

Abstract
Mammalian heart development requires multiple genetic networks, only some of which are becoming known in all their complexity. Substantial new information has become available thanks to an expanding toolkit that offers more and more mouse gene manipulation options, and that is taking the mouse closer to more powerful invertebrate genetic models. We review examples of recent data with a cardiac-lineage-based view of heart development, especially outflow tract and right ventricle. The medical significance of these studies is not only relevant to congenital heart disease, but also to to the biology of cardiac cell regeneration.
Keywords: Cardiac outflow tract, cardiogenic lineages, T-box, genetic control of heart development, review
1. Introduction
Congenital heart disease (CHD) represent the most common birth defects with a rate of about 1 in 100 livebirths and even higher in miscarriages [1, 2]. The last decade has seen great progress in our understanding of heart development, with the identification of a number of genes involved in cardiogenesis. Large-scale mutation screenings of CHD patients have identified several important genes but a major contribution has come the use of model systems, from the fruit fly to the mouse. In particular, the anatomical similarities of the human and mouse heart, the relatively close evolutionary relationship, and, most critically, the ameanability to genetic manipulation, have made the mouse an excellent model to study the genetics of heart development. In addition, the growing availability of genetic tools, such as Cre-drivers and specialized tools, have taken the mouse genetics well beyond the simple knockout strategy. Recently, on the wake of the discovery of the secondary or anterior heart field, more attention has been given to cardiac cell lineage in evaluating mutant phenotypes. Tissue- and time-based conditional gene ablation experiments have unveiled layers of complexity in the function of genes during mammalian cardiovascular development.
1.1. Mouse genetics tools
Many of the recent discoveries about mouse heart development would have been impossible using the standard knockout approach. This is mainly because, A) Genes that have a critical or vital role in early development cannot be studied at later developmental stages using germ-line mutants, B) The role of genes transiently expressed in a small population of cells may be underestimated or even unrecognized, especially if the transient expression domain is restricted to a narrow time window and if we don’t know the fate of that particular cell population.
Conditional, Cre-loxP based strategies and mRNA dosage manipulation using modified (hypomorphic) alleles can address most of these issues by allowing tissue and time specific ablation. While the Cre-loxP system has been available for many years, it is only recently that the portfolio of Cre drivers has become so substantial and largely available to allow creative and thorough approaches. In addition, it is becoming more and more common to use multiple Cre-drivers. This tendency is justified because there is no “perfect” Cre driver (by pattern, time of onset and level of Cre expression), and because the tissue specific requirement for a gene may be difficult to interpret if the gene is expressed in multiple, interacting tissues. Indeed, ablation of the gene on one or the other tissue may result, at least on surface, on similar downstream phenotypic consequences. The complementary experiment to tissue-specific ablation is tissue-specific re-activation. This is done by using an inactive allele that can be reactivated upon Cre-recombination. Ideally, these alleles should be null in the “inactive” state, and close to wild type, after recombination. The design of such alleles may be technically challenging. Last, but not least, the availability of ubiquitous, inducible Cre drivers has made time-based conditional ablation a reality. Because these Cre constructs can be easily induced in utero, the requirement of a gene can be tested at different developmental times.
The ease of cell fate mapping strategies has been critical for recent discoveries. Classic methods to study the fate of cells during development include physically labeling cells with a dye (or with a virus expressing a reporter gene), this method is technically challenging in a mammalian system, and uncommonly used. Another method is to use a transgenic vector (named laacZ) that, after spontaneous, random and rare recombination events activates a reporter gene [3]. This method requires the analysis of a very large number of embryos.
The most commonly used alternative, however, is the gene expression-based cell fate mapping in which a Cre (inducible or not) –encoding cDNA is knocked into the gene of interest or driven by a tissue-specific promoter-enhancer construct. Cre expression irreversibly activates a reporter allele so that cells expressing Cre, and their progeny, will be labeled. This is different from cell lineage analysis, which follows the progeny of individual cells. Gene expression-based cell fate follows the progeny of cells that express a particular gene, these cells may or may not be part of the same pedigree, and, during development, additional, unrelated cell populations may express the gene and thus join the ranks of traced cells. The use of inducible Cre constructs, allowing a pulse induction, ameliorates this limitation by restricting labeling to cells that express Cre within a specific time window. Limitations notwithstanding, expression-based cell fate analysis is a simple and powerful approach for mouse developmental biologists because it allows tracing of cells expressing the gene of interest in wild type and mutant genetic backgrounds. In addition, the method allows an easier detection of transient expression domains.
1.2. Principles of mouse heart morphogenesis
During mouse gastrulation, mesodermal cells destined to form the heart are located in the anterior region of the primitive streak [4, 5], then they migrate and coalesce anterior-laterally to form the cardiogenic regions now regarded as the primary heart field [6]. The two symmetric regions extend across the midline resulting in the cardiac crescent, where differentiated myocardial cells become detectable. The cardiac crescent fuses at the midline and form a heart tube, which connects with the body through a posterior inflow, or venous pole, and an anterior outflow, or arterial pole. The heart tube undergoes uneven growth and remodeling to form the primitive ventricles and atria. Growing, the heart tube turns rightward and brings the atria anterior and dorsal to the ventricles. At the venous pole, myocardial precursor cells mainly from the posterior region of the primary heart field are incorporated into the heart tube to support the formation and growth of the atria, while at the arterial pole, cells from the anterior (or second) heart field support the growth of the outflow tract (OFT, also referred to as conotruncus) and right ventricle. An additional, non-mesodermal cell population migrates from the neural crest into the heart via the pharyngeal arches and the OFT. These are named cardiac neural crest cells, and participate mainly to the septation of the heart and formation of the cardiac valves (reviewed in ref. [7]).
