

Abstract
Objective
Aging is associated with reduced tissue sensitivity to insulin. In women, these age-related changes may be accelerated by menopause. The effect of ovarian hormone deficiency on tissue insulin sensitivity in humans, however, has not been clearly defined. The purpose of this study was to evaluate the effect of suppression of endogenous ovarian hormone production on insulin-stimulated glucose disposal.
Design
Randomized, single-blind, placebo-controlled trial.
Setting
General Clinical Research Center.
Patients
Thirteen healthy, non-obese premenopausal women.
Interventions
Insulin-stimulated glucose disposal was determined by hyperinsulinemic (40 mU/m2/min) clamp during the early- to mid-follicular and mid-luteal phase of the menstrual cycle. Volunteers then received 2 months of treatment with the gonadotropin-releasing hormone agonist (GnRHa) leuprolide acetate (n=6) or placebo (n=7) and were retested.
Main Outcome Measure
Total, oxidative and non-oxidative insulin-stimulated glucose disposal.
Results
Because no effect of cycle phase was found on total, oxidative or non-oxidative glucose disposal, pre-treatment follicular and luteal phase values were averaged. Treatment with GnRHa had no effect on total glucose disposal (GnRHa: 10.6 ± 0.9 to 10.8 ± 0.9 vs. placebo: 10.2 ± 0.7 to 10.4 ± 1.0 mg/kg fat-free mass/min, P=0.99). Similarly, there was no effect of GnRHa administration on oxidative (GnRHa: 2.77 ± 0.58 to 3.89 ± 0.58 vs. placebo: 2.74 ± 0.42 to 3.33 ± 0.62 mg/kg fat-free mass/min, P=0.52; n=6 and 6, respectively) or non-oxidative (GnRHa: 7.82 ± 0.68 to 6.91 ± 0.66 vs. placebo: 7.94 ± 0.72 to 7.79 ± 0.99 mg/kg fat-free mass/min, P=0.59; n=6 and 6, respectively) components of glucose disposal.
Conclusions
Our results suggest that endogenous ovarian hormones do not regulate tissue responsiveness to insulin or intracellular pathways of glucose disposal.
Keywords: ovarian hormones, estrogen, insulin sensitivity, GnRH agonist
INTRODUCTION
Aging is characterized by a decline in tissue insulin sensitivity (1–2). In women, this decline may be accelerated following menopause (3). Some have hypothesized that ovarian hormone deficiency contributes to disturbances in glucose homeostasis (4). The role of ovarian hormones in regulating tissue insulin sensitivity, however, has not been clearly defined.
Studies in rodent models have consistently shown that ovarian hormone deficiency impairs insulin-stimulated glucose disposal and that replacement of ovarian hormones corrects insulin resistance (5–8). Whether ovarian hormones play a similar role in regulating insulin-stimulated glucose disposal in humans is less clear. Cross-sectional comparisons of pre- and postmenopausal women have shown either no difference (9) or increased insulin action in postmenopausal women (10). In further contrast, work in postmenopausal women has shown that resistance to the effects of insulin develops with increasing menopausal age (11). Studies examining the effect of hormone replacement therapy on glucose metabolism in postmenopausal women are equally divergent, with results showing increased (12–13), unchanged (14–18) and decreased insulin sensitivity (19–20). From these studies, it is clear that no conclusions can be drawn regarding the role, if any, that ovarian hormones play in the regulation of insulin-stimulated glucose disposal in women.
Our goal in the present study was to determine the physiological role of endogenous ovarian hormones on insulin-stimulated glucose disposal. To accomplish this objective, we measured glucose disposal under hyperinsulinemic clamp conditions in healthy, non-obese, premenopausal women during the early- to mid-follicular and mid-luteal phases of the menstrual cycle and following 2 months of treatment with either GnRHa or placebo. This experimental model permits us to compare the effects of an acute period of ovarian hormone deficiency (i.e., GnRHa group) versus the effects of normal cycling levels of the hormones (i.e., placebo group) on glucose disposal. Because animal studies suggest that ovarian hormones regulate glucose disposal through alterations in components of the non-oxidative pathway (7–8, 21), we further sought to define the effects of endogenous ovarian hormones on intracellular pathways of oxidative and non-oxidative glucose disposal. Our hypothesis was that ovarian hormone deficiency would reduce insulin-stimulated glucose disposal by decreasing flux through the non-oxidative pathway.
MATERIALS AND METHODS
Subjects
Thirteen healthy women ranging in age from 21 to 35 yr (mean ± SE; 27 ± 1 yr) were recruited. Women were non-obese (BMI<28 kg/m2; 22 ± 1 kg/m2), had a stable body weight (± 2 kg) for 6 months prior to study, were healthy based on medical history, physical exam and routine blood tests, were glucose tolerant (glucose <7.77 mmol/L 2 h following 75 g oral glucose load), had no history of tobacco use and were not on any medication that could affect ovarian/reproductive function or insulin sensitivity. All women were nulliparous, had not been exposed to any form of hormone-based contraceptive therapy for at least 6 months prior to study and reported having at least 2 spontaneous cycles in the 3 months prior to recruitment. The nature, purpose and possible risks of the study were explained to each subject before she gave written consent to participate. The experimental protocol was approved by the Committee on Human Research at the University of Vermont. Data from this cohort regarding the effects of GnRHa administration on protein turnover and blood flow have been published elsewhere (22–23). The primary goal of these studies was to assess the effect of GnRHa on protein metabolism, with the secondary goal of examining the effects of insulin-stimulated glucose disposal.
Experimental protocol
Each volunteer underwent an outpatient screening visit at which time medical history, physical examination, biochemical laboratory tests, an exercise stress test and an oral glucose tolerance test were performed. Volunteers that met the eligibility criteria were randomized by the study coordinator using a stratified block approach to receive the GnRHa leuprolide acetate (n=6; Lurpon Depot; 3.75 mg IM) or placebo (n=7; 0.9% saline). Groups were stratified for age and body mass index. All personnel performing metabolic testing on volunteers were blinded to treatment status. Prior to study, each volunteer’s menstrual cycle was monitored using a combination of menstrual diaries, ovulation prediction kits (Ovu-Quick One-Step; Quidel Co; San Diego, CA); and mid-luteal phase blood draws were used to discern cycle and phase length.
