

Abstract
The identification of the Philadelphia chromosome in cells from individuals with chronic myelogenous leukemia (CML) led to the recognition that the BCR-ABL tyrosine kinase causes CML. This in turn led to the development of imatinib mesylate, a clinically successful inhibitor of the BCR-ABL kinase. Incorporating the use of markers of BCR-ABL kinase inhibition into clinical trials led to the realization that imatinib-resistant kinase domain mutations are the major cause of relapse during imatinib therapy and the subsequent development of new inhibitors to treat CML patients. The development of imatinib validates an emerging paradigm in cancer, in which a tumor is defined by genetic abnormalities and effective therapies are developed that target events critical to the growth and survival of a specific tumor.
Introduction
The study of chronic myelogenous leukemia (CML) affords a unique opportunity to understand the process of cancer development. This was the first human cancer in which a consistent genetic abnormality was demonstrated to cause the disease. For CML, this is a shortened chromosome 22, which was first reported in 1960 (1). This chromosomal abnormality, referred to as the Philadelphia (Ph) chromosome, was later recognized to result from a reciprocal (9;22) translocation (2); that is, genetic material from the ends of chromosomes 9 and 22 is exchanged.
The molecular consequences of the (9;22) translocation are to fuse the ABL tyrosine kinase gene from chromosome 9 to the breakpoint cluster region (BCR) gene on chromosome 22 (3–5). The resulting fusion protein, BCR–v-abl Abelson murine leukemia viral oncogene homolog (BCR-ABL), functions as an oncogenic tyrosine kinase that causes CML (Figure 1) (6, 7). The oncogenic capabilities of BCR-ABL have been demonstrated in mouse models in which BCR-ABL expression is sufficient to cause leukemia (8–10).
Figure 1. The phenotype and genotype of CML.
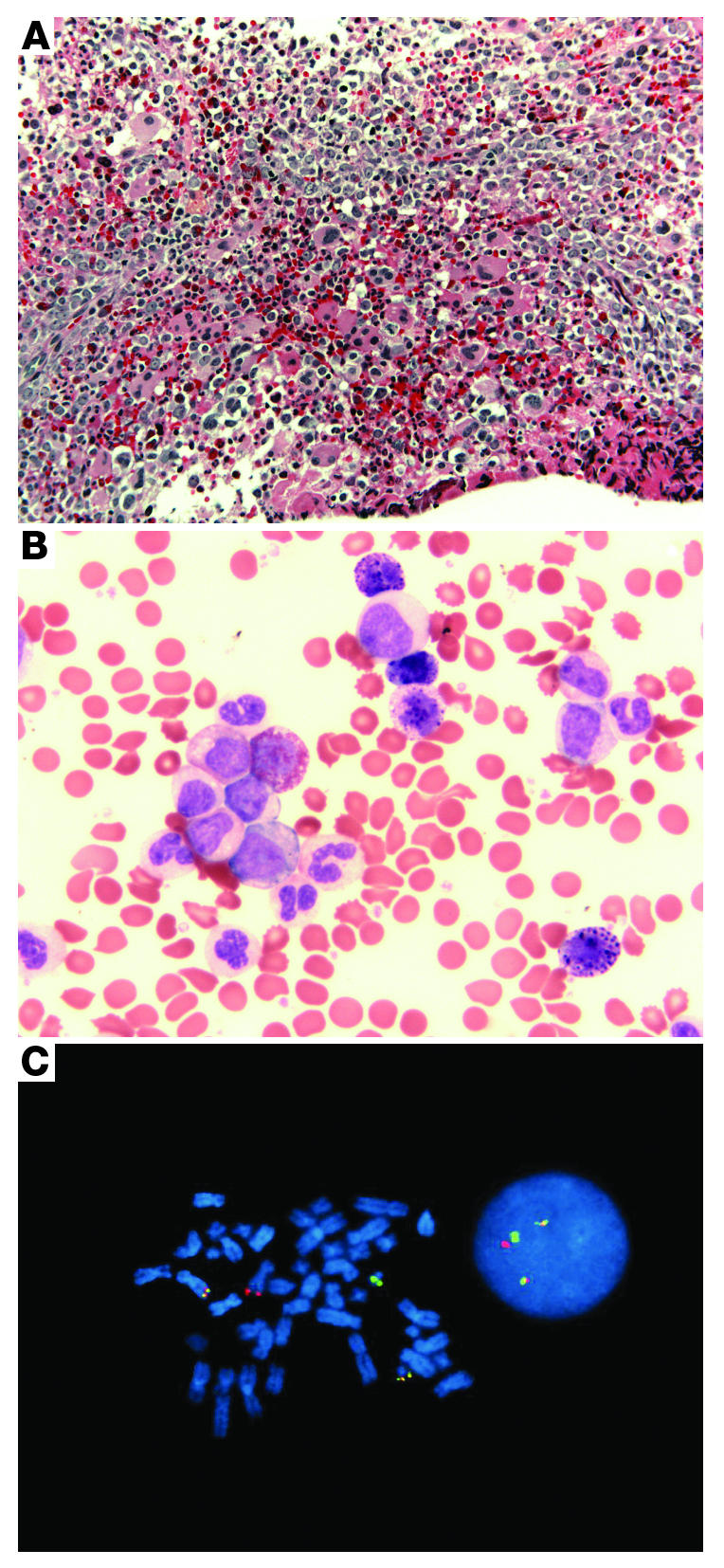
(A) A bone marrow biopsy from a patient with CML shows the typical hypercellularity with granulocytic and megakaryocytic hyperplasia (original magnification, ×200). (B) The peripheral blood is characterized by a full spectrum of myeloid cells, including immature myeloid cells with rare blasts. Basophilia is also observed (original magnification, ×630). (C) Dual-color, dual-fusion FISH displaying BCR-ABL signals in bone marrow cells in metaphase (left) and interphase (right). The red fluorescent probe is specific for ABL, while the green probe is specific for BCR. Yellow signals the presence of BCR-ABL and ABL-BCR fusions.
CML also functions as a dissectible model for other malignancies with clinically apparent phases that coincide with genetic progression of the disease. In the chronic phase of CML, the (9;22) translocation arises in a hematopoietic stem cell (11) and seems to be the sole genetic abnormality (12). The chronic phase is characterized by a massive increase in cells of the myeloid lineage, but these cells mature and function normally. Prior to recent treatment advances, the chronic phase lasted three to five years. Over time, the disease transforms into an invariably fatal acute leukemia (known as blast crisis) of either myeloid or lymphoid phenotype (13), often with a recognizable intermediate stage termed the accelerated phase. The accelerated phase and blast crisis are collectively referred to as advanced phase disease. Although it is clear that additional genetic abnormalities are responsible for blast crisis (14), the specific genetics lesions that cause disease progression are poorly characterized.
The clinical success of imatinib (also known as Gleevec, formerly STI571), an inhibitor of the BCR-ABL kinase, confirmed the critical dependence of CML cells on BCR-ABL tyrosine kinase activity for their survival, further establishing CML as a paradigm for the contributions of basic science to patient treatment. Here we will review the preclinical and clinical development of imatinib and discuss the challenges of treating patients with accelerated phase and blast crisis; the issue of imatinib resistance and the development of second-generation inhibitors of BCR-ABL will be discussed; and finally, the application of the imatinib paradigm to other malignancies will be reviewed.