Early events of cardiac mesoderm induction and specification have been extensively reviewed [8] and are thought to rely mainly on BMP and FGF signals from the adjacent endoderm. Subsequent morphogenetic processes are mainly regulated by a set of transcription factors belonging to the Nkx, GATA, HAND, T-box and MEF families as well as most if not all the major signaling systems (reviewed in ref. [9]).
2. Cardiogenic cell lineages
Although early experiments have suggested that the myocardial cells of the arterial pole are recruited from a source different from the pprimary heart field [10, 11], only recently, three studies have identified this source as the pharyngeal and splanchnic mesoderm [12–14], in chick and mouse. This source was named secondary or anterior heart field (SHF). The primary and secondary sources have common mesodermal progenitors, but probably diverged already at or soon after gastrulation (review in [15] and [16]). The primary source of cardiomyocyte precursors is mainly involved in the formation of the heart tube and contributes mostly to the left ventricle, part of the right ventricles and part of the atria of the mature heart. The secondary source contributes to the OFT, most of the right ventricle, and part of the atria.
Islet 1 (Isl1) encodes a LIM homeodomain transcription factor critical for the development of several organs, including the heart. In particular, Isl1 is required for the development of the heart regions contributed by the secondary heart field. Fate mapping using an Isl1Cre allele revealed that Isl1-traced cells contribute to the OFT, right ventricle, part of the left ventricle near the interventricular septum and a large portion of the atria [17]. Isl1 is probably one of the earliest markers of the SHF. Consistently, the distribution of Isl1-traced cells is one of the broadest reported for an SHF marker. Mef2c mutation also causes extensive abnormalities to the regions contributed by the SHF, similarly to Isl1 mutation. An Isl1-responsive enhancer of the Mef2c gene has been identified [18]. This enhancer was used to generate a Cre driver (Mef2c-AHFCre) that also labeled an extensive region of the heart similarly to Isl1Cre [19]. However, the population of SHF-derived cells is not homogeneous and can be dissected further using cell tracing with other markers. Fate mapping with an inducible Cre knocked into the Tbx1 locus (allele Tbx1mcm), revealed extensive labeling of the OFT but only partial labeling of the right ventricle [20, 21]. Earlier (E7.5 versus E8.5) activation of the inducible Cre of this allele revealed more extensive RV staining, suggesting a possible time-gradient in gene expression of SHF cells destined to the RV (earlier expressors) and to the OFT (later expressors). Whether a “simple” time delay mechanism in the activation of a transcription program is sufficient to determine where SHF cells are destined to, is unclear. Tbx1 mutation causes severe OFT defects and relatively mild RV defects, thus a much less severe heart phenotype than in Isl1 and Mef2c mutants. Timed ablation of Tbx1 showed a critical time window between E8.5 and E9.5 for its function in OFT development. We do not know the critical time for Isl1 and Mef2c function, but judging from the phenotypic abnormalities of the mutants, this is likely to be earlier than for Tbx1. Pitx2 is also express in the SHF and its mutation causes OFT defects. These defects are milder than those of Tbx1−/− mutants. Indeed, the OFT, although abnormal, is septated. Pitx2Cre-traced cells populate the OFT extensively and also populate a portion of the RV and LV [22, 23]. Overall, these observations suggest that the SHF “lineage”, defined as the Isl1-traced cell population, branches out into subpopulations identified by other transcription factors also important for their development (Fig. 1).
Fig. 1.
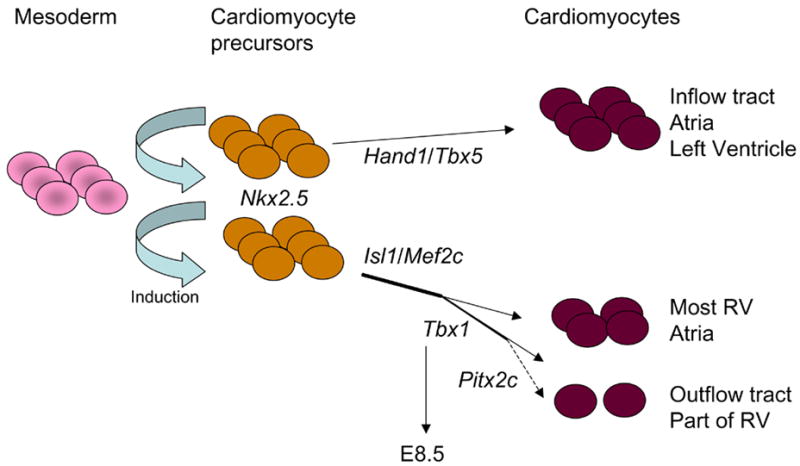
The two cell lineages that contribute to the heart.
Cell types derived from the SHF may not be limited to cardiomyocytes. Pulsed cell fate mapping using the Tbx1mcm allele revealed labeling of a subset of endothelial cells, too [19, 21], raising the question of whether the SHF may be a source for a selected population of endothelial cells in the heart. Further studies will be required to uderstand whether the progeny of an individual SHF mesodermal cell can give rise to cardiomyocytes and to endothelial cells, and perhaps other cell types populating the heart.