Each woman was tested on two occasions prior to treatment: during the early- to mid-follicular phase (cycle day 3 to 8) and mid-luteal phase (cycle day 19–25; assuming an average cycle length of 28 days). The order of pre-treatment metabolic testing with respect to cycle phase (follicular-luteal vs. luteal-follicular) was randomized. Following pre-treatment testing, GnRHa or placebo was administered by intramuscular injection during the mid-luteal phase. The second injection was given 30 d following the first injection in the GnRHa group and 25–32 d following the first injection in the placebo group, depending on the volunteer’s menstrual cycle length. Post-treatment metabolic testing was performed 60 d following the first injection in the GnRHa group. Post-treatment testing in the placebo group was performed in the phase of menstrual cycle that was tested second during pre-treatment testing approximately 50–64 days following the first injection. Directly preceding each bout of metabolic testing, volunteers were provided 3 d of a weight-maintenance, standardized diet (20% protein, 25% fat and 55% carbohydrate). The diet provided at least 200 g of carbohydrate per day.
Insulin-stimulated glucose disposal measurements were performed the morning following an overnight visit to the General Clinical Research Center. Volunteers were fasted after 1900 h the evening of admission. At approximately 0600 h, the subject was awakened and catheters were placed in an antecubital vein for infusion and retrograde in a dorsal hand vein of the contralateral arm and were kept patent with saline infusion. The hand was placed in a warming box to obtain arterialized-venous blood (24). Baseline blood and breath samples were taken and a primed (4.5 μmol/kg), continuous (4.5 μmol/kg/hr) infusion of [1-13C]leucine was started. The bicarbonate pool was primed (1.6 μmol/kg) with sodium [13C]bicarbonate. Leucine kinetic data were used to measure protein oxidation. At 210 min, insulin was started (40 mU/m2/min) with the goal of achieving circulating insulin levels that approximate postprandial levels. In addition, a constant rate (1.5 ml/kg/hr) infusion of 10% Aminosyn (Abbott Laboratories; Chicago, IL) was started. Glycemia was maintained by a variable rate infusion of 20% dextrose. Plasma glucose levels were monitored every 5 minutes and the dextrose infusion rate adjusted to maintain euglycemia. All infusions were stopped at 330 min except for the 20% dextrose infusion, which was continued and tapered until no longer required to maintain normal glycemia.
Blood and breath samples were drawn at 285, 300, 315 and 330 min for measurement of leucine oxidation during the clamp. Plasma was isolated and stored at −70°C until analysis. Oxygen consumption and carbon dioxide production rates were determined using the ventilated hood technique (DeltaTrac, Yorba Linda, CA). Indirect calorimetry measurements were not performed on one volunteer in the placebo group because of technical problems. Body composition measurements were performed directly following the clamp procedure.
Analytical methods
Plasma α-ketoisocaproate (KIC) enrichment was measured by electron impact ionization GCMS, as described previously (25). The enrichment of expired CO2 was measured by isotope ratio mass spectrometry (VG Sira II, Middlewich, Cheshire, UK). Plasma glucose concentrations were measured by a glucose analyzer (Yellow Springs Instruments; Yellow Springs, OH).
Calculations
Insulin-stimulated glucose disposal was calculated as the average glucose infusion rate (mg/min) during the last 30 min of the clamp. Based on prior studies from our laboratory showing that the degree of hyperinsulinemia reached with this rate of insulin infusion results in near complete suppression of endogenous glucose production in women (26), and the fact that variation in ovarian hormones have minimal effects on endogenous glucose production (20, 27–28), we assumed that endogenous glucose production was completely suppressed in these volunteers and was not affected by menstrual cycle phase (27) or GnRHa treatment (28).
To partition glucose disposal into oxidative and non-oxidative pathways, we measured carbohydrate (i.e., glucose) and fat oxidation using a combination of indirect calorimetry measurements and estimates of protein oxidation from [13C]leucine, as described previously (26). Leucine oxidation is derived from the rate of 13CO2 excretion into expired air (F13C; μmol/kg·hr−1), which is calculated as: FCO2 · ECO2 · 10/0.81, where FCO2 is the CO2 production rate (μmol/kg·hr−1), ECO2 is the enrichment of expired 13CO2, the constant 10 accounts for unit changes and the factor 0.81 accounts for the recovery of 13CO2 released from the bicarbonate pool (29).
During the clamp, the use of endogenous energy substrates will be decreased in favor of the use of exogenous substrates (30). This will alter 13CO2 excretion because of the different 13C abundances of endogenous and exogenous substrates (31). To account for this change, we measured 13CO2 excretion in a separate group of 6 young women (30 ± 1 yrs; 22 ± 1 kg/m2) undergoing an identical clamp protocol as described above, but without administration of 13C isotope tracers. Average 13CO2 enrichments derived from theses studies were used to correct 13CO2 enrichments for contribution from exogenously administered glucose and amino acids, as described previously (25). Dividing F13C by the rate of 13C infusion (i13C) gives the fraction of the tracer that is oxidized to CO2 (fox). To calculate leucine oxidation, fox was then multiplied by leucine appearance rate, which was calculated from standard equations (25) that were modified to account for the input of isotope tracer from two sources, as described previously (26). Leucine oxidation data were used to estimate protein oxidation, with the assumption that 8% of body protein is leucine. Protein oxidation data were used together with VO2 and VCO2 data to calculate carbohydrate and fat oxidation using standard equations (32). Non-oxidative glucose disposal was calculated as the difference between insulin-stimulated glucose disposal and carbohydrate oxidation.
