the lysates were kept at 4°C to prevent dephosphorylation of activated protein kinases. Further details of kinase assays are available from the manufacturer’s Web site (ERK-1/2, http://www.cellsignal.com/products/9800.html; p38, http://www.cellsignal.com/products/9820.html; and JNK, http://www.cellsignal.com/products/9810.html).
ICAM-1 cross-linking (positive control for signaling)
The addition of a primary anti-ICAM-1 MAb followed by a secondary MAb cross-links and clusters ICAM-1 receptors, resulting in activation and intracellular signaling [23]. Quiescent ECs were incubated at 37°C with 20 μg/mL mouse anti-ICAM-1 antibody (clone 6.5B5; Biosource) for 30 min. After washing with warm PBS, 60 μg/mL rabbit anti-mouse antibody was added and left for 30 min. This secondary antibody induced clustering and activation of ICAM-1 receptors [14]. The antibody solution was then removed, the cells were washed, and proteins were extracted as described above.
Immunoprecipitations and Western immunoblot analyses
Protein concentrations were measured using the DC protein assay kit (Bio-Rad) and double checked by running aliquot of cell lysate on SDS-PAGE. Equal amounts of protein were used in each kinase activation assay. The activities of p38, JNK, and ERK-1/2 MAPK were measured using phosphor-kinase assay kits (Cell Signaling) in accordance with the manufacturer’s instructions, with slight modifications. The principle of these nonradioactive assays is the immunoprecipitation of target proteins by specific antibodies followed by measurement of their activation using a specific kinase assay. Briefly, 200 μL of cell lysate was incubated with immobilized phospho-p38 MAPK (Thr180/Tyr182) antibodies, c-Jun fusion protein, and phospho-p44/42 MAPK (Thr202/Tyr204) antibodies (15 μL/200 μg of protein) overnight at 4°C. For the kinase assays, immunoprecipitated samples were resuspended in 50 μL of kinase buffer supplemented with 200 μmol/L ATP and 2 μg of ATF-2 fusion protein (for p38 MAPK), Elk-1 fusion protein (for ERK-1/2), or ATP only (for JNK) for 30 min at 30°C. After incubation, 2× Laemmli sample buffer was added to the reaction mixture and boiled for 5 min. The samples then were subjected to SDS-PAGE on 12.5% gels, proteins were transferred onto nitrocellulose membranes, and Western blotting was performed with phospho-ATF-2, phosph-c-Jun, or phospho-Elk-1 antibodies (1:10,000 dilution) and horseradish peroxidase (HRP)-conjugated anti-rabbit secondary antibody (1:2000) and HRP-conjugated anti-biotin antibody (1:10,000). Proteins on polyvinylidene fluoride membranes were detected using the ECL Advance Western Blotting Detection Kit (Amersham), and images were captured on x-ray film.
Static IE binding assays on HUVECs
Binding of P. falciparum-infected erythrocytes to human ECs on coverslips was performed as described elsewhere [24].
Band density measurement and statistical analysis
The density of each reaction band in the immunoblots was captured and analyzed using GeneTools software from SynGene (Synoptic). To standardize the results of 3 independent experiments and make them comparable, we used a ratio of each band density to a standard ICAM-1 cross-linking band run in each experiment. The numbers presented in the figures represent the means ± SDs. Statistical analysis was performed using Student’s t test with correction for multiple comparisons. The difference in the experimental groups was considered to be significant at P< .05.
RESULTS
Transient increase in cell-signaling activity in HUVECs after P. falciparum coculture
To investigate the time course of cell activation by P. falciparum, we performed coculture experiments for differing lengths of time. ERK-1/2, JNK, and p38 kinase activity were maximally increased after 15 min of incubation with P. falciparum culture (figure 1). This incubation period was therefore used in further experiments.
Figure 1.

Human umbilical vein endothelial cells (HUVECs) and human dermal microvascular endothelial cells (HDMECs) treated with tumor necrosis factor, then quiesced in low-serum medium overnight before incubation with ItG-infected erythrocytes (in low-serum medium) for the time period specified. The relative intensities represent the phosphorylation of extracellular signal-regulated kinase (ERK)-1/2, JNK, and p38 in cell lysates.