In contrast to the primary heart field, the SHF appears to continue feeding cells to the heart for a prolonged period, until relatively late in development (e.g. ~E9.5 for Tbx1-expressing cells). In addition, some cells expressing early SHF markers are still found in neonatal hearts. These cells have the characteristics of cardiomyocyte precursors and are capable of proliferate and differentiate into cardiomyocytes [24]. This is an exciting discovery that emphasizing the fundamental importance of understanding the developmental processes underlying cardiac lineage specification, expansion and differentiation in order to design potential cardiac tissue regeneration strategies. The processes involved in SHF development are easily accessible for study because they happen over a relatively long time, the tissue involved is well delimited, and there are a number of molecular and genetic tools to aide analysis.
2.1. The secondary heart field and congenital heart disease
The SHF contributes cells to a large portions of the heart, including regions often involved in congenital heart disease (CHD). Thus, it is predictable that disturbance of SHF development may cause CHD. Physical ablation of the SHF in chick causes cardiac defects [25], albeit surprisingly mild compared to those caused by mutation of genes coding for transcription factors central to development and viability of the second cardiogenic lineage in the mouse (e.g. Isl1, Foxh1, Mef2c, Tbx1). This may be because ablation may not remove the entire source of precursors or because of species-specific characteristics. Tbx1 has been the first CHD gene shown to be important for SHF development [21]. Tbx1 is needed for proliferation of SHF cells, before they differentiate into cardiomyocytes [21, 26]. NKX2.5 mutations are also associated with CHD [27]. However, NKX2.5 mutations in humans and Nkx2.5 loss of function in the mouse, have more diverse and broader phenotypic consequences than those expected from a specific SHF defect.
Thus, at this point the repertoire of genes involved in SHF development and also involved in CHD is very limited. Most likely, this is due to the still limited knowledge of gene mutations associated with CHD.
3. A putative genetic network in SHF development
3.1. Transcription factors
Most of the genes cited in the above section, have been “linked” to form a putative genetic network. A possible start point for this network is Isl1. This transcription factor is required for proliferation and survival of SHF cells and regulates other transcription factors, as well as signaling systems [28, 29]. Together with Gata4, Isl1 regulates Mef2c expression by interacting with a SHF-specific enhancer of the gene [18]. Interestingly, Foxh1 and Nkx2.5 can also regulate Mef2c gene expression in the SHF through interaction with a different enhancer of Mef2c [30]. Foxh1 is also critical for arterial pole and RV development. It remains to be understood if one or both of these enhancers are necessary for Mef2c function in SHF development. As it stands, the dyads Is1/Gata4 and Foxh1/Nkx2.5 appear to be at the top layer of hierarchical order controlling SHF development. The loss of function of any of these transcription factors has dramatic consequences for cardiovascular development. Whether or not Mef2c is the single integrating point of the first-tier factors is unknown. Mutation of the enhancers regulating these interactions on the Mef2c endogenous gene may provide an answer. In any case, it is clear that the loss of function of Mef2c results in phenotypic consequences similar to those of the first-tier factors (as far as the SHF is concerned). In turn, Mef2c activates Smyd1 [31]. Smyd1−/− animals have severe RV defects, while the OFT phenotype, although difficult to evaluate because of early lethality, did not appear nearly as severe as the one of Mef2c−/− mutants [32]. Thus, Smyd1 does not appear to be a major mediator of Mef2c function in the OFT. Consistent with this possibility, loss of function of Hand2, a putative target of Smyd1, mainly causes RV defects and is required for survival and expansion of RV cardiomyocytes [33]. Thus, the Mef2c -> Smyd1 -> Hand2 pathway appears to be a “lateral” branch of the SHF genetic network critical for the RV.
Isl1 can activate an Nkx2.5 enhancer synergistically with Tbx20 and Gata4 [34, 35]. A similar synergy is seen for a Mef2c enhancer [35]. Tbx20 is expressed in both the primary and the secondary heart field at early stages, and is enriched in the atria, outflow tract and valve primordia at later stages [34–38]. Tbx20 null mutants show reduced cell contribution from both heart fields to the heart, and chamber specification is abolished, arresting heart development at looping stage [34, 35, 39, 40]. Partial knockdown of the gene using RNA interference allowed the mutant embryos to develop until E13 [35]. In these embryos, the heart undergoes looping and chamber formation, however, the OFT and right ventricle development is impaired, resulting in persistent truncus arteriosus or double outlet right ventricle, and hypoplasitc right ventricle. These phenotypes indicate a dosage-dependent role of Tbx20 in OFT remodeling and right ventricle growth.
Links between the group Isl1-Foxh1-Mef2c and the next tier down (e.g. Tbx1) are only speculative at this point. One possibility is offered by the finding that Isl1 controls Shh signaling [28]. In turn, Shh signaling can regulate Tbx1 expression, probably through fork-head transcription factors [41]. While this is a rather tenuous “link”, from a phenotypic standpoint is not unreasonable because Shh−/− animals have OFT defects similar to those seen in Tbx1−/− animals [42], at least on surface. Foxc1 and Foxc2, possible mediators of Shh signal, can regulate Tbx1 expression through fox binding sites of the Tbx1 gene [33, 43]. Foxc1;Foxc2 combined mutants show reduced expression of Tbx1 and have severe OFT defects not unlike those seen in Tbx1−/− mutants [44].