Body composition
Body mass was measured on a metabolic scale (Scale-Tronix, Inc.; Wheaton, IL). Fat mass, fat-free mass and bone mineral mass were measured by dual energy x-ray absorptiometry using a GE Lunar Prodigy densitometer (GE Lunar Co, Madison, WI). Bone mass data are not reported.
Abdominal adiposity was measured between the L1 and L4 vertebral bodies using the Region of Interest option of the software following the general approach of Glickman et al. (33), with minor modifications. The inferior boundary was set at the top of the iliac crest and the superior boundary at the T12/L1 intervertebral space. The lateral boundaries were drawn to include all soft tissue in the abdominal region. To standardize the cutpoint placements within the same volunteer, the aforementioned transverse sections were determined on the scan that provided the best visualization of the T12/L1 intervertebral space and the distance between the superior and inferior cutpoints was measured to the nearest millimeter. Using the transverse section on the iliac crest as a starting point, this distance was then used to set the superior cutpoint for the analysis of all subsequent scans. All scans were analyzed by the same technician who was blinded to the volunteer’s treatment status. Analysis was performed using the Prodigy Version 5.60.003 software for body composition analysis.
Statistics
Normality was confirmed for all variables using the Shapiro-Wilk test prior to analysis. Unpaired t tests were used to compare baseline characteristics between groups. Paired t tests were used to compare data between pretreatment follicular and luteal phase measurements in the entire cohort. Because no effect of cycle phase was found on glucose disposal measures, follicular and luteal phase measurements performed prior to treatment were averaged and are referred to as ‘pre-treatment’ values. A repeated measures analysis of variance (RMANOVA) model was then used to detect group X time interaction effects with treatment group (GnRHa vs. placebo) as the between subjects factor and time (pre-treatment vs. post-treatment) as the within-subjects factor. All analyses were conducted with SPSS software (SPSS v 12.0; Chicago, IL).
RESULTS
Ovarian suppression (estradiol <50 pg/ml) was confirmed in 5 out of 6 volunteers randomized to GnRHa treatment 10 d following the initial injection. In one volunteer, ovarian suppression was confirmed at 16 d following the initial injection. Ovarian suppression was again confirmed in all women randomized to GnRHa treatment at the post-treatment study with circulating estrogen levels <50 pg/ml (range 14–38 pg/ml).
Body composition measurements are shown in Table 1. There was no significant effect of menstrual cycle phase on body composition (n=13). Thus, data were pooled for all subsequent analyses. There were no differences in body size/composition between the GnRHa and placebo groups at baseline. Moreover, there were no group by time interaction effects observed for any body composition variable.
Table 1.
Body composition.
| Follicular | Luteal | Post-treatment | ||||
|---|---|---|---|---|---|---|
| GnRHa | Placebo | GnRHa | Placebo | GnRHa | Placebo | |
| Weight (kg) | 62.9 ± 2.6 | 59.7 ± 3.0 | 62.7 ± 2.5 | 59.9 ± 3.2 | 62.2 ± 2.4 | 59.3 ± 2.9 |
| Fat mass (kg) | 20.6 ± 2.1 | 16.2 ± 1.5 | 20.6 ± 2.3 | 16.1 ± 1.7 | 20.7 ± 2.1 | 15.9 ± 1.1 |
| Fat-free mass (kg) | 40.7 ± 1.0 | 41.8 ± 2.3 | 40.3 ± 1.0 | 41.8 ± 2.5 | 40.0 ± 1.4 | 41.7 ± 2.5 |
| Percent fat | 33.3 ± 2.3 | 27.9 ± 2.0 | 33.5 ± 2.7 | 27.6 ± 2.5 | 33.8 ± 2.5 | 27.8 ± 1.7 |
| Intra-abdominal fat (kg) | 1.58 ± 0.21 | 1.34 ± 0.20 | 1.61 ± 0.25 | 1.36 ± 0.22 | 1.64 ± 0.23 | 1.36 ± 0.15 |
Data are mean ± SEM.
Indirect calorimetry and substrate oxidation data during the clamp are shown in Table 2. There was no significant effect of menstrual cycle phase on any variable (n=12). There were no differences in baseline values nor were there any group by time interaction effects for any of the measures, except protein oxidation (P=0.02).
Table 2.
Indirect calorimetry data under euglycemic-hyperinsulinemic conditions.
| Pre-treatment | Post-treatment | |||
|---|---|---|---|---|
| GnRHa | Placebo | GnRHa | Placebo | |
| VO2 (ml/min) | 236 ± 5 | 231 ± 9 | 230 ± 7 | 230 ± 3 |
| VCO2 (ml/min) | 197 ± 6 | 195 ± 9 | 203 ± 8 | 201 ± 8 |
| Protein oxidation (mg/min) | 34 ± 5 | 33 ± 3 | 34 ± 6 | 46 ± 4 a |
| Carbohydrate oxidation (mg/min) | 114 ± 24 | 119 ± 18 | 155 ± 24 | 142 ± 25 |
| Fat oxidation (mg/min) | 54 ± 11 | 49 ± 6 | 34 ± 9 | 33 ± 9 |
Data are mean ± SEM. VO2, oxygen consumption, VCO2, carbon dioxide production.
P=0.02 for group by time interaction
Figure 1 shows total, oxidative and non-oxidative glucose disposal measurements in the follicular and luteal phases of the menstrual cycle. There were no differences in total (10.7 ± 0.6 vs. 10.0 ± 0.7 mg/kg fat-free mass/min, P=0.45; n=13), oxidative (2.85 ± 0.45 vs. 2.61 ± 0.38 mg/kg fat-free mass/min, P=0.66; n=12), or non-oxidative glucose disposal (8.11 ± 0.63 vs. 7.67 ± 0.75 mg/kg fat-free mass/min, P=0.66; n=12) between the follicular and luteal phases, respectively. Because of the absence of menstrual cycle phase effects, pretreatment data were pooled.
Figure 1.