Beginning of the imatinib era
Imatinib is a small organic molecule synthesized for the purpose of protein kinase inhibition (15, 16). It has activity against all the ABL tyrosine kinases, including BCR-ABL, cellular homologue of the Abelson murine leukemia viral oncogene product (c-ABL), v-ABL, and Abelson-related gene (ARG) (15–17). In addition to the ABL tyrosine kinases, other kinases inhibited by imatinib are PDGFR-α, PDGFR-β, and KIT (15, 16, 18). Given the critical role of tyrosine kinases in the regulation of cell growth and known activation in several cancers, such as BCR-ABL in CML and HER2 overexpression in breast cancer, it was hypothesized that specific inhibitors of these protein kinases might be effective anticancer agents (19).
Beginning in the late 1980s, scientists at Ciba-Geigy (now Novartis) performed high-throughput screens of chemical libraries searching for compounds with kinase inhibitory activity. From this time-consuming approach, a lead compound was identified; its inhibitory activity against PDGFR was optimized by synthesizing a series of chemically related compounds and analyzing their relationship between structure and activity. The most potent molecules in the series were dual inhibitors of the PDGFR and ABL kinases. Of the several compounds generated from this program, imatinib emerged as the lead compound for clinical development because of its superior in vitro selectivity against CML cells and its drug-like properties, including pharmacokinetic and formulation properties (20).
In a critical set of preclinical experiments, imatinib specifically killed cell lines expressing BCR-ABL and did not kill the parental cell lines from which they were derived (16). Imatinib also inhibited tumor formation by cells expressing BCR-ABL in vivo (16). In colony-forming assays of peripheral blood or bone marrow from patients with CML, imatinib caused a 92%–98% decrease in the number of colonies expressing BCR-ABL that formed (16). These experiments suggested that cells expressing BCR-ABL become addicted to that oncogene and are dependent on it not only for proliferation but also for survival. Thus, inhibition of the BCR-ABL tyrosine kinase resulted in apoptosis instead of merely reverting the cells to normal. A mouse model with inducible BCR-ABL expression has further extended this notion by demonstrating full dependence on oncogene expression in maintaining leukemia (21).
In 1998, a phase I clinical trial with imatinib was begun (22). This initial phase I trial was a dose escalation trial that enrolled 83 patients with chronic phase CML. Their clinical features were typical of the disease and were as follows: their ages ranged from 19–76 years, and they had a median time from diagnosis of 3.8 years. At study entry, their white blood cell counts ranged from 9,400 to 199,000/mm3 (normal is 4,000 to 11,000/mm3), and the Ph chromosome was present in all leukemic cells in each patient. As this was a phase I clinical trial, all patients were required to have received prior therapy with the current standard of care, interferon-α, which was either no longer controlling their disease (n = 70) or was poorly tolerated (n = 13). Interferon-α therapy had previously been shown to prolong survival by a median of 20 months over the natural disease course of 3 to 5 years (23). However, the mechanism of action of interferon-α is poorly understood, and its side effects are significant. Therefore, the patients enrolled in the phase I clinical trial of imatinib had late chronic phase CML with no standard treatment options available. Although there was optimism at the beginning of the trial based on the promising preclinical experiments, there was also concern that blockade of the normal, ubiquitously expressed c-ABL, PDGFR, and/or KIT would lead to unforeseen toxicities. After 29 patients were enrolled, therapeutic doses of 300 mg or more per day were reached, and 53 of 54 patients achieved a complete hematologic response (normal blood counts with a white blood cell count of less than 10,000/mm3 and platelet counts of less than 450,000/mm3). Side effects of imatinib were relatively minimal (22). Based on the results of the phase I trial, the use of imatinib was expanded to large international phase II and phase III clinical trials.
Cumulative experience with imatinib
The experience with imatinib has yielded a wealth of information, including data from a randomized clinical trial with five years of follow-up (the International Randomized Study of Interferon and STI571 [IRIS] study) (23), a crystal structure of the ABL kinase domain in complex with imatinib (24, 25), and important insights into the mechanism of imatinib resistance. The IRIS trial was initiated in 2000 for patients newly diagnosed with CML in the chronic phase (26). Initially designed as a comparison of imatinib to interferon-α plus cytarabine, the substantial superiority of imatinib resulted in study results being disclosed early and most patients being crossed over to the imatinib arm. Accordingly, this study is now a long-term follow-up study of patients who received imatinib as initial therapy, with the median follow-up now being 5 years. The overall survival at 5 years is 89% (23). Previous experience found 5-year survival rates for patients treated with interferon-α plus cytarabine to be 68%–70% (27, 28). An estimated 93% of imatinib-treated patients remain free from disease progression to the accelerated phase or blast crisis (26). An additional 6% of patients have shown some evidence of loss of response to imatinib, but their disease has not progressed to the accelerated phase or blast crisis (26). Most of the side effects of imatinib are mild to moderate, with the most common being edema, muscle cramps, diarrhea, nausea, skin rashes, and myelosuppression. Thus, imatinib therapy has substantially increased survival for patients with CML while simultaneously greatly decreasing side effects.
Imatinib was the first small-molecule kinase inhibitor tested in patients, and its use has illuminated some fundamental characteristics of CML biology as well as helped to establish guiding principles for kinase inhibitor use in general. Most patients on imatinib therapy achieve a complete cytogenetic response (26), which is defined as the absence of the Ph chromosome in 20 bone marrow cells in metaphase. Despite the high frequency of complete cytogenetic responses, BCR-ABL transcripts are detectable in most patients by RT-PCR. Even patients in whom BCR-ABL is undetectable by this technique (classified as a complete molecular response) typically relapse if imatinib therapy is discontinued (29, 30). Therefore, a picture of disease response has emerged in which BCR-ABL kinase inhibition triggers apoptosis in differentiated cells and halts progenitor cell proliferation but does not eliminate hematopoietic stem cells that harbor the translocation (31, 32). The kinetics of these effects fit a biphasic mathematical model based on BCR-ABL transcript reduction in patients (33). A rapid initial decline occurs as differentiated cells undergo apoptosis, followed by a slower decline associated with progenitor cell turnover. Whether the hematopoietic stem cells expressing BCR-ABL are resistant to imatinib or continue to be slowly eliminated over time is not clear. Of interest, another mathematical model has recently been reported that also features a biphasic imatinib response but instead raises the possibility of an additional slow decline in the number of hematopoietic stem cells expressing BCR-ABL when these cells enter the cell cycle (34). Whether hematopoietic stem cells expressing BCR-ABL are resistant to imatinib or are sensitive only upon cell division is not clear. Insights into the mechanism that allows leukemic stem cells to persist during therapy would be an important contribution to the understanding of leukemogenesis and kinase inhibitor treatment.
Imatinib in advanced Ph chromosome–positive leukemia
Soon after the first trial of imatinib for the treatment of chronic-phase CML was initiated, the efficacy of imatinib in treating two more challenging manifestations of BCR-ABL–positive leukemia — blast crisis CML and Ph chromosome–positive acute lymphoblastic leukemia (ALL) — was investigated (35). Despite high response rates in these clinical trials, it quickly became apparent that most responses were short lived (35). Hematologic responses were seen in 55% (n = 38) of patients with blast crisis CML of myeloid phenotype and in 70% (n = 20) of patients with lymphoid blast crisis CML or Ph chromosome–positive ALL. Subsequent findings in larger phase II clinical trials confirmed these observations (36). Hematologic responses were observed in 52% of patients (n = 260), with a median response duration of 10 months (36). Of note, 48% of patients in this trial developed new cytogenetic abnormalities during treatment, demonstrating clonal evolution. The short duration of response highlighted the necessity of combination therapy in combating resistance. Recent studies of patients with Ph chromosome–positive ALL treated with combinations of imatinib and a standard chemotherapy have shown high response rates and suggest improved durability of responses as compared with previous clinical trials (37–40). As specific genetic abnormalities are identified in the more aggressive manifestations of Ph chromosome–positive disease (blast crisis CML and Ph chromosome–positive ALL), these will also become candidates for targeted therapeutic agents.