Tbx1 is required for OFT development in humans and mice [45]. Recent data indicate that mesodermal expression of Tbx1 is necessary and sufficient (in a Tbx1 mutant background) to support normal septation, growth and alignment of the OFT [26]. Loss of function in the mesoderm causes substantial reduction of cell proliferation in the splanchnic and pharyngeal mesoderm already at E8.5, i.e. before anatomical abnormalities become apparent [26]. The interaction between Tbx1 and the FGF signaling will be discussed later. Microarray analyses have identified a number of genes altered in mutants. Perhaps of particular interest for the scope of this review, is the downregulation of genes coding for enzymes that degrade retinoic acid (RA) [46]. In addition, RA may negatively regulate Tbx1 expression [47] in a chick model. Furthermore, reduction of endogenous RA may partially rescue some of the Tbx1+/−;Crkl+/− mutant abnormalities [48] in mice. The latter results are exciting, but need to be extended in order to evaluate the full significance of RA reduction for the OFT phenotype of Tbx1−/− mutants.
Tbx1 may regulate other transcription factors known to play a role in the SHF. Pitx2 expression is partially overlapping with Tbx1 in the SHF. Most (~60%) Tbx1+/−; Pitx2+/− mutants suffer severe cardiac defects and die soon after birth [49]. The authors suggested that Tbx1 may regulate Pitx2 expression via a T-Box binding element (TBE) in an enhancer of the Pitx2 gene, close to an Nkx2.5 binding site. Using a luciferase assay, Tbx1 and Nkx2.5 were able to synergistically activate the Pitx2 enhancer, in a TBE-dependent manner. Hence the hypothesis that Tbx1 and Nkx2.5 interact to regulate the expression of Pitx2 in the SHF. The expression of Pitx2 is indeed reduced in the SHF of Tbx1−/− mutants [49]. Tbx1 is required in the Nkx2.5 and in the mesodermal domain of expression for OFT development [21, 26]. Interestingly, however, Nkx2.5 and Tbx1 do not interact in vivo [21, 26].
3.2. Signaling systems
Isl1 loss of function is associated with downregulation of ligands of several signaling systems, FGF, BMP, SHH [17, 28]. However, Isl1 expression is downregulated in embryos that lack Fgf8 in the mesoderm [50]. Suggesting an “upstream” role for the FGF signaling. Early ablation of Fgf8 in the mesoderm, using the Mesp1Cre driver, causes severe heart tube and OFT formation defects. However, a later and more restricted mesodermal ablation, driven by Mef2c-AHFCre, only resulted in OFT alignment defects. Whether this dramatic difference is due to different timing or different pattern of ablation, remains to be established using a timed-ablation approach. Nevertheless, it is clear that Fgf8 has multiple functions in the SHF. A complex and extensive set of tissue-specific ablation experiments led to propose that endodermal expression of Fgf8 is required for septation of the OFT, while expression in the SHF is required for OFT alignment [50]. This is an attractive and “straightforward” interpretation of the data, also consistent with the finding that the splanchnic mesoderm is the critical tissue targeted by Fgf8 signal during SHF development [51]. Consistent with this view, Tbx1 loss of function, is required for septation and alignment of the OFT, and is required for Fgf8 expression in the splanchnic/SHF mesoderm and endoderm [26, 52]. However, mesodermal deletion of Tbx1, which ablates Fgf8 expression in the splanchnic mesoderm but not in the endoderm, is sufficient to cause OFT and alignment defects. Conversely, reactivation of Tbx1 expression in the mesoderm of mutant embryos is sufficient to rescue both the septation and alignment defects. Thus, Fgf8 expression is not linearly linked with the Tbx1 mutant phenotype [26]. Indeed, an Fgf8 cDNA knocked into the Tbx1 locus does not rescue the OFT defects of Tbx1 null embryos [53]. A possible explanation of these results is if Tbx1 is not only involved in the regulation of ligand expression, which is now fairly solidly established [26, 52, 54, 55], but also in the regulation of the of FGF signal transduction, in some yet to be established manner. The noted downregulation of Fgfr1 expression in the Tbx1−/− embryos [50] is a clue that needs to be followed up.
Recent data obtained in chick, add another element to Fgf8 regulation in OFT morphogenesis, that is neural crest-derived cells (NCCs). NCCs can regulate, negatively, Fgf8 signaling. Neural crest ablation causes OFT defects and excessive Fgf8 signal in the SHF. A block of the Fgf8 signal can significantly ameliorate the OFT phenotype in neural crest-ablated chick embryos [56]. The exact molecular mechanisms underlying this phenomenon are not yet clear, but these results should inspire caution when interpreting the OFT phenotype of mutants where both the SHF and NCCs are affected. This is the case for most if not all the mutants discussed here.
The other signaling system widely studied in the context of OFT development is the BMP signaling [57]. BMP signaling may need to be suppressed in the early phases of OFT development. Chordin encodes one of the antagonists of BMP proteins [58] expressed early in mouse embryogenesis. Homozygous loss of Chordin in mice causes OFT septation defects very similar to those caused by Tbx1 mutation [59]. In situ hybridization revealed that the expression of Tbx1 and Fgf8 in the pharyngeal area was reduced or absent in Chordin null mutants. Hence the suggestion that Chordin may function upstream of Tbx1. However, the molecular cascade by which Chordin regulates Tbx1 expression is unknown.
BMP signaling is also required later in OFT morphogenesis, probably through a cross-talk between the NCCs migrated into the OFT (that express BMP receptors) and the muscle layer (that express ligands). This is supported by BMP receptor modification or tissue-specific gene ablation experiments [60–62], as well al by tissue specific ablation of the gene encoding the ligand Bmp4 [63].
An attempt to integrate the genetic interactions, established or suggested by the available data, is shown in Fig. 2.