Comparison of signaling activity in HUVECs after coincubation with P. falciparum parasite clones with different ICAM-1 binding affinities
ItG-infected erythrocytes (high avidity for ICAM-1) stimulated significantly similarly higher JNK, p38, and ERK-1/2 MAPK activity than did A4-infected erythrocytes (moderate avidity for ICAM-1) (P ≤ .05) for all 3 proteins tested, which in turn stimulated significantly higher MAPK activity than did C24-infected erythrocytes (no ICAM-1 binding) (P ≤ .03) (figure 2). Signaling activity of both ItG-IE and A4-IE could be blocked using the adhesion blocking anti-ICAM-1 MAb15.2, which is known to inhibit IE cytoadherence via ICAM-1 [25].
Figure 2.

Signaling activities by human umbilical vein endothelial cells (HUVECs) as a ratio of intercellular adhesion molecule (ICAM)-1 cross-linking caused by different parasite lines (C24, ItG, and A4) and red blood cells (RBCs) in the absence and presence of blocking monoclonal antibody (MAb) 15.2 and with MAb15.2 on its own (15.2). The response of HUVECs homozygous for the ICAM-1Kilifi allele (Kilifi) with reduced ICAM-1 adhesion is also shown. The protocols for determining the phosphorylation of specific kinases is described in Materials and Methods and is plotted as the ratio of ICAM-1 cross-linking. Mock-incubated uninfected RBCs were used as a control. P values for specific comparisons are indicated on each graph.
Comparison of signaling response in HUVEC expressing different ICAM-1Kilifi genotypes
An ICAM-1 polymorphism called ICAM-1Kilifi has been described in the region encoding the binding site for PfEMP-1 [26]. This allele is common in sub-Saharan Africa (gene frequency, 0.3) but has not been found in white persons. The polymorphism alters the protein structure and reduces IE binding in vitro among other functional changes [27]. HUVECs obtained from umbilical cords from babies of known ICAM-1Kilifi genotype were used to determine how ICAM-1-mediated signaling varied with ICAM-1 genotype, and hence, the binding avidity to IEs. Figure 3 shows that in vitro IE binding was significantly reduced to ICAM-1Kilifi homozygous, compared with ICAM-1Ref homozygous, HUVECs, which confirms findings previously obtained using IE binding to ICAM-1Kilifi and ICAM-1Ref proteins [28]. MAPK activity was significantly reduced in ICAM-1Kilifi homozygous HUVECs when cocultured with ItG -infected IE (figure 2), in accordance with reduced binding via ICAM-1. MAPK signaling in ICAM-1 heterozygous HUVECs was comparable to that in ICAM-1Ref homozygous HUVECs.
Figure 3.
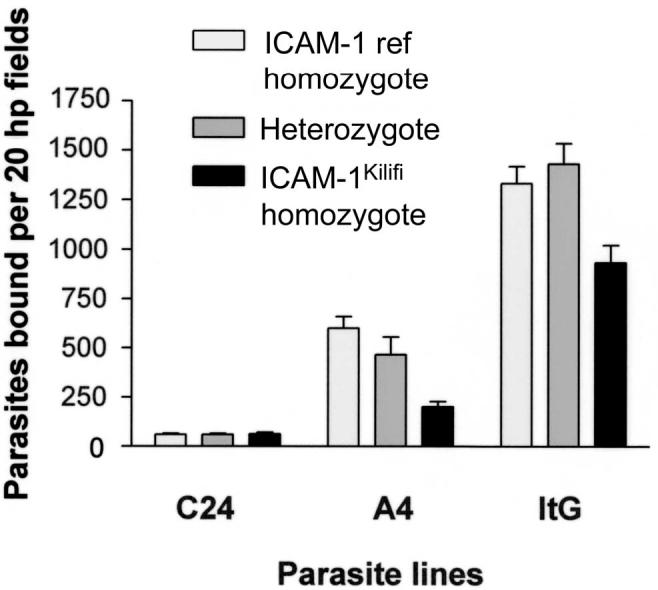
Differential effect of binding to human umbilical vein endothelial cells (intercellular adhesion molecule [ICAM]-1Kilifi homozygous, heterozygous, and reference [ref] genotypes) with Plasmodium falciparum lines ItG, A4, and C24. hp, high power.