The association between genomic instability and CML progression has been recognized for many years. In 1976, it was documented that the leukemic cells of approximately 80% of patients in accelerated phase or blast crisis harbored additional recurrent chromosomal abnormalities (41). An interesting question is whether BCR-ABL itself has a causal role in the generation of secondary genetic abnormalities. BCR-ABL has been observed to translocate to the nucleus and disrupt signaling integral to checkpoint signaling from the ATR kinase following DNA damage, a checkpoint pathway that is essential for maintenance of genomic integrity (42). It has also been reported that BCR-ABL induces reactive oxygen species, which cause DNA double-strand breaks and point mutations (43–45). By either mechanism, it can be postulated that BCR-ABL is responsible for, or at least contributes to, the genomic instability that results in secondary abnormalities leading to blast crisis. This hypothesis would bode well for the BCR-ABL–centric approach that dominates current clinical efforts, as it suggests that kinase inhibition in CML might prevent or delay the development of blast crisis. This hypothesis is consistent with the clinical data showing that few patients have progressed to the advanced phases of the disease while undergoing imatinib therapy.
The genetic complexity of blast crisis CML implicates a multi-stage pathogenesis (14). BCR-ABL promotes the excess proliferation of myeloid cells during chronic phase and, as noted, may contribute to genetic instability, which leads to the additional mutations that drive disease progression. These additional mutations produce a block in cell differentiation or disable the key tumor suppressor pathways of p53 or Rb. Possible culprits that would block differentiation are transcription factors that are also commonly disrupted in acute myelogenous leukemia (AML). For example, the (3;21) translocation (observed in approximately 2% of patients with blast crisis CML) fuses the AML1 (RUNX1) core binding factor to the EVI1 transcriptional repressor (46). AML1 is essential to the differentiation of hematopoietic cells, and the AML1-EVI1 fusion protein blocks this process (47). In mice, coexpression of BCR-ABL and AML1-EVI1 results in AML (48). Although the cases of CML patients with AML1-EVI1 are few, it represents a general mechanism for disease progression. A recent study has examined the characteristics of the CML-initiating cells in advanced disease (49). By comparing cells from patients in blast crisis with those from individuals with chronic phase CML, it was found that granulocyte-macrophage progenitors from patients with blast crisis but not chronic phase CML displayed self-renewal capability (49). These data suggest that cells that normally do not have the capacity for self renewal can adopt stem cell–like properties during disease progression to blast crisis.
The challenge to optimizing therapy: imatinib resistance
Although most patients with chronic phase CML treated with imatinib have well controlled disease, some patients have relapsed and/or progressed to accelerated phase or blast crisis (26). Furthermore, most patients with advanced phases of CML initially respond to imatinib but subsequently relapse (35, 36). Therefore, the mechanisms of relapse emerged as a major question. The first insights into relapse mechanisms came from assays that evaluated BCR-ABL kinase inhibition. BCR-ABL is a constitutively active tyrosine kinase in which the N-terminal domains of the BCR signaling protein are joined to nearly the entire c-ABL kinase (50, 51). The functional consequence is the dysregulation of c-ABL by two intramolecular effects (Figure 2). The first is a loss-of-function effect in which an N-terminal region of c-ABL, referred to as the Cap domain (see Figure 2), is partially lost from the BCR-ABL fusion gene. This N-terminal Cap region is an important component of the intramolecular apparatus that inhibits the kinase activity of c-ABL (52, 53). The second effect is a gain-of-function resulting from the presence of a coiled-coil domain in BCR that functions to dimerize or tetramerize BCR-ABL (54). This domain is essential for the transforming properties of BCR-ABL (55–57). Collectively, these two effects yield a kinase that is constitutively phosphorylated (52, 56). The result is the alteration of the tightly regulated c-ABL kinase into a predominantly active kinase that phosphorylates numerous intracellular proteins (50). These phosphorylated proteins activate signaling pathways that control cell proliferation, including the PI3K, RAS, and STAT5 pathways (58–62). BCR-ABL also protects cells from apoptosis by upregulating BCLXL and by phosphorylation and inactivation of the proapoptotic molecule BAD (63, 64). One of the most heavily tyrosine phosphorylated proteins in CML patient samples is the SH2-SH3 domain–containing adaptor protein CRKL (65), and the level of CRKL phosphorylation was used in the clinical trials of imatinib to monitor the activity of the BCR-ABL kinase (22).
Figure 2. A model of the domains of the c-ABL and BCR-ABL proteins displays the functional consequences of the translocation.

(A) In c-ABL, the P-loop and the activation loop surround the active site for substrate phosphorylation (shaded). Under normal conditions c-ABL is inactive, with the SH2 and SH3 domains bound to the kinase domain, restricting its kinase activity (53). This conformation is stabilized by the cap domain, which is anchored to the kinase domain by a myristoyl group (jagged line) (52, 53). (B) In BCR-ABL, the myristoyl group of the cap domain is lost and is replaced by BCR, shown for simplicity as a single globular domain. This is presumed to destabilize binding of SH2 and SH3 to the kinase domain (56). The N terminus of BCR, the coiled-coil domain, forms a helical dimer (that also tetramerizes) and tethers individual BCR-ABL proteins together (54). This allows for transphosphorylation (P; yellow circles) of the kinase, which promotes the kinase to adopt an active conformation with a significant outward twist of the activation loop. (C) Imatinib binds to the active site of the kinase domain, freezing it in the inactive conformation, preventing BCR-ABL activation (25).
In evaluating mechanisms of relapse, a major breakthrough occurred with the observation that patients who relapsed displayed reactivation of BCR-ABL signaling, as assessed by CRKL phosphorylation. The reactivation of BCR-ABL was found to occur either by point mutation or gene amplification (66). Ensuing research found that the most common mechanism of BCR-ABL reactivation, occurring in 76 of 144 (53%) relapsed patients in the initial cohorts studied, was point mutation of the kinase domain (66–71). A spectrum of point mutations throughout the kinase domain has subsequently been characterized to confer decreased sensitivity to imatinib (Figure 2).
The understanding of relapses while on imatinib therapy has been further aided by the solution of the crystal structure of the ABL kinase domain in complex with imatinib. This crystal structure showed that imatinib bound the kinase in its inactive conformation (24, 25). Although the active conformations of different kinases share many common features, the inactive conformations of each have greater differences (25). This helps explain the specificity of imatinib. The most common imatinib-resistant mutations cluster around the active site cleft of the kinase domain and seem to have one of two effects on the ABL kinase domain–imatinib complex. The first is observed when the amino acid substitution occurs at contact points for imatinib binding (70). The characteristic mutation of this category is the change of threonine to the more bulky isoleucine at residue 315 in the heart of the imatinib-binding site. This was the first mutation described (66) and has proven to be the most difficult to treat. Other mutants at contact points for imatinib are F317L and F359V (70). The second category of kinase domain mutations is hypothesized to have conformational effects on the kinase, either favoring the active conformation to which imatinib cannot bind or decreasing the flexibility of the P-loop such that the conformational changes required for imatinib to bind this region cannot be adopted (70). These mutations include those in the P-loop, which bridges the ATP-binding pocket of the kinase domain (M244V, G250E, Q252H, Y253F/H, and E255K/V) and the activation loop (H396R/P). These mutations, along with M351T and E355G, are the most common in patients who relapse (72).