Fig. 2.
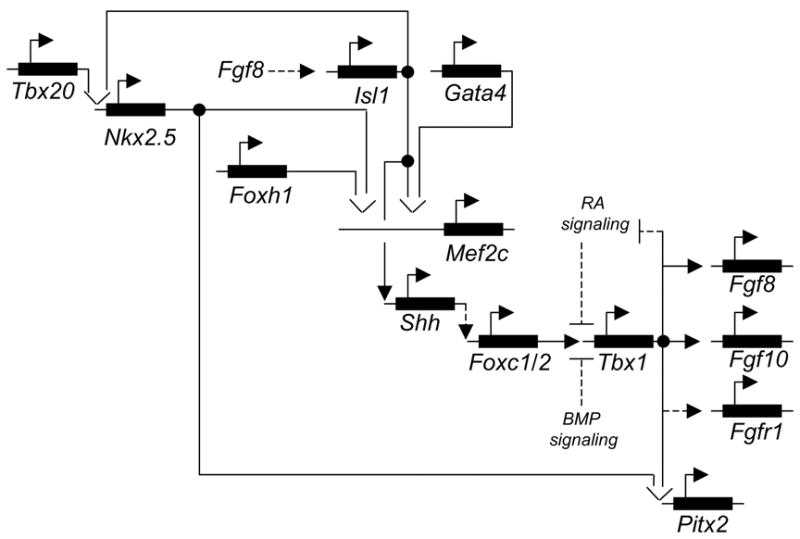
A putative genetic network operating in the second heart lineage. Dashed lines indicate the most speculative links.
4. Concluding remarks
While it is still unclear whether or not the notion of cardiac lineage will provide a key for resolving genetic networks, cardiogenic cell lineage distinction provides at least an element for higher resolution analysis of heart development. The same can be said for the identification of critical tissues, source or receivers of signals, and expressors of critical transcription factors. Furthermore, identification of critical time windows for gene function will provide an additional dimension for integration of functional data. Currently, is not always possible to integrate tissue-specific requirement data generated by different groups, because of the use of different Cre drivers. This situation is improving, though, as certain Cre drivers are used more and more commonly. Concerning time-specific requirements, there are just not enough data yet. As reagents are currently available, it is auspicable that more and more timed-ablation data will be produced for critical cardiogenic genes.
While a number of transcription factors and signaling molecules critical for SHF development are known, there are significant gaps in our understanding of molecular and cell biology mechanisms that guide SHF cells, in their expansion migration and differentiation processes, from the splanchnic and pharyngeal mesoderm into the heart. For example, one of the cell phenotypes most commonly cited as abnormal in SHF mutants, is cell proliferation and, perhaps to a lesser extent, cell survival. Thus, an important issue to address is what keeps cardiomyocyte progenitors in the SHF proliferating and thus able to provide cells to the heart for a relatively long developmental time. This issue is relevant not only for research in congenital heart disease but also in cardiac regeneration.
Acknowledgments
The authors wish to apologize for the many relevant references that have not been cited in the interest of space. Research in the laboratory of the authors is funded by the National Institutes of Health, USA, and by the Telethon Foundation, Italy.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Hoffman JI. Incidence of congenital heart disease: I. Postnatal incidence. Pediatr Cardiol. 1995;16:103–13. doi: 10.1007/BF00801907. [DOI] [PubMed] [Google Scholar]
- 2.Hoffman JI. Incidence of congenital heart disease: II. Prenatal incidence. Pediatr Cardiol. 1995;16:155–65. doi: 10.1007/BF00794186. [DOI] [PubMed] [Google Scholar]
- 3.Meilhac SM, Esner M, Kelly RG, Nicolas JF, Buckingham ME. The clonal origin of myocardial cells in different regions of the embryonic mouse heart. Dev Cell. 2004;6:685–98. doi: 10.1016/s1534-5807(04)00133-9. [DOI] [PubMed] [Google Scholar]
- 4.Garcia-Martinez V, Schoenwolf GC. Primitive-streak origin of the cardiovascular system in avian embryos. Dev Biol. 1993;159:706–19. doi: 10.1006/dbio.1993.1276. [DOI] [PubMed] [Google Scholar]
- 5.Tam PP, Parameswaran M, Kinder SJ, Weinberger RP. The allocation of epiblast cells to the embryonic heart and other mesodermal lineages: the role of ingression and tissue movement during gastrulation. Development. 1997;124:1631–42. doi: 10.1242/dev.124.9.1631. [DOI] [PubMed] [Google Scholar]
- 6.Rosenquist GC. Location and movements of cardiogenic cells in the chick embryo: the heart-forming portion of the primitive streak. Dev Biol. 1970;22:461–75. doi: 10.1016/0012-1606(70)90163-6. [DOI] [PubMed] [Google Scholar]
- 7.Hutson MR, Kirby ML. Neural crest and cardiovascular development: a 20-year perspective. Birth Defects Res C Embryo Today. 2003;69:2–13. doi: 10.1002/bdrc.10002. [DOI] [PubMed] [Google Scholar]
- 8.Brand T. Heart development: molecular insights into cardiac specification and early morphogenesis. Dev Biol. 2003;258:1–19. doi: 10.1016/s0012-1606(03)00112-x. [DOI] [PubMed] [Google Scholar]
- 9.Cripps RM, Olson EN. Control of cardiac development by an evolutionarily conserved transcriptional network. Dev Biol. 2002;246:14–28. doi: 10.1006/dbio.2002.0666. [DOI] [PubMed] [Google Scholar]
- 10.Viragh S, Challice CE. Origin and differentiation of cardiac muscle cells in the mouse. J Ultrastruct Res. 1973;42:1–24. doi: 10.1016/s0022-5320(73)80002-4. [DOI] [PubMed] [Google Scholar]