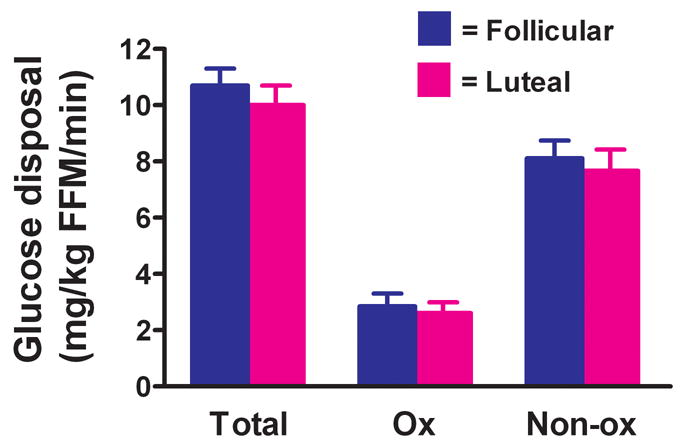
Effect of menstrual cycle phase on total (n=13), oxidative (Ox; n=12) and non-oxidative (NonOx; n=12) insulin-stimulated glucose disposal. Glucose disposal data are expressed per kg of fat-free mass (FFM). Values are mean ± SEM.
The effect of ovarian suppression with GnRHa on total, oxidative and non-oxidative glucose disposal is shown in Figure 2. Treatment with GnRHa had no effect on total glucose disposal (GnRHa: 10.6 ± 0.9 to 10.8 ± 0.9 vs. placebo: 10.2 ± 0.7 to 10.4 ± 1.0 mg/kg fat-free mass/min, P=0.99; n=6 and n=7, respectively). Similarly, there was no effect of GnRHa administration on oxidative (GnRHa: 2.77 ± 0.58 to 3.89 ± 0.58 vs. placebo: 2.74 ± 0.42 to 3.33 ± 0.62 mg/kg fat-free mass/min, P=0.52; n=6 and n=6, respectively) or non-oxidative (GnRHa: 7.82 ± 0.68 to 6.91 ± 0.66 vs. placebo: 7.94 ± 0.72 to 7.79 ± 0.99 mg/kg fat-free mass/min, P=0.59; n=6 and n=6, respectively) components of glucose disposal.
Figure 2.
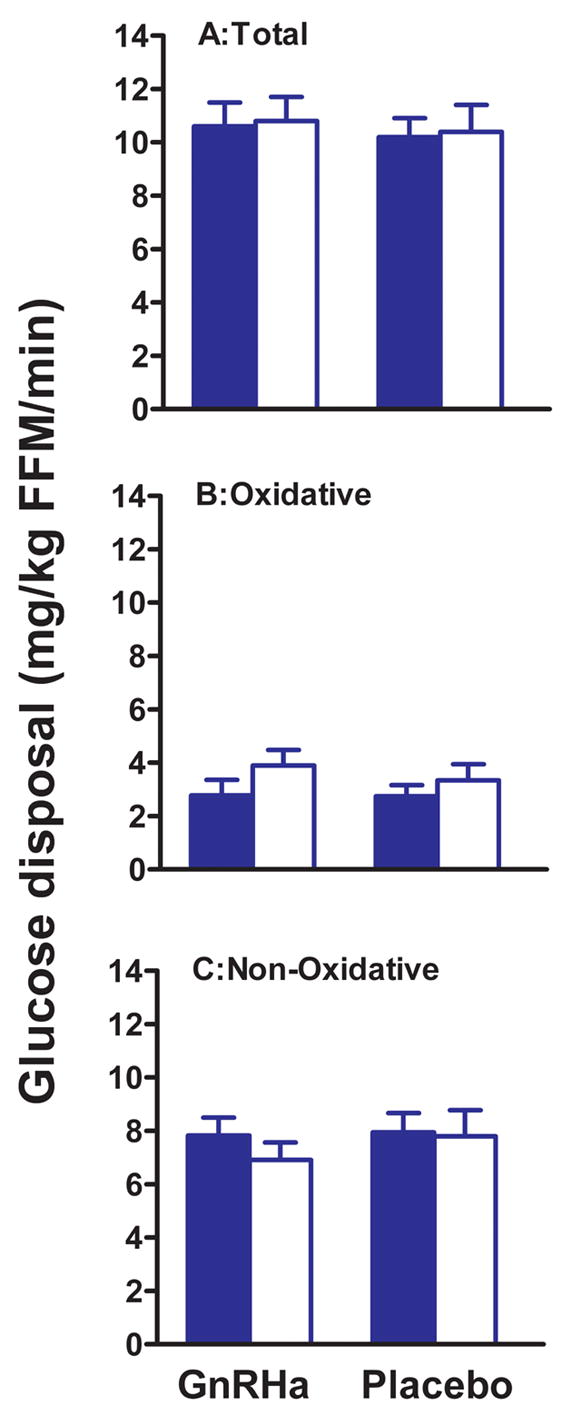
Effect of GnRHa or placebo administration on total (A) oxidative (B) and non-oxidative (C) insulin-stimulated glucose disposal. For oxidative and non-oxidative disposal, n=6 for the placebo group. Pre-treatment values are filled bars and post-treatment values open bars. Pre-treatment values are the average of follicular and luteal phase measurements. Glucose disposal data are expressed per kg of fat-free mass (FFM). Values are mean ± SEM.
Group mean and individual values for circulating glucose and insulin levels under fasting and euglycemic-hyperinsulinemic conditions are shown in Figure 3. Ovarian suppression with GnRHa had no effect on fasting glucose levels (GnRHa: 75.0 ± 1.3 to 73.7 ± 0.9 vs. placebo: 73.8 ± 1.0 to 72.4 ± 0.8 mg/dl, P=0.99) or glucose levels during the euglycemic-hyperinsulinemic clamp (GnRHa: 82.1 ± 1.1 to 80.8 ± 0.9 vs. placebo: 83.7 ± 0.9 to 84.6 ± 1.4 mg/dl, P=0.07). Treatment with GnRHa also had no effect on fasting insulin levels (GnRHa: 12.5 ± 1.0 to 12.2 ± 1.1 vs. placebo: 11.3 ± 0.5 to 13.5 ± 1.1 μIU/ml, P=0.17) or insulin levels during the euglycemic-hyperinsulinemic clamp (GnRHa: 134 ± 6 to 122 ± 7 vs. placebo: 128 ± 5 to 124 ± 6 μIU/ml, P=0.38; n=6 and n=6, respectively, for post-treatment).