An issue that emerges from the study of patients who relapse while on imatinib therapy is whether imatinib induces BCR-ABL kinase domain mutations or whether the mutations are preexisting. Thus far, the clinical data is consistent with mutations preexisting therapy, with imatinib therapy selecting for resistant clones (26). For example, in patients with advanced disease, imatinib-resistant clones have been detected prior to the initiation of therapy (73). Although mutations are rarely detected in patients with chronic phase CML, it is notable that the rate of disease progression peaks in the first two years during imatinib therapy and then trends downward (26). If mutations precede imatinib therapy, the rate of emergence of resistance to the drug would depend on the size of the mutant clone at the start of therapy and its doubling time. As most mutated and unmutated BCR-ABL clones have similar doubling times (74), it would be predicted that patients harboring a mutant clone that is below the level of detection by PCR should be at highest risk of relapse over the first years of therapy. This prediction is consistent with the clinical data (26).
The second generation of BCR-ABL inhibitors
Shortly after the identification of imatinib-resistant mutations, several new inhibitors joined imatinib in the clinic. These inhibitors represent two different structural classes. The first class is exemplified by dasatinib (also known as Sprycel, formerly BMS-354825), a broad-spectrum kinase inhibitor that was developed as an inhibitor of SRC family kinases (75). One of the tyrosine kinase families that dasatinib inhibits are the ABL kinases, including c-ABL and BCR-ABL (75). It is more than 100-fold more potent than imatinib against the ABL kinases and retains this low nanomolar inhibitory activity against the common imatinib-resistant mutations, with the exception of the T315I mutation (76). Crystallography and nuclear magnetic resonance have each shown striking differences between dasatinib and imatinib (77, 78). These studies show that dasatinib binds at the active site of the ABL kinase domain in a vastly different manner to imatinib. Specifically, it binds the active conformation of ABL exclusively as opposed to the inactive conformation, to which imatinib binds (Figure 3) (77, 78). Therefore, dasatinib would not be susceptible to resistance caused by mutations that favor the active conformation of the ABL kinase domain. In addition, there is no distortion of the P-loop upon binding, as occurs with imatinib (78). Lastly, due to its smaller size, there are fewer direct contact points, perhaps affording less opportunity for resistant mutations. However, one direct contact point is T315, which explains the resistance of T315I to dasatinib.
Figure 3. Magnified views into the active sites of the ABL kinase domain in complex with imatinib (A) and dasatinib (B).

The two compounds have very different modes of binding to the kinase, with dasatinib more confined to the ATP-binding pocket than imatinib. In addition, imatinib binds the inactive and dasatinib the active conformations, with opposite orientations of the catalytic Glu-Phe-Gly amino acid residues (shown in light blue stick format). In the dasatinib structure, the glutamic acid of the Glu-Phe-Gly motif that coordinates a Mg2+ ion during catalysis is oriented properly for catalysis, whereas in the imatinib structure this residue points away from the active site. Both inhibitors reside in close proximity to the T315 residue. The side chain atoms of the residues susceptible to resistant mutations for each inhibitor are shown in green and inhibitor atoms shown in yellow stick format (nitrogen, dark blue; oxygen, red; sulfur, orange; chlorine, hot pink). The P-loop (residues 244–255) is shown in magenta and the activation loop (residues 381–402) is shown in light blue. (A) PDB entry 1IEP (24). (B) PDB entry 2GQG (78). The figure was created using PyMol (http://pymol.sourceforge.net/).
Another kinase inhibitor, nilotinib (also known as AMN107), was developed by modifying the chemical structure of imatinib to optimize potency against the ABL kinases. In preclinical testing, nilotinib showed 10- to 30-fold increased potency over imatinib against the major resistant mutants, except T315I (79). Nilotinib retains the kinase specificity profile of imatinib, with inhibition confined to ABL kinases, ARG, Kit, and PDGFR kinases. Nilotinib, in contrast to dasatinib, binds the kinase domain of ABL kinases in the inactive conformation (79).
Dasatinib and nilotinib have progressed rapidly through clinical trials, and dasatinib has been FDA approved for imatinib-resistant patients with CML. In a phase I clinical trial of dasatinib in patients with CML with imatinib resistance or intolerance, complete hematologic responses were achieved in 37 of 40 patients with chronic phase CML (80). Although it is still early, the responses are durable in 95% of such patients, with a median follow-up of 12 months (80). Similar results have been observed with nilotinib (81). Response rates of patients with advanced phase disease are also quite high, but similar to the experience with imatinib, relapses are commonplace in this population. Furthermore, dasatinib and nilotinib both seem well tolerated, although each have unique side effects. For example, pleural effusions occur in some patients treated with dasatinib, and elevated liver enzymes and prolongation of QT interval corrected for rate (QTc) occur in some patients treated with nilotinib (81–84).
In the clinical trials of dasatinib and nilotinib, one of the most common mechanisms of resistance has been the emergence of clones with the T315I mutation in BCR-ABL (82–85). Therefore, T315I remains a recurring problem for all the current ABL kinase inhibitors. Despite this challenge, preclinical T315I inhibitors have been reported, and at least one has been administered to patients (86–88). The aurora kinase inhibitor MK-0457 (VX-680) was observed to have affinity for the T315I mutant (89). As MK-0457 was already in clinical trials for patients with solid tumors, its testing in patients with CML was expedited. Although responses have been observed in patients with the T315I mutation (86), it is not clear whether these represent a specific effect or are due to antiproliferative effects from aurora kinase inhibition. Other preclinical compounds have also been reported to have specific activity against T315I and might soon be tested clinically (87, 88). One could imagine a scenario in which combinations of inhibitors could be used to circumvent resistance entirely, although persistence of hematopoietic stem cells expressing BCR-ABL is still probable with this approach.
Application of the imatinib paradigm to other malignancies
A key lesson learned from imatinib is that the discovery of the primary genetic abnormality in a malignancy, coupled with the development of an agent that targets that abnormality, can lead to therapeutic success. With that in mind, a major goal for cancer research should be to reclassify all forms of cancer according to their genotypes, rather than being classified solely on histology. Genotypic classification of malignancy is further supported by the successful use of imatinib in diseases such as gastrointestinal stromal tumors and hypereosinophilic syndrome. Both of these have an imatinib-susceptible kinase at their core (90, 91). As more cancers are genetically classified, those cancers will become amenable to hypothesis-driven testing of targeted therapeutic approaches.
The introduction of imatinib has generated a great deal of excitement about kinase-targeted therapy for the treatment for human cancer. Several prominent examples of oncogenic kinases associated with major malignancies are known, and researchers and clinicians are moving quickly to target these abnormalities. For example, the use of EGFR inhibitors to treat patients with non–small-cell lung cancer has shown remarkable response when correlated to EGFR mutation status (92, 93). In AML, fms-related tyrosine kinase 3 (FLT3) mutations are present in up to one-third of patients, but additional mutations are required to cause the disease (94). Clinical data with FLT3 inhibitors has shown that some patients respond, but it is not likely that these inhibitors can be used as monotherapy because of the genetic complexity of AML (95–97). Ongoing clinical trials are examining FLT3 inhibitors as a component of combination therapy. By contrast, myeloproliferative disorders such as polycythemia vera have become prime candidates for kinase inhibitor monotherapy after it was observed that a high frequency of individuals with this disease have a V617F-activating mutation in the JAK2 kinase (98–100). It is hoped that treating these myeloproliferative disorders at early stages, akin to the chronic phase of CML, will have similar results to those obtained with imatinib.