- 11.de la Cruz MV, Sanchez Gomez C, Arteaga MM, Arguello C. Experimental study of the development of the truncus and the conus in the chick embryo. J Anat. 1977;123:661–86. [PMC free article] [PubMed] [Google Scholar]
- 12.Waldo KL, Kumiski DH, Wallis KT, Stadt HA, Hutson MR, Platt DH, et al. Conotruncal myocardium arises from a secondary heart field. Development. 2001;128:3179–88. doi: 10.1242/dev.128.16.3179. [DOI] [PubMed] [Google Scholar]
- 13.Mjaatvedt CH, Nakaoka T, Moreno-Rodriguez R, Norris RA, Kern MJ, Eisenberg CA, et al. The outflow tract of the heart is recruited from a novel heart-forming field. Dev Biol. 2001;238:97–109. doi: 10.1006/dbio.2001.0409. [DOI] [PubMed] [Google Scholar]
- 14.Kelly RG, Brown NA, Buckingham ME. The arterial pole of the mouse heart forms from Fgf10-expressing cells in pharyngeal mesoderm. Dev Cell. 2001;1:435–40. doi: 10.1016/s1534-5807(01)00040-5. [DOI] [PubMed] [Google Scholar]
- 15.Buckingham M, Meilhac S, Zaffran S. Building the mammalian heart from two sources of myocardial cells. Nat Rev Genet. 2005;6:826–35. doi: 10.1038/nrg1710. [DOI] [PubMed] [Google Scholar]
- 16.Abu-Issa R, Waldo K, Kirby ML. Heart fields: one, two or more? Dev Biol. 2004;272:281–5. doi: 10.1016/j.ydbio.2004.05.016. [DOI] [PubMed] [Google Scholar]
- 17.Cai CL, Liang X, Shi Y, Chu PH, Pfaff SL, Chen J, et al. Isl1 identifies a cardiac progenitor population that proliferates prior to differentiation and contributes a majority of cells to the heart. Dev Cell. 2003;5:877–89. doi: 10.1016/s1534-5807(03)00363-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dodou E, Verzi MP, Anderson JP, Xu SM, Black BL. Mef2c is a direct transcriptional target of ISL1 and GATA factors in the anterior heart field during mouse embryonic development. Development. 2004;131:3931–42. doi: 10.1242/dev.01256. [DOI] [PubMed] [Google Scholar]
- 19.Verzi MP, McCulley DJ, De Val S, Dodou E, Black BL. The right ventricle, outflow tract, and ventricular septum comprise a restricted expression domain within the secondary/anterior heart field. Dev Biol. 2005;287:134–45. doi: 10.1016/j.ydbio.2005.08.041. [DOI] [PubMed] [Google Scholar]
- 20.Xu H, Cerrato F, Baldini A. Timed mutation and cell-fate mapping reveal reiterated roles of Tbx1 during embryogenesis, and a crucial function during segmentation of the pharyngeal system via regulation of endoderm expansion. Development. 2005;132:4387–95. doi: 10.1242/dev.02018. [DOI] [PubMed] [Google Scholar]
- 21.Xu H, Morishima M, Wylie JN, Schwartz RJ, Bruneau BG, Lindsay EA, et al. Tbx1 has a dual role in the morphogenesis of the cardiac outflow tract. Development. 2004;131:3217–27. doi: 10.1242/dev.01174. [DOI] [PubMed] [Google Scholar]
- 22.Liu C, Liu W, Palie J, Lu MF, Brown NA, Martin JF. Pitx2c patterns anterior myocardium and aortic arch vessels and is required for local cell movement into atrioventricular cushions. Development. 2002;129:5081–91. doi: 10.1242/dev.129.21.5081. [DOI] [PubMed] [Google Scholar]
- 23.Ai D, Liu W, Ma L, Dong F, Lu MF, Wang D, et al. Pitx2 regulates cardiac left-right asymmetry by patterning second cardiac lineage-derived myocardium. Dev Biol. 2006;296:437–49. doi: 10.1016/j.ydbio.2006.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Laugwitz KL, Moretti A, Lam J, Gruber P, Chen Y, Woodard S, et al. Postnatal isl1+ cardioblasts enter fully differentiated cardiomyocyte lineages. Nature. 2005;433:647–53. doi: 10.1038/nature03215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ward C, Stadt H, Hutson M, Kirby ML. Ablation of the secondary heart field leads to tetralogy of Fallot and pulmonary atresia. Dev Biol. 2005;284:72–83. doi: 10.1016/j.ydbio.2005.05.003. [DOI] [PubMed] [Google Scholar]
- 26.Zhang Z, Huynh T, Baldini A. Mesodermal expression of Tbx1 is necessary and sufficient for pharyngeal arch and cardiac outflow tract development. Development. 2006;133:3587–3595. doi: 10.1242/dev.02539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Benson DW, Silberbach GM, Kavanaugh-McHugh A, Cottrill C, Zhang Y, Riggs S, et al. Mutations in the cardiac transcription factor NKX2.5 affect diverse cardiac developmental pathways. J Clin Invest. 1999;104:1567–73. doi: 10.1172/JCI8154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lin L, Bu L, Cai CL, Zhang X, Evans S. Isl1 is upstream of sonic hedgehog in a pathway required for cardiac morphogenesis. Dev Biol. 2006;295:756–63. doi: 10.1016/j.ydbio.2006.03.053. [DOI] [PubMed] [Google Scholar]