Cellular MAPK activity in HDMECs
HDMECs provide a more complicated model because they support both ICAM-1 and CD36 adhesion of IEs. IEs commonly bind to HDMECs via ICAM-1 and CD36, and these cell-surface receptors are known to act synergistically [29]. Therefore, blocking antibodies to these receptors were used to assess their relative contribution to cell activation.
C24-infected erythrocytes bind HDMECs via CD36 but not ICAM-1, whereas ItG-IEs bind to both CD36 and ICAM-1, although the latter appears to be critical under flow conditions [17]. Both C24 and ItG induced significant signaling activity, compared with uninfected RBC controls (figure 4). Preventing cytoadherence with MAbs to CD36 and ICAM-1 (both singly and in combination) reduced MAPK activity (P = .02) (figure 4). C24-stimulated JNK activity was significantly reduced by CD36-blocking MAbs (P = .02) but not by ICAM-1-blocking MAbs alone (data not shown), whereas ItG-stimulated JNK activity was unaffected by anti-CD36 MAb but was significantly reduced by anti-ICAM-1 MAbs. For p38 and ERK-1/2, although blocking conditions that reduced cytoadherence to HDMECs tended to reduce kinase activation, this was only statistically significant for ItG when both CD36 and ICAM-1 blocking MAbs were used.
Figure 4.
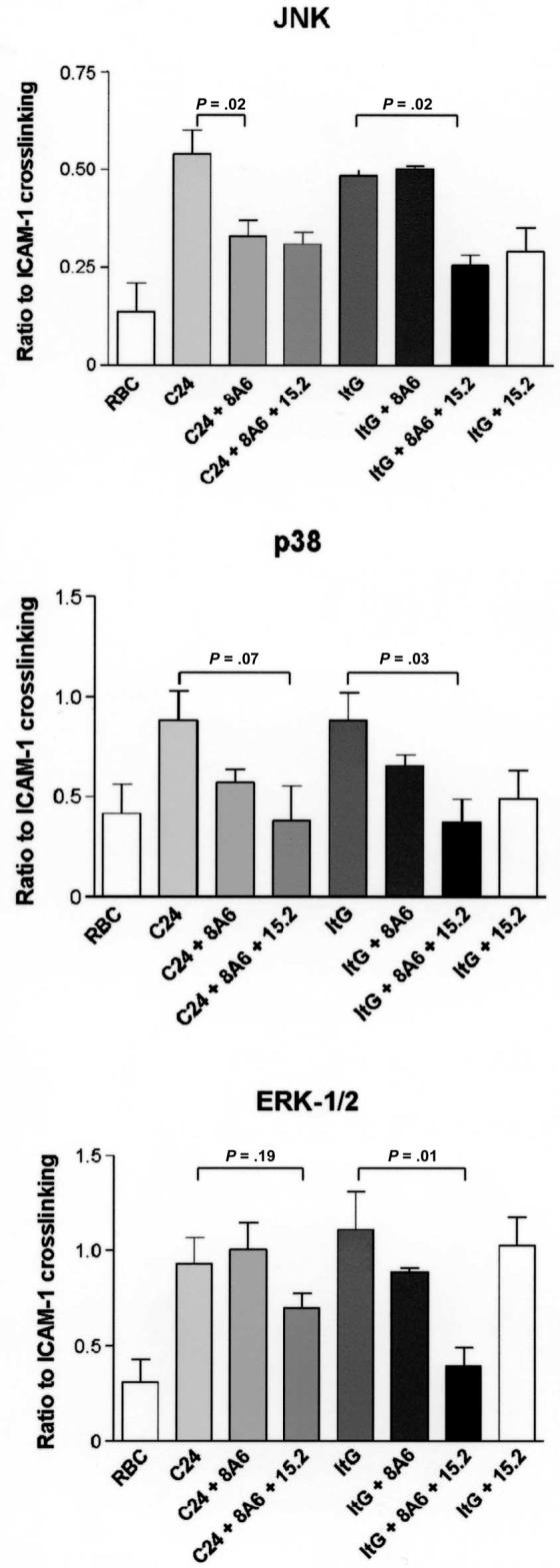
Signaling activities by human dermal microvascular endothelial cells as a ratio of intercellular adhesion molecule (ICAM)-1 cross-linking caused by different parasite lines (C24 and ItG) and mock-incubated uninfected red blood cells (RBCs) in the absence and presence of blocking monoclonal antibodies 15.2 and 8A6. P values for specific comparisons are indicated on each graph.