Conclusion: an outlook for the 21st century
In closing, it is worth remembering the impact four decades of discovery have had on patients with CML by recounting a patient’s story. One patient seen at the Oregon Health & Science University is a 55-year-old male who was diagnosed in 1997 with chronic phase CML. He was subsequently treated with hydroxyurea and interferon-α and underwent an autologous stem cell transplant in 1998. Over the following year, his cytogenetics displayed an increasing percentage of Ph chromosome–positive metaphases, despite treatment with interferon-α. In late 2000, 20 of 20 metaphases were Ph chromosome positive, and he had an additional copy of the Ph chromosome along with trisomy 8. Based on these findings, he was enrolled in a clinical trial of imatinib and rapidly achieved a complete cytogenetic response. After two years of therapy, he lacked detectable BCR-ABL transcripts by quantitative RT-PCR. In 2003, he had cytogenetic relapse and subsequent detection of the H396R mutant of BCR-ABL. This development made him a prime candidate for a second-generation inhibitor, as both nilotinib and dasatinib are predicted to have high efficacy in patients with this mutation. Over the last 3 years, he has been treated with dasatinib and has maintained a major cytogenetic response. In February of 2007, this male reached a full decade of living with CML, during which he has been able to continue to work and maintain his normal lifestyle, whereas previously, survival beyond five years of patients with CML was uncommon. Ultimately, our greatest goal is to continue this kind of tangible improvement in cancer therapies. We now look forward to further discoveries during the 21st century that will continue the tradition of translation of basic research findings such as the discovery of the Ph chromosome into clinically meaningful therapies.
Acknowledgments
We are grateful to Jeffrey Tyner, Thomas O’Hare, and Christopher Eide for helpful comments on the manuscript. We also thank Susan Olson and Helen Lawce for the FISH micrograph and Marc Loriaux for providing the pathology micrographs.
Footnotes
Nonstandard abbreviations used: ABL, v-abl Abelson murine leukemia viral oncogene homolog; ALL, acute lymphoblastic leukemia; AML, acute myelogenous leukemia; ARG, Abelson-related gene; BCR, breakpoint cluster region; c-ABL, cellular homologue of the Abelson murine leukemia viral oncogene product; CML, chronic myelogenous leukemia; Ph, Philadelphia (chromosome).
Conflict of interest: The authors have declared that no conflict of interest exists.
Citation for this article: J. Clin. Invest. 116:2067–2074 (2007). doi:10.1172/JCI31988.
References
- 1.Nowell P.C., Hungerford D.A. Chromosome studies on normal and leukemic human leukocytes. J. Natl. Cancer Inst. 1960;25:85–109. [PubMed] [Google Scholar]
- 2.Rowley J.D. Letter: A new consistent chromosomal abnormality in chronic myelogenous leukaemia identified by quinacrine fluorescence and Giemsa staining. Nature. 1973;243:290–293. doi: 10.1038/243290a0. [DOI] [PubMed] [Google Scholar]
- 3.Collins S.J., Kubonishi I., Miyoshi I., Groudine M.T. Altered transcription of the c-abl oncogene in K-562 and other chronic myelogenous leukemia cells. Science. 1984;225:72–74. doi: 10.1126/science.6587568. [DOI] [PubMed] [Google Scholar]
- 4.Gale R.P., Canaani E. An 8-kilobase abl RNA transcript in chronic myelogenous leukemia. Proc. Natl. Acad. Sci. U. S. A. 1984;81:5648–5652. doi: 10.1073/pnas.81.18.5648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shtivelman E., Lifshitz B., Gale R.P., Canaani E. Fused transcript of abl and bcr genes in chronic myelogenous leukaemia. Nature. 1985;315:550–554. doi: 10.1038/315550a0. [DOI] [PubMed] [Google Scholar]
- 6.Ben-Neriah Y., Daley G.Q., Mes-Masson A.M., Witte O.N., Baltimore D. The chronic myelogenous leukemia-specific P210 protein is the product of the bcr/abl hybrid gene. Science. 1986;233:212–214. doi: 10.1126/science.3460176. [DOI] [PubMed] [Google Scholar]
- 7.Davis R.L., Konopka J.B., Witte O.N. Activation of the c-abl oncogene by viral transduction or chromosomal translocation generates altered c-abl proteins with similar in vitro kinase properties. Mol. Cell. Biol. 1985;5:204–213. doi: 10.1128/mcb.5.1.204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Daley G.Q., Van Etten R.A., Baltimore D. Induction of chronic myelogenous leukemia in mice by the P210bcr/abl gene of the Philadelphia chromosome. Science. 1990;247:824–830. doi: 10.1126/science.2406902. [DOI] [PubMed] [Google Scholar]
- 9.Heisterkamp N., et al. Acute leukaemia in bcr/abl transgenic mice. Nature. 1990;344:251–253. doi: 10.1038/344251a0. [DOI] [PubMed] [Google Scholar]
- 10.Kelliher M.A., McLaughlin J., Witte O.N., Rosenberg N. Induction of a chronic myelogenous leukemia-like syndrome in mice with v-abl and BCR/ABL. Proc. Natl. Acad. Sci. U. S. A. 1990;87:6649–6653. doi: 10.1073/pnas.87.17.6649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fialkow P.J., Denman A.M., Jacobson R.J., Lowenthal M.N. Chronic myelocytic leukemia. Origin of some lymphocytes from leukemic stem cells. J. Clin. Invest. 1978;62:815–823. doi: 10.1172/JCI109193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Michor F., Iwasa Y., Nowak M.A. The age incidence of chronic myeloid leukemia can be explained by a one-mutation model. Proc. Natl. Acad. Sci. U. S. A. 2006;103:14931–14934. doi: 10.1073/pnas.0607006103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kantarjian H.M., et al. Chronic myelogenous leukemia in blast crisis. Analysis of 242 patients. Am. J. Med. 1987;83:445–454. doi: 10.1016/0002-9343(87)90754-6. [DOI] [PubMed] [Google Scholar]
- 14.Calabretta B., Perrotti D. The biology of CML blast crisis. Blood. 2004;103:4010–4022. doi: 10.1182/blood-2003-12-4111. [DOI] [PubMed] [Google Scholar]
- 15.Buchdunger E., et al. Inhibition of the Abl protein-tyrosine kinase in vitro and in vivo by a 2-phenylaminopyrimidine derivative. Cancer Res. 1996;56:100–104. [PubMed] [Google Scholar]
- 16.Druker B.J., et al. Effects of a selective inhibitor of the Abl tyrosine kinase on the growth of Bcr-Abl positive cells. Nat. Med. 1996;2:561–566. doi: 10.1038/nm0596-561. [DOI] [PubMed] [Google Scholar]
- 17.Okuda K., Weisberg E., Gilliland D.G., Griffin J.D. ARG tyrosine kinase activity is inhibited by STI571. Blood. 2001;97:2440–2448. doi: 10.1182/blood.v97.8.2440. [DOI] [PubMed] [Google Scholar]