- 29.Yang L, Cai CL, Lin L, Qyang Y, Chung C, Monteiro RM, et al. Isl1Cre reveals a common Bmp pathway in heart and limb development. Development. 2006;133:1575–85. doi: 10.1242/dev.02322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.von Both I, Silvestri C, Erdemir T, Lickert H, Walls JR, Henkelman RM, et al. Foxh1 is essential for development of the anterior heart field. Dev Cell. 2004;7:331–45. doi: 10.1016/j.devcel.2004.07.023. [DOI] [PubMed] [Google Scholar]
- 31.Phan D, Rasmussen TL, Nakagawa O, McAnally J, Gottlieb PD, Tucker PW, et al. BOP, a regulator of right ventricular heart development, is a direct transcriptional target of MEF2C in the developing heart. Development. 2005;132:2669–78. doi: 10.1242/dev.01849. [DOI] [PubMed] [Google Scholar]
- 32.Gottlieb PD, Pierce SA, Sims RJ, Yamagishi H, Weihe EK, Harriss JV, et al. Bop encodes a muscle-restricted protein containing MYND and SET domains and is essential for cardiac differentiation and morphogenesis. Nat Genet. 2002;31:25–32. doi: 10.1038/ng866. [DOI] [PubMed] [Google Scholar]
- 33.Yamagishi H, Yamagishi C, Nakagawa O, Harvey RP, Olson EN, Srivastava D. The combinatorial activities of Nkx2.5 and dHAND are essential for cardiac ventricle formation. Dev Biol. 2001;239:190–203. doi: 10.1006/dbio.2001.0417. [DOI] [PubMed] [Google Scholar]
- 34.Stennard FA, Harvey RP. T-box transcription factors and their roles in regulatory hierarchies in the developing heart. Development. 2005;132:4897–910. doi: 10.1242/dev.02099. [DOI] [PubMed] [Google Scholar]
- 35.Takeuchi JK, Mileikovskaia M, Koshiba-Takeuchi K, Heidt AB, Mori AD, Arruda EP, et al. Tbx20 dose-dependently regulates transcription factor networks required for mouse heart and motoneuron development. Development. 2005;132:2463–74. doi: 10.1242/dev.01827. [DOI] [PubMed] [Google Scholar]
- 36.Kraus F, Haenig B, Kispert A. Cloning and expression analysis of the mouse T-box gene Tbx18. Mech Dev. 2001;100:83–6. doi: 10.1016/s0925-4773(00)00494-9. [DOI] [PubMed] [Google Scholar]
- 37.Lincoln J, Alfieri CM, Yutzey KE. Development of heart valve leaflets and supporting apparatus in chicken and mouse embryos. Dev Dyn. 2004;230:239–50. doi: 10.1002/dvdy.20051. [DOI] [PubMed] [Google Scholar]
- 38.Plageman TF, Jr, Yutzey KE. Differential expression and function of Tbx5 and Tbx20 in cardiac development. J Biol Chem. 2004;279:19026–34. doi: 10.1074/jbc.M314041200. [DOI] [PubMed] [Google Scholar]
- 39.Singh MK, Petry M, Haenig B, Lescher B, Leitges M, Kispert A. The T-box transcription factor Tbx15 is required for skeletal development. Mech Dev. 2005;122:131–44. doi: 10.1016/j.mod.2004.10.011. [DOI] [PubMed] [Google Scholar]
- 40.Cai CL, Zhou W, Yang L, Bu L, Qyang Y, Zhang X, et al. T-box genes coordinate regional rates of proliferation and regional specification during cardiogenesis. Development. 2005;132:2475–87. doi: 10.1242/dev.01832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yamagishi H, Maeda J, Hu T, McAnally J, Conway SJ, Kume T, et al. Tbx1 is regulated by tissue-specific forkhead proteins through a common Sonic hedgehog-responsive enhancer. Genes Dev. 2003;17:269–81. doi: 10.1101/gad.1048903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Washington Smoak I, Byrd NA, Abu-Issa R, Goddeeris MM, Anderson R, Morris J, et al. Sonic hedgehog is required for cardiac outflow tract and neural crest cell development. Dev Biol. 2005;283:357–72. doi: 10.1016/j.ydbio.2005.04.029. [DOI] [PubMed] [Google Scholar]
- 43.Maeda J, Yamagishi H, McAnally J, Yamagishi C, Srivastava D. Tbx1 is regulated by forkhead proteins in the secondary heart field. Dev Dyn. 2006;235:701–10. doi: 10.1002/dvdy.20686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Seo S, Kume T. Forkhead transcription factors, Foxc1 and Foxc2, are required for the morphogenesis of the cardiac outflow tract. Dev Biol. 2006;296:421–36. doi: 10.1016/j.ydbio.2006.06.012. [DOI] [PubMed] [Google Scholar]
- 45.Baldini A. Dissecting contiguous gene defects: TBX1. Curr Opin Genet Dev. 2005;15:279–84. doi: 10.1016/j.gde.2005.03.001. [DOI] [PubMed] [Google Scholar]
- 46.Ivins S, Lammerts van Beuren K, Roberts C, James C, Lindsay E, Baldini A, et al. Microarray analysis detects differentially expressed genes in the pharyngeal region of mice lacking Tbx1. Dev Biol. 2005 doi: 10.1016/j.ydbio.2005.06.026. [DOI] [PubMed] [Google Scholar]