Figure 3.
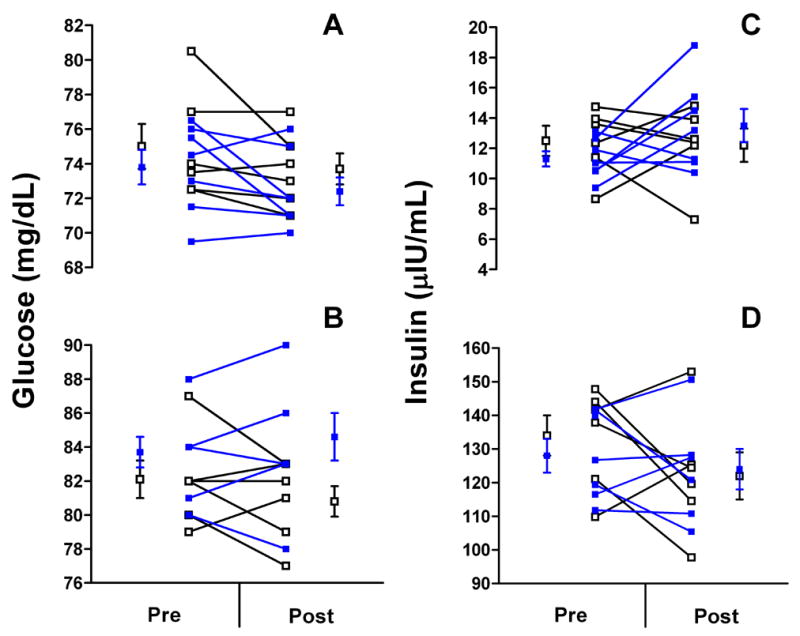
Group mean and individual values for circulating glucose and insulin levels under fasting (A and C) and during the hyperinsulinemic clamp (B and D) in women randomized to GnRHa (n=6; blue squares) or placebo (n=7; open squares). Pre, pre-treatment; Post, post-treatment. Note: three volunteers in the placebo group in Panel B have pre- and post-treatment values of 84 and 86 mg/dL, respectively, and n=6 for placebo for Panel D.
DISCUSSION
The present study examined the hypothesis that endogenous ovarian steroid hormones regulate whole-body insulin-stimulated glucose uptake through their effects on non-oxidative pathways. To address this hypothesis, we measured glucose disposal under hyperinsulinemic conditions in healthy, non-obese, premenopausal women during the early- to mid- follicular and mid-luteal phases of the menstrual cycle and following 2 months of treatment with either GnRHa or placebo. Our results indicate that total, oxidative and non-oxidative glucose disposal are not affected by ovarian hormone suppression with GnRHa.
Our study demonstrates that a short-term deficiency of endogenous ovarian hormones does not influence tissue insulin responsiveness in non-obese, adult women. The present results agree with data from Dumesic et al. (28), who examined the effect of 3 months of leuprolide acetate therapy on glucose disposal in hyperandrogenic, anovulatory women and healthy controls. An important caveat to this earlier study, however, was that both hyperandrogenic women and controls were obese (mean BMI=35 kg/m2). Because total and abdominal obesity are important modulators of insulin sensitivity (34–35), it is possible that the absence of an effect of ovarian hormone deficiency on glucose disposal was due to the confounding effect of adiposity. In other words, ovarian hormone deficiency may not affect insulin action in obese populations above and beyond the inhibitory effect of adiposity (i.e., floor effect). Our results extend these findings to show that, in the absence of the confounding effect of obesity, GnRHa-induced ovarian hormone deficiency has no effect on insulin-stimulated glucose disposal.
The effect of ovarian hormones on glucose metabolism may not be apparent when only ‘total’ glucose disposal is considered. For instance, there may be reciprocal changes in non-oxidative and oxidative glucose disposal that effectively result in no net change in total glucose disposal. To account for this possibility, we also measured oxidative and non-oxidative components of glucose disposal. In accord with total glucose disposal, neither oxidative nor non-oxidative pathways of glucose disposal were altered by GnRHa administration. Thus, our data suggest that ovarian hormone deficiency does not alter intracellular pathways of glucose disposal.
Our results, together with the findings of Dumesic et al. (28) and studies performed during different phases of the menstrual cycle (27, 36–37), argue against a role for endogenous ovarian hormones in the regulation of tissue insulin sensitivity in adult women. This conclusion is at odds with animal studies showing that ovariectomy promotes tissue insulin resistance (5–8). Differences between human and animal studies may be explained by species differences in the effect of sex steroids on tissue insulin action and/or glucose metabolism. In addition, an important caveat to these animal studies is that ovariectomy stimulates food intake and body fat accumulation (38–39), which has been shown to reduce insulin-stimulated glucose disposal by decreasing flux through non-oxidative pathways (40). In contrast, we did not observe changes in total or central adiposity with GnRHa administration (Table 1). Thus, species differences in the effect of ovarian hormones on energy homeostasis may account for divergent findings with respect to insulin action.