DISCUSSION
The sequestration of mature P. falciparum IEs appears to be necessary but not sufficient to cause CM and severe malaria [3, 30]. The key mechanism in sequestration is cytoadherence—usually the binding between PfEMP-1 and one of numerous host EC surface receptors [31]. Subsequent changes in the microcirculation are thought to result in high concentrations of inflammatory mediators and metabolites adjacent to disruptions in the BBB. Using an in vitro coculture model, we have shown that IEs increase the activity of ERK-1/2, p38, and JNK MAPKs in both HUVECs and HDMECs. The increase in cell signaling is dependent on cytoadherence but does not appear to rely solely on ICAM-1.
In vitro models suggested that IEs can induce the expression of surface receptors and nitric oxide and induce apoptosis in ECs [32-34]. These studies have suggested contact is required but did not detail the effect of cytoadherence. For both EC types, coculture with P. falciparum resulted in a significant increase in MAPK activity. This activation was transient, peaking within 15 min of parasite binding and returning to base levels within 1 h. This signaling activity was, in some cases, higher than that achieved by ICAM-1 cross-linking. Cytoadherence blocking antibodies consistently reduced MAPK activity in both HUVECs and HDMECs. Our findings suggest that the cell activation is not dependant on IE-ICAM-1 receptor binding alone, because C24 parasites, which do not bind to ICAM-1, also induced activation in HDMECs at a level comparable to that induced by ItG (high ICAM-1 binding avidity). C24-mediated signaling was prevented by CD36-blocking antibodies but unaffected by antibodies that block binding to ICAM-1. In short, it appears that C24 parasites activate HDMECs via CD36 binding and ItG parasites via ICAM-1 binding. However, the induction of signaling is not entirely a simple case of binding translating into activity. In HDMECs, ItG-derived signaling (JNK and p38) is inhibited by blocking ICAM-1-dependent adhesion with MAb15.2 but not significantly by the anti-CD36 MAb8A6, even though ItG shows relatively strong interactions with CD36. Thus, there appears to be some specificity in the ability of different parasite variants to activate signaling pathways using different EC adhesion receptors, but differential MAPK activation is also seen—for example, ItG/ERK-1/2, where signaling is not blocked by MAb15.2 on its own but p38 and JNK signaling are.
Although our coculture model is not an accurate representation of the sequestration occurring within a cerebral postcapillary venule, we have designed and validated a useful model for the study of signaling in ECs in response to IE binding. It is important to note that this is a static model and therefore may have important differences in parasite-receptor kinetics, compared with the situation under flow in vivo. Future work will need to investigate signaling mechanisms in different ECs under static and ex vivo flow conditions. We did not use brain endothelium, which quickly loses its specialized phenotype during in vitro culture. Recently published work using brain microvascular endothelium has indicated that signaling pathways are also involved in these cells [35], as well as in a rodent model of CM [36].
Cytoadherence and sequestration occur commonly within populations in areas where malaria is endemic, including in many individuals with chronic, asymptomatic infection. It is possible that signaling requires a threshold level of adhesion or that the pathogenic processes attributable to signaling require an amplitude and duration of signaling that is only achieved under specific conditions. One of these might be the avidity of interaction between host receptors and IEs, so that the number of IEs binding would not directly translate into disease but rather the nature of that adhesion. Differences in avidity of binding by laboratory and patient isolates is seen for both CD36 and ICAM-1 (A.C., data not shown), and our results clearly demonstrate that ICAM-1 is not the only receptor mediating signaling in malaria. Indeed, other studies have already shown that CD36 is important in terms of Src-dependent signaling on HDMECs [32]. The situation on brain endothelium is further complicated by the role of platelets in mediating adhesion to ECs, which do not normally display CD36 expression [37]. In addition, further work using coculture between HUVECs and IEs (S.C., data not shown) has demonstrated that at least some components of EC activation are PfEMP-1 independent.
We suggest that postadhesive signaling processes are activated during malaria infection; however, we have not demonstrated here whether they are pathological. Although there is a strong rationale for the involvement of these types of processes that result in disruptions to the endothelial barrier, some signaling pathways may protect ECs. Activation of MAPK pathways may result in cell proliferation or protect ECs against apoptotic stimuli in vitro. Parasites that bind to and then kill ECs may recirculate and be removed by the immune system. It is feasible that a successful parasite is able to sequester to an EC and then prolong its survival to facilitate transmission by influencing protective, antiapoptotic intracellular processes. However, should factors such as the sequestered parasite load and the up-regulation of endothelial receptors result in increased and sustained activation of intracellular signals, this could result in severe symptoms.