- 18.Buchdunger E., et al. Abl protein-tyrosine kinase inhibitor STI571 inhibits in vitro signal transduction mediated by c-kit and platelet-derived growth factor receptors. J. Pharmacol. Exp. Ther. 2000;295:139–145. [PubMed] [Google Scholar]
- 19.Levitzki A., Gazit A. Tyrosine kinase inhibition: an approach to drug development. Science. 1995;267:1782–1788. doi: 10.1126/science.7892601. [DOI] [PubMed] [Google Scholar]
- 20.Deininger M., Buchdunger E., Druker B.J. The development of imatinib as a therapeutic agent for chronic myeloid leukemia. Blood. 2005;105:2640–2653. doi: 10.1182/blood-2004-08-3097. [DOI] [PubMed] [Google Scholar]
- 21.Huettner C.S., Zhang P., Van Etten R.A., Tenen D.G. Reversibility of acute B-cell leukaemia induced by BCR-ABL1. Nat. Genet. 2000;24:57–60. doi: 10.1038/71691. [DOI] [PubMed] [Google Scholar]
- 22.Druker B.J., et al. Efficacy and safety of a specific inhibitor of the BCR-ABL tyrosine kinase in chronic myeloid leukemia. N. Engl. J. Med. 2001;344:1031–1037. doi: 10.1056/NEJM200104053441401. [DOI] [PubMed] [Google Scholar]
- 23.Silver R.T., et al. An evidence-based analysis of the effect of busulfan, hydroxyurea, interferon, and allogeneic bone marrow transplantation in treating the chronic phase of chronic myeloid leukemia: developed for the American Society of Hematology. Blood. 1999;94:1517–1536. [PubMed] [Google Scholar]
- 24.Nagar B., et al. Crystal structures of the kinase domain of c-Abl in complex with the small molecule inhibitors PD173955 and imatinib (STI-571). Cancer Res. 2002;62:4236–4243. [PubMed] [Google Scholar]
- 25.Schindler T., et al. Structural mechanism for STI-571 inhibition of abelson tyrosine kinase. Science. 2000;289:1938–1942. doi: 10.1126/science.289.5486.1938. [DOI] [PubMed] [Google Scholar]
- 26.Druker B.J., et al. Five-year follow-up of patients receiving imatinib for chronic myeloid leukemia. N. Engl. J. Med. 2006;355:2408–2417. doi: 10.1056/NEJMoa062867. [DOI] [PubMed] [Google Scholar]
- 27.Baccarani M., et al. A randomized study of interferon-alpha versus interferon-alpha and low-dose arabinosyl cytosine in chronic myeloid leukemia. Blood. 2002;99:1527–1535. doi: 10.1182/blood.v99.5.1527. [DOI] [PubMed] [Google Scholar]
- 28.Guilhot F., et al. Interferon alfa-2b combined with cytarabine versus interferon alone in chronic myelogenous leukemia. French Chronic Myeloid Leukemia Study Group. N. Engl. J. Med. 1997;337:223–229. doi: 10.1056/NEJM199707243370402. [DOI] [PubMed] [Google Scholar]
- 29.Cortes J., O’Brien S., Kantarjian H. Discontinuation of imatinib therapy after achieving a molecular response. Blood. 2004;104:2204–2205. doi: 10.1182/blood-2004-04-1335. [DOI] [PubMed] [Google Scholar]
- 30.Higashi T., et al. Imatinib mesylate-sensitive blast crisis immediately after discontinuation of imatinib mesylate therapy in chronic myelogenous leukemia: report of two cases. Am. J. Hematol. 2004;76:275–278. doi: 10.1002/ajh.20096. [DOI] [PubMed] [Google Scholar]
- 31.Graham S.M., et al. Primitive, quiescent, Philadelphia-positive stem cells from patients with chronic myeloid leukemia are insensitive to STI571 in vitro. Blood. 2002;99:319–325. doi: 10.1182/blood.v99.1.319. [DOI] [PubMed] [Google Scholar]
- 32.Holtz M.S., et al. Imatinib mesylate (STI571) inhibits growth of primitive malignant progenitors in chronic myelogenous leukemia through reversal of abnormally increased proliferation. Blood. 2002;99:3792–3800. doi: 10.1182/blood.v99.10.3792. [DOI] [PubMed] [Google Scholar]
- 33.Michor F., et al. Dynamics of chronic myeloid leukaemia. Nature. 2005;435:1267–1270. doi: 10.1038/nature03669. [DOI] [PubMed] [Google Scholar]
- 34.Roeder I., et al. Dynamic modeling of imatinib-treated chronic myeloid leukemia: functional insights and clinical implications. Nat. Med. 2006;12:1181–1184. doi: 10.1038/nm1487. [DOI] [PubMed] [Google Scholar]
- 35.Druker B.J., et al. Activity of a specific inhibitor of the BCR-ABL tyrosine kinase in the blast crisis of chronic myeloid leukemia and acute lymphoblastic leukemia with the Philadelphia chromosome. N. Engl. J. Med. 2001;344:1038–1042. doi: 10.1056/NEJM200104053441402. [DOI] [PubMed] [Google Scholar]
- 36.Sawyers C.L., et al. Imatinib induces hematologic and cytogenetic responses in patients with chronic myelogenous leukemia in myeloid blast crisis: results of a phase II study. Blood. 2002;99:3530–3539. doi: 10.1182/blood.v99.10.3530. [DOI] [PubMed] [Google Scholar]
- 37.de Labarthe A., et al. Imatinib combined with induction or consolidation chemotherapy in patients with de novo Philadelphia chromosome-positive acute lymphoblastic leukemia: results of the GRAAPH-2003 study. Blood. 2007;109:1408–1413. doi: 10.1182/blood-2006-03-011908. [DOI] [PubMed] [Google Scholar]
- 38.Thomas D.A., et al. Outcome with the hyper-CVAD and imatinib mesylate regimen as frontline therapy for adult Philadelphia (Ph) positive acute lymphocytic leukemia (ALL) [abstract]. Blood (ASH Annual Meeting Abstracts). 2006;108:284. [Google Scholar]
- 39.Wassmann B., et al. Alternating versus concurrent schedules of imatinib and chemotherapy as front-line therapy for Philadelphia-positive acute lymphoblastic leukemia (Ph+ ALL). Blood. 2006;108:1469–1477. doi: 10.1182/blood-2005-11-4386. [DOI] [PubMed] [Google Scholar]
- 40.Yanada M., et al. High complete remission rate and promising outcome by combination of imatinib and chemotherapy for newly diagnosed BCR-ABL-positive acute lymphoblastic leukemia: a phase II study by the Japan Adult Leukemia Study Group. J. Clin. Oncol. 2006;24:460–466. doi: 10.1200/JCO.2005.03.2177. [DOI] [PubMed] [Google Scholar]
- 41.Mitelman F., Levan G., Nilsson P.G., Brandt L. Non-random karyotypic evolution in chronic myeloid leukemia. Int. J. Cancer. 1976;18:24–30. doi: 10.1002/ijc.2910180105. [DOI] [PubMed] [Google Scholar]
- 42.Dierov J., Dierova R., Carroll M. BCR/ABL translocates to the nucleus and disrupts an ATR-dependent intra-S phase checkpoint. Cancer Cell. 2004;5:275–285. doi: 10.1016/s1535-6108(04)00056-x. [DOI] [PubMed] [Google Scholar]
- 43.Koptyra M., et al. BCR/ABL kinase induces self-mutagenesis via reactive oxygen species to encode imatinib resistance. Blood. 2006;108:319–327. doi: 10.1182/blood-2005-07-2815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nowicki M.O., et al. BCR/ABL oncogenic kinase promotes unfaithful repair of the reactive oxygen species-dependent DNA double-strand breaks. Blood. 2004;104:3746–3753. doi: 10.1182/blood-2004-05-1941. [DOI] [PubMed] [Google Scholar]
- 45.Sattler M., et al. The BCR/ABL tyrosine kinase induces production of reactive oxygen species in hematopoietic cells. J. Biol. Chem. 2000;275:24273–24278. doi: 10.1074/jbc.M002094200. [DOI] [PubMed] [Google Scholar]