The short-term nature of our study influences the interpretation of our findings. Our model of ovarian suppression with GnRHa is not representative of the hormonal changes that accompany the normal menopause. In this respect, our findings are limited to the physiological effects of a short-term period of ovarian hormone deficiency on insulin-stimulated glucose metabolism. However, we specifically chose to study the effect of GnRHa over a short period of time (i.e., 2 months) to minimize the effects of ovarian suppression on other physiological systems that could influence glucose disposal measurements. In this respect, our study design permits a focused examination of the effects of ovarian hormone deficiency, per se, on insulin-stimulated glucose disposal. Importantly, this period of study should be more than sufficient to allow for any alterations in gene transcription and protein expression of insulin signaling pathways and glucose metabolism machinery in target tissues. On the other hand, the study period may not be sufficient to observe changes in insulin sensitivity related to the secondary effects of ovarian hormones on other physiological systems, such as what might occur during the normal menopause. For example, ovarian hormone deficiency associated with the menpause has been hypothesized to cause accumulation of body fat and redistribution of fat towards the abdominal compartment (41–42). With prolonged periods of ovarian hormone deficiency, such changes in body fat levels and distribution may develop and could promote insulin resistance. Indeed, a significant proportion of the age-related decline in insulin sensitivity is believed to be caused by changes in the size and distribution of adipose tissue (43–45). In this context, our conclusions are limited to the short-term effect of ovarian suppression on glucose metabolism. Nevertheless, our results do not support a direct effect of ovarian hormones on tissue insulin sensitivity or intracellular pathways of glucose metabolism.
We should acknowledge that our results could be influenced by the inclusion of amino acids during the clamp. Previous studies have shown that infusion of amino acids reduces whole-body and skeletal muscle glucose uptake in response to insulin (46–47), although it is important to note that this finding is not unanimous (48–49). For several reasons, we believe that any effect of amino acids on glucose disposal in our study was minimal. First, the effect of amino acids to inhibit glucose disposal (46) is similar mechanistically to the effects of fatty acids on glucose disposal (50). Although substrate-induced insulin resistance has been demonstrated in men, recent studies suggest it does not occur in women (51). Nonetheless, one could argue that if females are afforded protection against substrate-induced insulin resistance by ovarian hormones (51), women treated with GnRHa group could become susceptible to an effect of amino acids on glucose disposal. This possibility is unlikely, however, since there were no change in insulin-stimulated glucose disposal in either group with treatment. Second, rates of glucose disposal reported in the present study are very similar to those reported in healthy premenopausal women by our laboratory (9) and others (51–52). In comparison with these studies, we find no evidence for a profound reduction in insulin-stimulated glucose disposal reported with amino acid administration reported by others (46). Thus, we favor the interpretation that any effect of the amino acid infusion on insulin-stimulated glucose disposal in the present study was minimal.
In conclusion, our study showed that endogenous ovarian hormone suppression brought about by GnRHa administration in healthy, non-obese women does not alter total, oxidative or non-oxidative glucose disposal. These findings, along with prior studies (27, 28, 36, 37), support the hypothesis that endogenous ovarian hormones do not regulate tissue insulin sensitivity or intracellular pathways of glucose disposal in adult women.
Acknowledgments
The authors would like to thank all the participants who volunteered their time for this study. This work was supported by grants from the NIH (AG-021602; RR-00109).
Footnotes
Grant support: NIH AG-021602 and NIH RR-00109
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Rowe JR, Minaker KD, Pallotta JA, Flier JS. Characterization of the insulin resistance of aging. J Clin Invest. 1983;71:1581–7. doi: 10.1172/JCI110914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Defronzo RA. Glucose intolerance and aging. Evidence for tissue insensitivity to insulin. Diabetes. 1979;28:1095–101. doi: 10.2337/diab.28.12.1095. [DOI] [PubMed] [Google Scholar]
- 3.Wu SI, Chou P, Tsai ST. The impact of years since menopause on the development of impaired glucose tolerance. J Clin Epidemiol. 2001;54:117–20. doi: 10.1016/s0895-4356(00)00284-5. [DOI] [PubMed] [Google Scholar]
- 4.Godsland IF. Influence of female sex steroids on glucose metabolism and insulin action: human studies. J Intern Med. 1996;240:25–49. [PubMed] [Google Scholar]
- 5.Puah JA, Bailey CJ. Effect of ovarian hormones on glucose metabolism in mouse soleus muscle. Endocrinol. 1985;117:1336–40. doi: 10.1210/endo-117-4-1336. [DOI] [PubMed] [Google Scholar]
- 6.Ahmed-Sorour H, Bailey CJ. Role of ovarian hormones in the long-term control of glucose homeostasis. Hormone Res. 1980;13:396–403. doi: 10.1159/000179307. [DOI] [PubMed] [Google Scholar]