We have described how IEs that are able to adhere to surface receptors on ECs result in the activation of cell processes known to influence EC barrier function, transcription factors, and cell survival and that these activation processes can, in part, be attributable to the nature of the adhesive interaction. These, along with many other factors occurring within the cerebral vasculature, may help to explain the pathophysiology of CM and other manifestations of severe disease. However, we also suggest that the pathology of severe disease in malaria is complex and that a direct association with the amplitude of MAPK phosphorylation may not wholly reflect the situation in vivo, which is supported by our results with ICAM-1Kilifi homozygous HUVECs showing reduced signaling in our coculture experiments but that have, in some studies, been associated with poor clinical outcome [26].
Acknowledgments
We thank John Barnwell, for his generous gift of monoclonal antibody 8A6; Gareth Turner, for his support and encouragement throughout this project; and the mothers in the Kilifi District General Hospital, for the donation of umbilical cords for human umbilical vein endothelial cell preparation. This article is published with the permission of the director of the Kenya Medical Research Institute.
Footnotes
Financial support: Wellcome Trust (Training Fellowship GR066684MA to N.J. and Functional Genomics Initiative on Malaria grant GR066742MA to A.C.).
Potential conflicts of interest: none reported.
References
- 1.van der Heyde HC, Nolan J, Combes V, Gramaglia I, Grau GE. A unified hypothesis for the genesis of cerebral malaria: sequestration, inflammation and hemostasis leading to microcirculatory dysfunction. Trends Parasitol. 2006;22:503–8. doi: 10.1016/j.pt.2006.09.002. [DOI] [PubMed] [Google Scholar]
- 2.Pongponratn E, Turner GD, Day NP, et al. An ultrastructural study of the brain in fatal Plasmodium falciparum malaria. Am J Trop Med Hyg. 2003;69:345–59. [PubMed] [Google Scholar]
- 3.Taylor TE, Fu WJ, Carr RA, et al. Differentiating the pathologies of cerebral malaria by postmortem parasite counts. Nat Med. 2004;10:143–5. doi: 10.1038/nm986. [DOI] [PubMed] [Google Scholar]
- 4.Newbold C, Warn P, Black G, et al. Receptor-specific adhesion and clinical disease in Plasmodium falciparum. Am J Trop Med Hyg. 1997;57:389–98. doi: 10.4269/ajtmh.1997.57.389. [DOI] [PubMed] [Google Scholar]
- 5.Rogerson SJ, Tembenu R, Dobano C, Plitt S, Taylor TE, Molyneux ME. Cytoadherence characteristics of Plasmodium falciparum-infected erythrocytes from Malawian children with severe and uncomplicated malaria. Am J Trop Med Hyg. 1999;61:467–72. doi: 10.4269/ajtmh.1999.61.467. [DOI] [PubMed] [Google Scholar]
- 6.Medana IM, Turner GD. Human cerebral malaria and the blood-brain barrier. Int J Parasitol. 2006;36:555–68. doi: 10.1016/j.ijpara.2006.02.004. [DOI] [PubMed] [Google Scholar]
- 7.Hoefen RJ, Berk BC. The role of MAP kinases in endothelial activation. Vascul Pharmacol. 2002;38:271–3. doi: 10.1016/s1537-1891(02)00251-3. [DOI] [PubMed] [Google Scholar]
- 8.Bogatcheva NV, Dudek SM, Garcia JG, Verin AD. Mitogen-activated protein kinases in endothelial pathophysiology. J Investig Med. 2003;51:341–52. doi: 10.1136/jim-51-06-30. [DOI] [PubMed] [Google Scholar]
- 9.Aplin AE, Howe AK, Juliano RL. Cell adhesion molecules, signal transduction and cell growth. Curr Opin Cell Biol. 1999;11:737–44. doi: 10.1016/s0955-0674(99)00045-9. [DOI] [PubMed] [Google Scholar]