- 47.Roberts C, Ivins SM, James CT, Scambler PJ. Retinoic acid down-regulates Tbx1 expression in vivo and in vitro. Dev Dyn. 2005;232:928–38. doi: 10.1002/dvdy.20268. [DOI] [PubMed] [Google Scholar]
- 48.Guris DL, Duester G, Papaioannou VE, Imamoto A. Dose-Dependent Interaction of Tbx1 and Crkl and Locally Aberrant RA Signaling in a Model of del22q11 Syndrome. Dev Cell. 2006;10:81–92. doi: 10.1016/j.devcel.2005.12.002. [DOI] [PubMed] [Google Scholar]
- 49.Nowotschin S, Liao J, Gage PJ, Epstein JA, Campione M, Morrow BE. Tbx1 affects asymmetric cardiac morphogenesis by regulating Pitx2 in the secondary heart field. Development. 2006;133:1565–73. doi: 10.1242/dev.02309. [DOI] [PubMed] [Google Scholar]
- 50.Park EJ, Ogden LA, Talbot A, Evans S, Cai CL, Black BL, et al. Required, tissue-specific roles for Fgf8 in outflow tract formation and remodeling. Development. 2006;133:2419–33. doi: 10.1242/dev.02367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ilagan R, Abu-Issa R, Brown D, Yang YP, Jiao K, Schwartz RJ, et al. Fgf8 is required for anterior heart field development. Development. 2006;133:2435–45. doi: 10.1242/dev.02408. [DOI] [PubMed] [Google Scholar]
- 52.Vitelli F, Taddei I, Morishima M, Meyers EN, Lindsay EA, Baldini A. A genetic link between Tbx1 and Fibroblast Growth Factor signaling. Development. 2002;129:4605–4611. doi: 10.1242/dev.129.19.4605. [DOI] [PubMed] [Google Scholar]
- 53.Vitelli F, Zhang Z, Huynh T, Sobotka A, Mupo A, Baldini A. Fgf8 expression in the Tbx1 domain causes skeletal abnormalities and modifies the aortic arch but not the outflow tract phenotype of Tbx1 mutants. Dev Biol. 2006;295:559–70. doi: 10.1016/j.ydbio.2006.03.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Brown CB, Wenning JM, Lu MM, Epstein DJ, Meyers EN, Epstein JA. Cre-mediated excision of Fgf8 in the Tbx1 expression domain reveals a critical role for Fgf8 in cardiovascular development in the mouse. Dev Biol. 2004;267:190–202. doi: 10.1016/j.ydbio.2003.10.024. [DOI] [PubMed] [Google Scholar]
- 55.Hu T, Yamagishi H, Maeda J, McAnally J, Yamagishi C, Srivastava D. Tbx1 regulates fibroblast growth factors in the anterior heart field through a reinforcing autoregulatory loop involving forkhead transcription factors. Development. 2004;131:5491–502. doi: 10.1242/dev.01399. [DOI] [PubMed] [Google Scholar]
- 56.Hutson MR, Zhang P, Stadt HA, Sato AK, Li YX, Burch J, et al. Cardiac arterial pole alignment is sensitive to FGF8 signaling in the pharynx. Dev Biol. 2006;295:486–97. doi: 10.1016/j.ydbio.2006.02.052. [DOI] [PubMed] [Google Scholar]
- 57.Chen D, Zhao M, Mundy GR. Bone morphogenetic proteins. Growth Factors. 2004;22:233–41. doi: 10.1080/08977190412331279890. [DOI] [PubMed] [Google Scholar]
- 58.Piccolo S, Sasai Y, Lu B, De Robertis EM. Dorsoventral patterning in Xenopus: inhibition of ventral signals by direct binding of chordin to BMP-4. Cell. 1996;86:589–98. doi: 10.1016/s0092-8674(00)80132-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Bachiller D, Klingensmith J, Shneyder N, Tran U, Anderson R, Rossant J, et al. The role of chordin/Bmp signals in mammalian pharyngeal development and DiGeorge syndrome. Development. 2003;130:3567–78. doi: 10.1242/dev.00581. [DOI] [PubMed] [Google Scholar]
- 60.Delot EC, Bahamonde ME, Zhao M, Lyons KM. BMP signaling is required for septation of the outflow tract of the mammalian heart. Development. 2003;130:209–20. doi: 10.1242/dev.00181. [DOI] [PubMed] [Google Scholar]
- 61.Stottmann RW, Choi M, Mishina Y, Meyers EN, Klingensmith J. BMP receptor IA is required in mammalian neural crest cells for development of the cardiac outflow tract and ventricular myocardium. Development. 2004;131:2205–18. doi: 10.1242/dev.01086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kaartinen V, Dudas M, Nagy A, Sridurongrit S, Lu MM, Epstein JA. Cardiac outflow tract defects in mice lacking ALK2 in neural crest cells. Development. 2004;131:3481–90. doi: 10.1242/dev.01214. [DOI] [PubMed] [Google Scholar]
- 63.Liu W, Selever J, Wang D, Lu MF, Moses KA, Schwartz RJ, et al. Bmp4 signaling is required for outflow-tract septation and branchial-arch artery remodeling. Proc Natl Acad Sci U S A. 2004;101:4489–94. doi: 10.1073/pnas.0308466101. [DOI] [PMC free article] [PubMed] [Google Scholar]