- 7.Kumagai S, Holmang A, Björntorp P. The effects of oestrogen and progesterone on insulin sensitivity in female rats. Acta Physiol Scand. 1993;149:91–7. doi: 10.1111/j.1748-1716.1993.tb09596.x. [DOI] [PubMed] [Google Scholar]
- 8.Rincon J, Holmang A, Wahlstrom EO, Lonnroth P, Björntorp P, Zierath JR, Wallberg-Henriksson H. Mechanisms behind insulin resistance in rat skeletal muscle after oophorectomy and additional testosterone treatment. Diabetes. 1996;45:615–21. doi: 10.2337/diab.45.5.615. [DOI] [PubMed] [Google Scholar]
- 9.Toth MJ, Sites CK, Eltabbakh GH, Poehlman ET. Effect of menopausal status on insulin-stimulated glucose disposal: comparison of middle-aged premenopausal and early postmenopausal women. Diabetes Care. 2000;23:801–6. doi: 10.2337/diacare.23.6.801. [DOI] [PubMed] [Google Scholar]
- 10.Walton C, Godsland IF, Proudler AJ, Wynn V, Stevenson JC. The effects of the menopause on insulin sensitivity, secretion and elimination in non-obese, healthy women. Eur J Clin Invest. 1993;23:466–73. doi: 10.1111/j.1365-2362.1993.tb00792.x. [DOI] [PubMed] [Google Scholar]
- 11.Proudler AJ, Felton CV, Stevenson JC. Ageing and the response of plasma insulin, glucose and C-peptide concentrations to intravenous glucose in postmenopausal women. Clin Sci. 1992;83:489–94. doi: 10.1042/cs0830489. [DOI] [PubMed] [Google Scholar]
- 12.O’Sullivan AJ, Ho KKY. A comparison of the effects of oral and transdermal estrogen replacement on insulin sensitivity in postmenopausal women. J Clin Endocrinol Metab. 1995;80:1783–8. doi: 10.1210/jcem.80.6.7775623. [DOI] [PubMed] [Google Scholar]
- 13.Cagnacci A, Soldani R, Carriero PL, Paoletti AM, Fioretti P, Melis GB. Effects of low doses of transdermal 17β-estradiol on carbohydrate metabolism in postmenopausal women. J Clin Endocrinol Metab. 1992;74:1396–40. doi: 10.1210/jcem.74.6.1317387. [DOI] [PubMed] [Google Scholar]
- 14.Lasco A, Alvaro S, Frisina N, Di Benedetto A, Denuzzo G, Cucinotta D. Long-term transdermal estrogen therapy improves lipid profile but not insulin resistance in healthy postmenopausal women. Diabetes Care. 2000;23:422–4. doi: 10.2337/diacare.23.3.422. [DOI] [PubMed] [Google Scholar]
- 15.Duncan AC, Lyall H, Roberts RN, Petrie JR, Perera MJ, Monaghan S, Hart DM, Connell JM, Lumsden MA. The effect of estradiol and a combined estradiol/progestagen preparation on insulin sensitivity in healthy postmenopausal women. J Clin Endocrinol Metab. 1999;84:2402–7. doi: 10.1210/jcem.84.7.5836. [DOI] [PubMed] [Google Scholar]
- 16.Cucinelli F, Paparella P, Soranna L, Barini A, Cinque B, Mancuso S, Lanzone A. Differential effect of transdermal estrogen plus progestagen replacement therapy on insulin metabolism in postmenopausal women: relation to their insulinemic secretion. Eur J Endocrinol. 1999;140:215–223. doi: 10.1530/eje.0.1400215. [DOI] [PubMed] [Google Scholar]
- 17.Mattiasson I, Rendell M, Tornquist C, Jeppsson S, Hulthen UL. Effects of estrogen replacement therapy on abdominal fat compartments as related to glucose and lipid metabolism in early postmenopausal women. Horm Metab Res. 2002;34:583–8. doi: 10.1055/s-2002-35420. [DOI] [PubMed] [Google Scholar]
- 18.Walker RJ, Lewis-Barned NJ, Sutherland WH, Goulding A, Edwards EA, deJong SA, Gold E, Walker HL. The effects of sequential combined oral 17β-estradiol and norethisterone acetate on insulin sensitivity and body composition in healthy postmenopausal women: a randomized single blind placebo-controlled study. Menopause. 2001;8:27–32. doi: 10.1097/00042192-200101000-00006. [DOI] [PubMed] [Google Scholar]
- 19.Kimmerle R, Heinemann L, Heise T, Bender R, Weyer C, Hirshberger S, Berger M. Influence of continuous combined estradiol-norethisterone acetate preparations on insulin sensitivity in postmenopausal nondiabetic women. Menopause. 1999;6:36–42. [PubMed] [Google Scholar]
- 20.Sites CK, L’Hommedieu GD, Toth MJ, Brochu M, Cooper BC, Fairhurst PA. The effect of hormone replacement therapy on body composition, body fat distribution, and insulin sensitivity in menopausal women: a randomized, double-blind, placebo-controlled trial. J Clin Endocrinol Metab. 2005;90:2701–7. doi: 10.1210/jc.2004-1479. [DOI] [PubMed] [Google Scholar]
- 21.Beckett T, Tchernof A, Toth MJ. Effect of ovariectomy and estradiol replacement on skeletal muscle enzyme activity in female rats. Metabolism. 2002;51:1397–401. doi: 10.1053/meta.2002.35592. [DOI] [PubMed] [Google Scholar]
- 22.Toth MJ, Sites CK, Matthews DE, Casson PR. Ovarian suppression with gonadotropin-releasing hormone agonist reduces whole body protein turnover in women. Am J Physiol. doi: 10.1152/ajpendo.00600.2005. in press. [DOI] [PubMed] [Google Scholar]
- 23.Cooper BC, Sites CK, Fairhurst PA, Toth MJ. Evidence against a role for ovarian hormones in the regulation of blood flow. Fertil Steril. doi: 10.1016/j.fertnstert.2006.01.017. in press. [DOI] [PubMed] [Google Scholar]
- 24.Abumrad NN, Rabin D, Diamond MP, Lacy WW. Use of a heated superficial hand vein as an alternative site for the measurement of amino acid concentrations and for the study of glucose and alanine kinetics in man. Metab Clin Exp. 1981;20:936–40. doi: 10.1016/0026-0495(81)90074-3. [DOI] [PubMed] [Google Scholar]
- 25.Toth MJ, Tchernof A, Rosen CJ, Matthews DE, Poehlman ET. Regulation of protein metabolism in middle-aged, premenopausal women: roles of adiposity and estradiol. J Clin Endocr Metab. 2000;85:1382–7. doi: 10.1210/jcem.85.4.6533. [DOI] [PubMed] [Google Scholar]