- 10.Rosales C, Juliano RL. Signal transduction by cell adhesion receptors in leukocytes. J Leukoc Biol. 1995;57:189–98. doi: 10.1002/jlb.57.2.189. [DOI] [PubMed] [Google Scholar]
- 11.Juliano RL. Signal transduction by cell adhesion receptors and the cytoskeleton: functions of integrins, cadherins, selectins, and immunoglobulin-superfamily members. Annu Rev Pharmacol Toxicol. 2002;42:283–323. doi: 10.1146/annurev.pharmtox.42.090401.151133. [DOI] [PubMed] [Google Scholar]
- 12.Wang Q, Pfeiffer GR, II, Stevens T, Doerschuk CM. Lung microvascular and arterial endothelial cells differ in their responses to intercellular adhesion molecule-1 ligation. Am J Respir Crit Care Med. 2002;166:872–7. doi: 10.1164/rccm.2201007. [DOI] [PubMed] [Google Scholar]
- 13.Lee SJ, Drabik K, Van Wagoner NJ, et al. ICAM-1-induced expression of proinflammatory cytokines in astrocytes: involvement of extracellular signal-regulated kinase and p38 mitogen-activated protein kinase pathways. J Immunol. 2000;165:4658–66. doi: 10.4049/jimmunol.165.8.4658. [DOI] [PubMed] [Google Scholar]
- 14.Etienne S, Adamson P, Greenwood J, Strosberg AD, Cazaubon S, Couraud PO. ICAM-1 signaling pathways associated with Rho activation in microvascular brain endothelial cells. J Immunol. 1998;161:5755–61. [PubMed] [Google Scholar]
- 15.Lawson C, Ainsworth M, Yacoub M, Rose M. Ligation of ICAM-1 on endothelial cells leads to expression of VCAM-1 via a nuclear factor-kappaB-independent mechanism. J Immunol. 1999;162:2990–6. [PubMed] [Google Scholar]
- 16.Roberts DJ, Craig AG, Berendt AR, et al. Rapid switching to multiple antigenic and adhesive phenotypes in malaria. Nature. 1992;357:689–92. doi: 10.1038/357689a0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gray C, McCormick C, Turner G, Craig A. ICAM-1 can play a major role in mediating P. falciparum adhesion to endothelium under flow. Mol Biochem Parasitol. 2003;128:187–93. doi: 10.1016/s0166-6851(03)00075-6. [DOI] [PubMed] [Google Scholar]
- 18.Trager W, Jensen JB. Human malaria parasites in continuous culture. Science. 1976;193:673–5. doi: 10.1126/science.781840. [DOI] [PubMed] [Google Scholar]
- 19.Goodyer ID, Johnson J, Eisenthal R, Hayes DJ. Purification of maturestage Plasmodium falciparum by gelatine flotation. Ann Trop Med Parasitol. 1994;88:209–11. doi: 10.1080/00034983.1994.11812859. [DOI] [PubMed] [Google Scholar]
- 20.Jaffe EA, Nachman RL, Becker CG, Minick CR. Culture of human endothelial cells derived from umbilical veins: identification by morphologic and immunologic criteria. J Clin Invest. 1973;52:2745–56. doi: 10.1172/JCI107470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jenkins NE, Mwangi TW, Kortok M, Marsh K, Craig AG, Williams TN. A polymorphism of intercellular adhesion molecule-1 is associated with a reduced incidence of nonmalarial febrile illness in Kenyan children. Clin Infect Dis. 2005;41:1817–9. doi: 10.1086/498156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.van de Stolpe A, van der Saag PT. Intercellular adhesion molecule-1. J Mol Med. 1996;74:13–33. doi: 10.1007/BF00202069. [DOI] [PubMed] [Google Scholar]
- 23.Durieu-Trautmann O, Chaverot N, Cazaubon S, Strosberg AD, Couraud PO. Intercellular adhesion molecule 1 activation induces tyrosine phosphorylation of the cytoskeleton-associated protein cortactin in brain microvessel endothelial cells. J Biol Chem. 1994;269:12536–40. [PubMed] [Google Scholar]
- 24.Berendt AR, Simmons DL, Tansey J, Newbold CI, Marsh K. Intercellular adhesion molecule-1 is an endothelial cell adhesion receptor for Plasmodium falciparum. Nature. 1989;341:57–9. doi: 10.1038/341057a0. [DOI] [PubMed] [Google Scholar]