- 46.Mitani K., et al. Generation of the AML1-EVI-1 fusion gene in the t(3;21)(q26;q22) causes blastic crisis in chronic myelocytic leukemia. EMBO J. 1994;13:504–510. doi: 10.1002/j.1460-2075.1994.tb06288.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tanaka T., et al. Dual functions of the AML1/Evi-1 chimeric protein in the mechanism of leukemogenesis in t(3;21) leukemias. Mol. Cell. Biol. 1995;15:2383–2392. doi: 10.1128/mcb.15.5.2383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cuenco G.M., Ren R. Cooperation of BCR-ABL and AML1/MDS1/EVI1 in blocking myeloid differentiation and rapid induction of an acute myelogenous leukemia. Oncogene. 2001;20:8236–8248. doi: 10.1038/sj.onc.1205095. [DOI] [PubMed] [Google Scholar]
- 49.Jamieson C.H., et al. Granulocyte-macrophage progenitors as candidate leukemic stem cells in blast-crisis CML. N. Engl. J. Med. 2004;351:657–667. doi: 10.1056/NEJMoa040258. [DOI] [PubMed] [Google Scholar]
- 50.Deininger M.W., Goldman J.M., Melo J.V. The molecular biology of chronic myeloid leukemia. Blood. 2000;96:3343–3356. [PubMed] [Google Scholar]
- 51.Konopka J.B., Watanabe S.M., Witte O.N. An alteration of the human c-abl protein in K562 leukemia cells unmasks associated tyrosine kinase activity. Cell. 1984;37:1035–1042. doi: 10.1016/0092-8674(84)90438-0. [DOI] [PubMed] [Google Scholar]
- 52.Hantschel O., et al. A myristoyl/phosphotyrosine switch regulates c-Abl. Cell. 2003;112:845–857. doi: 10.1016/s0092-8674(03)00191-0. [DOI] [PubMed] [Google Scholar]
- 53.Nagar B., et al. Structural basis for the autoinhibition of c-Abl tyrosine kinase. Cell. 2003;112:859–871. doi: 10.1016/s0092-8674(03)00194-6. [DOI] [PubMed] [Google Scholar]
- 54.Zhao X., Ghaffari S., Lodish H., Malashkevich V.N., Kim P.S. Structure of the Bcr-Abl oncoprotein oligomerization domain. Nat. Struct. Biol. 2002;9:117–120. doi: 10.1038/nsb747. [DOI] [PubMed] [Google Scholar]
- 55.McWhirter J.R., Galasso D.L., Wang J.Y. A coiled-coil oligomerization domain of Bcr is essential for the transforming function of Bcr-Abl oncoproteins. Mol. Cell. Biol. 1993;13:7587–7595. doi: 10.1128/mcb.13.12.7587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Smith K.M., Yacobi R., Van Etten R.A. Autoinhibition of Bcr-Abl through its SH3 domain. Mol. Cell. 2003;12:27–37. doi: 10.1016/S1097-2765(03)00274-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Young M.A., et al. Structure of the kinase domain of an imatinib-resistant Abl mutant in complex with the Aurora kinase inhibitor VX-680. Cancer Res. 2006;66:1007–1014. doi: 10.1158/0008-5472.CAN-05-2788. [DOI] [PubMed] [Google Scholar]
- 58.Ilaria R.L., Jr., Van Etten R.A. P210 and P190(BCR/ABL) induce the tyrosine phosphorylation and DNA binding activity of multiple specific STAT family members. J. Biol. Chem. 1996;271:31704–31710. doi: 10.1074/jbc.271.49.31704. [DOI] [PubMed] [Google Scholar]
- 59.Klejman A., et al. The Src family kinase Hck couples BCR/ABL to STAT5 activation in myeloid leukemia cells. EMBO J. 2002;21:5766–5774. doi: 10.1093/emboj/cdf562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sawyers C.L., McLaughlin J., Witte O.N. Genetic requirement for Ras in the transformation of fibroblasts and hematopoietic cells by the Bcr-Abl oncogene. J. Exp. Med. 1995;181:307–313. doi: 10.1084/jem.181.1.307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Skorski T., et al. Transformation of hematopoietic cells by BCR/ABL requires activation of a PI-3k/Akt-dependent pathway. EMBO J. 1997;16:6151–6161. doi: 10.1093/emboj/16.20.6151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Skorski T., et al. Phosphatidylinositol-3 kinase activity is regulated by BCR/ABL and is required for the growth of Philadelphia chromosome-positive cells. Blood. 1995;86:726–736. [PubMed] [Google Scholar]
- 63.Horita M., et al. Blockade of the Bcr-Abl kinase activity induces apoptosis of chronic myelogenous leukemia cells by suppressing signal transducer and activator of transcription 5-dependent expression of Bcl-xL. J. Exp. Med. 2000;191:977–984. doi: 10.1084/jem.191.6.977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Salomoni P., Condorelli F., Sweeney S.M., Calabretta B. Versatility of BCR/ABL-expressing leukemic cells in circumventing proapoptotic BAD effects. Blood. 2000;96:676–684. [PubMed] [Google Scholar]
- 65.Oda T., et al. Crkl is the major tyrosine-phosphorylated protein in neutrophils from patients with chronic myelogenous leukemia. . J. Biol. Chem. 1994;269:22925–22928. [PubMed] [Google Scholar]
- 66.Gorre M.E., et al. Clinical resistance to STI-571 cancer therapy caused by BCR-ABL gene mutation or amplification. Science. 2001;293:876–880. doi: 10.1126/science.1062538. [DOI] [PubMed] [Google Scholar]
- 67.Barthe C., Cony-Makhoul P., Melo J.V., Mahon J.R. Roots of clinical resistance to STI-571 cancer therapy. Science. 2001;293:2163. doi: 10.1126/science.293.5538.2163a. [DOI] [PubMed] [Google Scholar]
- 68.Branford S., et al. High frequency of point mutations clustered within the adenosine triphosphate-binding region of BCR/ABL in patients with chronic myeloid leukemia or Ph-positive acute lymphoblastic leukemia who develop imatinib (STI571) resistance. Blood. 2002;99:3472–3475. doi: 10.1182/blood.v99.9.3472. [DOI] [PubMed] [Google Scholar]
- 69.Hochhaus A., et al. Molecular and chromosomal mechanisms of resistance to imatinib (STI571) therapy. Leukemia. 2002;16:2190–2196. doi: 10.1038/sj.leu.2402741. [DOI] [PubMed] [Google Scholar]
- 70.Shah N.P., et al. Multiple BCR-ABL kinase domain mutations confer polyclonal resistance to the tyrosine kinase inhibitor imatinib (STI571) in chronic phase and blast crisis chronic myeloid leukemia. Cancer Cell. 2002;2:117–125. doi: 10.1016/s1535-6108(02)00096-x. [DOI] [PubMed] [Google Scholar]
- 71.von Bubnoff N., Schneller F., Peschel C., Duyster J. BCR-ABL gene mutations in relation to clinical resistance of Philadelphia-chromosome-positive leukaemia to STI571: a prospective study. Lancet. 2002;359:487–491. doi: 10.1016/S0140-6736(02)07679-1. [DOI] [PubMed] [Google Scholar]
- 72.Hughes T., et al. Monitoring CML patients responding to treatment with tyrosine kinase inhibitors: review and recommendations for harmonizing current methodology for detecting BCR-ABL transcripts and kinase domain mutations and for expressing results. Blood. 2006;108:28–37. doi: 10.1182/blood-2006-01-0092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Roche-Lestienne C., et al. Several types of mutations of the Abl gene can be found in chronic myeloid leukemia patients resistant to STI571, and they can pre-exist to the onset of treatment. Blood. 2002;100:1014–1018. doi: 10.1182/blood.v100.3.1014. [DOI] [PubMed] [Google Scholar]
- 74.Griswold I.J., et al. Kinase domain mutants of Bcr-Abl exhibit altered transformation potency, kinase activity, and substrate utilization, irrespective of sensitivity to imatinib. Mol. Cell. Biol. 2006;26:6082–6093. doi: 10.1128/MCB.02202-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Lombardo L.J., et al. Discovery of N-(2-chloro-6-methyl- phenyl)-2-(6-(4-(2-hydroxyethyl)- piperazin-1-yl)-2-methylpyrimidin-4- ylamino)thiazole-5-carboxamide (BMS-354825), a dual Src/Abl kinase inhibitor with potent antitumor activity in preclinical assays. J. Med. Chem. 2004;47:6658–6661. doi: 10.1021/jm049486a. [DOI] [PubMed] [Google Scholar]
- 76.Shah N.P., et al. Overriding imatinib resistance with a novel ABL kinase inhibitor. Science. 2004;305:399–401. doi: 10.1126/science.1099480. [DOI] [PubMed] [Google Scholar]
- 77.Manely P.W., et al. Bcr-Abl binding modes of dasatinib, imatinib and nilotinib: an NMR study [abstract]. Blood (ASH Annual Meeting Abstracts). 2006;108:747. [Google Scholar]
- 78.Tokarski J.S., et al. The structure of dasatinib (BMS-354825) bound to activated ABL kinase domain elucidates its inhibitory activity against imatinib-resistant ABL mutants. Cancer Res. 2006;66:5790–5797. doi: 10.1158/0008-5472.CAN-05-4187. [DOI] [PubMed] [Google Scholar]
- 79.Weisberg E., et al. Characterization of AMN107, a selective inhibitor of native and mutant Bcr-Abl. Cancer Cell. 2005;7:129–141. doi: 10.1016/j.ccr.2005.01.007. [DOI] [PubMed] [Google Scholar]
- 80.Talpaz M., et al. Dasatinib in imatinib-resistant Philadelphia chromosome-positive leukemias. N. Engl. J. Med. 2006;354:2531–2541. doi: 10.1056/NEJMoa055229. [DOI] [PubMed] [Google Scholar]
- 81.Kantarjian H., et al. Nilotinib in imatinib-resistant CML and Philadelphia chromosome-positive ALL. N. Engl. J. Med. 2006;354:2542–2551. doi: 10.1056/NEJMoa055104. [DOI] [PubMed] [Google Scholar]
- 82.Cortes J., et al. Dasatinib induces complete hematologic and cytogenetic responses in patients with imatinib-resistant or -intolerant chronic myeloid leukemia in blast crisis. Blood. 2007;109:3207–3213. doi: 10.1182/blood-2006-09-046888. [DOI] [PubMed] [Google Scholar]
- 83.Guilhot F., et al. Dasatinib induces significant hematologic and cytogenetic responses in patients with imatinib-resistant or -intolerant chronic myeloid leukemia in accelerated phase. Blood. 2007;109:4143–4150. doi: 10.1182/blood-2006-09-046839. [DOI] [PubMed] [Google Scholar]
- 84.Hochhaus A., et al. Dasatinib induces notable hematologic and cytogenetic responses in chronic-phase chronic myeloid leukemia after failure of imatinib therapy. Blood. 2007;109:2303–2309. doi: 10.1182/blood-2006-09-047266. [DOI] [PubMed] [Google Scholar]
- 85.Hochhaus A., et al. Hematologic and cytogenetic response dynamics to nilotinib (AMN07) depend on the type of BCR-ABL mutations in patients with chronic myelogenous leukemia (CML) after imatinib failure. Blood (ASH Annual Meeting Abstracts). 2006;108:749. [Google Scholar]
- 86.Giles F.J., et al. MK-0457, a novel kinase inhibitor, is active in patients with chronic myeloid leukemia or acute lymphocytic leukemia with the T315I BCR-ABL mutation. Blood. 2007;109:500–502. doi: 10.1182/blood-2006-05-025049. [DOI] [PubMed] [Google Scholar]
- 87.O’Hare T., et al. Inhibition of T315I Bcr-Abl and other imatinib-resistant Bcr-Abl mutants by the selective Abl kinase inhibitor SGX70393 [abstract]. Blood (ASH Annual Meeting Abstracts). 2006;108:1373. [Google Scholar]
- 88.Shakespeare W.C., et al. Orally active inhibitors of the imatinib resistant Bcr-Abl mutant T315I [abstract]. Blood (ASH Annual Meeting Abstracts). 2006;108:2180. [Google Scholar]
- 89.Carter T.A., et al. Inhibition of drug-resistant mutants of ABL, KIT, and EGF receptor kinases. Proc. Natl. Acad. Sci. U. S. A. 2005;102:11011–11016. doi: 10.1073/pnas.0504952102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Corless C.L., Fletcher J.A., Heinrich M.C. Biology of gastrointestinal stromal tumors. J. Clin. Oncol. 2004;22:3813–3825. doi: 10.1200/JCO.2004.05.140. [DOI] [PubMed] [Google Scholar]
- 91.Pardanani A., Tefferi A. Imatinib targets other than bcr/abl and their clinical relevance in myeloid disorders. Blood. 2004;104:1931–1939. doi: 10.1182/blood-2004-01-0246. [DOI] [PubMed] [Google Scholar]
- 92.Lynch T.J., et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N. Engl. J. Med. 2004;350:2129–2139. doi: 10.1056/NEJMoa040938. [DOI] [PubMed] [Google Scholar]
- 93.Paez J.G., et al. Genome coverage and sequence fidelity of phi29 polymerase-based multiple strand displacement whole genome amplification. Nucleic Acids Res. 2004;32:e71. doi: 10.1093/nar/gnh069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Gilliland D.G., Griffin J.D. The roles of FLT3 in hematopoiesis and leukemia. Blood. 2002;100:1532–1542. doi: 10.1182/blood-2002-02-0492. [DOI] [PubMed] [Google Scholar]
- 95.Fiedler W., et al. A phase 1 study of SU11248 in the treatment of patients with refractory or resistant acute myeloid leukemia (AML) or not amenable to conventional therapy for the disease. Blood. 2005;105:986–993. doi: 10.1182/blood-2004-05-1846. [DOI] [PubMed] [Google Scholar]
- 96.Smith B.D., et al. Single-agent CEP-701, a novel FLT3 inhibitor, shows biologic and clinical activity in patients with relapsed or refractory acute myeloid leukemia. Blood. 2004;103:3669–3676. doi: 10.1182/blood-2003-11-3775. [DOI] [PubMed] [Google Scholar]
- 97.Stone R.M., et al. Patients with acute myeloid leukemia and an activating mutation in FLT3 respond to a small-molecule FLT3 tyrosine kinase inhibitor, PKC412. Blood. 2005;105:54–60. doi: 10.1182/blood-2004-03-0891. [DOI] [PubMed] [Google Scholar]
- 98.Baxter E.J., et al. Acquired mutation of the tyrosine kinase JAK2 in human myeloproliferative disorders. Lancet. 2005;365:1054–1061. doi: 10.1016/S0140-6736(05)71142-9. [DOI] [PubMed] [Google Scholar]
- 99.James C., et al. A unique clonal JAK2 mutation leading to constitutive signalling causes polycythaemia vera. Nature. 2005;434:1144–1148. doi: 10.1038/nature03546. [DOI] [PubMed] [Google Scholar]
- 100.Levine R.L., et al. Activating mutation in the tyrosine kinase JAK2 in polycythemia vera, essential thrombocythemia, and myeloid metaplasia with myelofibrosis. Cancer Cell. 2005;7:387–397.. doi: 10.1016/j.ccr.2005.03.023. [DOI] [PubMed] [Google Scholar]