- 25.Berendt AR, McDowall A, Craig AG, et al. The binding site on ICAM-1 for Plasmodium falciparum-infected erythrocytes overlaps, but is distinct from, the LFA-1-binding site. Cell. 1992;68:71–81. doi: 10.1016/0092-8674(92)90207-s. [DOI] [PubMed] [Google Scholar]
- 26.Fernandez-Reyes D, Craig AG, Kyes SA, et al. A high frequency African coding polymorphism in the N-terminal domain of ICAM-1 predisposing to cerebral malaria in Kenya. Hum Mol Genet. 1997;6:1357–60. doi: 10.1093/hmg/6.8.1357. [DOI] [PubMed] [Google Scholar]
- 27.Craig A, Fernandez-Reyes D, Mesri M, et al. A functional analysis of a natural variant of intercellular adhesion molecule-1 (ICAM-1Kilifi) Hum Mol Genet. 2000;9:525–30. doi: 10.1093/hmg/9.4.525. [DOI] [PubMed] [Google Scholar]
- 28.Adams S, Turner GD, Nash GB, Micklem K, Newbold CI, Craig AG. Differential binding of clonal variants of Plasmodium falciparum to allelic forms of intracellular adhesion molecule 1 determined by flow adhesion assay. Infect Immun. 2000;68:264–9. doi: 10.1128/iai.68.1.264-269.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.McCormick CJ, Craig A, Roberts D, Newbold CI, Berendt AR. Intercellular adhesion molecule-1 and CD36 synergize to mediate adherence of Plasmodium falciparum-infected erythrocytes to cultured human microvascular endothelial cells. J Clin Invest. 1997;100:2521–9. doi: 10.1172/JCI119794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.MacPherson GG, Warrell MJ, White NJ, Looareesuwan S, Warrell DA. Human cerebral malaria: a quantitative ultrastructural analysis of parasitized erythrocyte sequestration. Am J Pathol. 1985;119:385–401. [PMC free article] [PubMed] [Google Scholar]
- 31.Kyes S, Horrocks P, Newbold C. Antigenic variation at the infected red cell surface in malaria. Annu Rev Microbiol. 2001;55:673–707. doi: 10.1146/annurev.micro.55.1.673. [DOI] [PubMed] [Google Scholar]
- 32.Yipp BG, Robbins SM, Resek ME, Baruch DI, Looareesuwan S, Ho M. Src-family kinase signaling modulates the adhesion of Plasmodium falciparum on human microvascular endothelium under flow. Blood. 2003;101:2850–7. doi: 10.1182/blood-2002-09-2841. [DOI] [PubMed] [Google Scholar]
- 33.Pino P, Vouldoukis I, Dugas N, et al. Induction of the CD23/nitric oxide pathway in endothelial cells downregulates ICAM-1 expression and decreases cytoadherence of Plasmodium falciparum-infected erythrocytes. Cell Microbiol. 2004;6:839–48. doi: 10.1111/j.1462-5822.2004.00406.x. [DOI] [PubMed] [Google Scholar]
- 34.Pino P, Vouldoukis I, Kolb JP, et al. Plasmodium falciparum-infected erythrocyte adhesion induces caspase activation and apoptosis in human endothelial cells. J Infect Dis. 2003;187:1283–90. doi: 10.1086/373992. [DOI] [PubMed] [Google Scholar]
- 35.Tripathi AK, Sullivan DJ, Stins MF. Plasmodium falciparum-infected erythrocytes increase intercellular adhesion molecule 1 expression on brain endothelium through NF-kappaB. Infect Immun. 2006;74:3262–70. doi: 10.1128/IAI.01625-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lu Z, Serghides L, Patel SN, et al. Disruption of JNK2 decreases the cytokine response to Plasmodium falciparum glycosylphosphatidylinositol in vitro and confers protection in a cerebral malaria model. J Immunol. 2006;177:6344–52. doi: 10.4049/jimmunol.177.9.6344. [DOI] [PubMed] [Google Scholar]
- 37.Wassmer SC, Lepolard C, Traore B, Pouvelle B, Gysin J, Grau GE. Platelets reorient Plasmodium falciparum-infected erythrocyte cytoadhesion to activated endothelial cells. J Infect Dis. 2004;189:180–9. doi: 10.1086/380761. [DOI] [PubMed] [Google Scholar]