- 26.Toth MJ, Sites CK, Cefalu WT, Matthews DE, Poehlman ET. Determinants of insulin-stimulated glucose disposal in middle-aged, premenopausal women. Am J Physiol Endocrinol Metab. 2001;281:E113–21. doi: 10.1152/ajpendo.2001.281.1.E113. [DOI] [PubMed] [Google Scholar]
- 27.Diamond MP, Jacob R, Connolly-Diamond M, DeFronzo RA. Glucose metabolism during the menstrual cycle. Assessment with the euglycemic, hyperinsulinemic clamp. J Reprod Med. 1993;38:417–21. [PubMed] [Google Scholar]
- 28.Dumesic DA, Nielsen MF, Abbott DH, Eisner JR, Nair KS, Rizza RA. Insulin action during variable hyperglycemic-hyperinsulinemic infusions in hyperandrogenic anovulatory patients and healthy women. Fertil Steril. 1999;72:458–66. doi: 10.1016/s0015-0282(99)00265-4. [DOI] [PubMed] [Google Scholar]
- 29.Allsop JR, Wolfe RR, Burke JF. Tracer priming the bicarbonate pool. 1978;45:137–9. doi: 10.1152/jappl.1978.45.1.137. [DOI] [PubMed] [Google Scholar]
- 30.Wolfe RR, Shaw JHF, Nadel ER, Wolfe MH. Effect of substrate intake and physiological state on background 13CO2 enrichment. J Appl Physiol. 1984;56:230–4. doi: 10.1152/jappl.1984.56.1.230. [DOI] [PubMed] [Google Scholar]
- 31.Schoeller DA, Klein PD, Watkins JB, Heim T, MacLean WC. 13C abundances of nutrients and the effect of variations in 13C isotopic abundances of test meals formulated for 13CO2 breath tests. Am J Clin Nutr. 1980;33:2375–85. doi: 10.1093/ajcn/33.11.2375. [DOI] [PubMed] [Google Scholar]
- 32.Simonson DC, DeFronzo RA. Indirect calorimetry: methodological and interpretative problems. Am J Physiol Endocrinol Metab. 1990;258:E399–412. doi: 10.1152/ajpendo.1990.258.3.E399. [DOI] [PubMed] [Google Scholar]
- 33.Glickman SG, Marn CS, Supiano MA, Dengel DR. Validity and reliability of dual-energy X-ray absortiometry for the assessment of abdominal adiposity. J Appl Physiol. 2004;97:509–14. doi: 10.1152/japplphysiol.01234.2003. [DOI] [PubMed] [Google Scholar]
- 34.Ross R, Freeman J, Hudson R, Janssen I. Abdominal obesity, muscle composition, and insulin resistance in premenopausal women. J Clin Endocrinol Metab. 2002;87:5044–51. doi: 10.1210/jc.2002-020570. [DOI] [PubMed] [Google Scholar]
- 35.Abate N, Haffner SM, Garg A, Peshock RM, Grundy SM. Sex steroid hormones, upper body obesity, and insulin resistance. J Clin Endocrinol Metab. 2002;87:4522–7. doi: 10.1210/jc.2002-020567. [DOI] [PubMed] [Google Scholar]
- 36.Toth EL, Suthijumroon A, Crockford PM, Ryan EA. Insulin action does not change during the menstrual cycle in normal women. J Clin Endocrinol Metab. 1987;64:74–80. doi: 10.1210/jcem-64-1-74. [DOI] [PubMed] [Google Scholar]
- 37.Yki-Jarvinen H. Insulin sensitivity during the menstrual cycle. J Clin Endocrinol Metab. 1984;59:350–3. doi: 10.1210/jcem-59-2-350. [DOI] [PubMed] [Google Scholar]
- 38.Richard D. Effect of ovarian hormones on energy balance and brown adipose tissue thermogenesis. Am J Physiol Renal Physiol. 1986;250:R245–9. doi: 10.1152/ajpregu.1986.250.2.R245. [DOI] [PubMed] [Google Scholar]
- 39.Chen Y, Heiman ML. Increased weight gain after ovariectomy is not a consequence of leptin resistance. Am J Physiol Endocrinol Metab. 2001;280:E315–22. doi: 10.1152/ajpendo.2001.280.2.E315. [DOI] [PubMed] [Google Scholar]
- 40.Banerjee S, Saenger P, Hu M, Chen W, Barzilai N. Fat accretion and the regulation of insulin-mediated glycogen synthesis after puberty in rats. Am J Physiol Renal Physiol. 1997;273:R1534–9. doi: 10.1152/ajpregu.1997.273.4.R1534. [DOI] [PubMed] [Google Scholar]
- 41.Toth MJ, Tchernof A, Sites CK, Poehlman ET. Effect of menopausal status on body composition and abdominal fat distribution. Int J Obesity. 2000;24:226–31. doi: 10.1038/sj.ijo.0801118. [DOI] [PubMed] [Google Scholar]
- 42.Tchernof A, Desmeules A, Richard C, LaBerge P, Daris M, Mailloux J, Rhéaume C, DuPont P. Ovarian hormone status and abdominal visceral adipose tissue metabolism. J Clin Endocrinol Metab. 2004;89:3425–30. doi: 10.1210/jc.2003-031561. [DOI] [PubMed] [Google Scholar]
- 43.Ferrannini E, Vichi S, Beck-Nielsen H, Laasku M, Paolisso G, Smith U European Group for the Study of Insulin Resistance (EGIR) Insulin action and age. Diabetes. 1996;45:947–53. doi: 10.2337/diab.45.7.947. [DOI] [PubMed] [Google Scholar]
- 44.Kohrt WM, Kirwan JP, Staten MA, Bourey RE, King DS, Holloszy JO. Insulin resistance in aging is related to abdominal obesity. Diabetes. 1993;42:273–81. [PubMed] [Google Scholar]
- 45.Basu R, Breda E, Oberg AL, Powell CC, Man CD, Basu A, Vittone JL, Klee GG, Arora P, Jensen MD, Toffolo G, Cobelli C, Rizza RA. Mechanisms of the age-associated deterioration in glucose tolerance. Contribution of alterations in insulin secretion, action and clearance. Diabetes. 2003;52:1738–48. doi: 10.2337/diabetes.52.7.1738. [DOI] [PubMed] [Google Scholar]
- 46.Krebs M, Krssak M, Bernroider E, Anderwald C, Brehm A, Meyerspeer M, Nowotny P, Roth E, Walhausl W, Roden M. Mechanism of amino acid-induced skeletal muscle insulin resistance in humans. Diabetes. 2002;51:599–605. doi: 10.2337/diabetes.51.3.599. [DOI] [PubMed] [Google Scholar]
- 47.Pisters PWT, Restifo NP, Cersosimo E, Brennan MF. The effects of euglycemic hyperinsulinemia and amino acid infusion on regional and whole body glucose disposal in man. Metab Clin Exp. 1991;40:59–65. doi: 10.1016/0026-0495(91)90193-z. [DOI] [PubMed] [Google Scholar]
- 48.Buckspan R, Hoxworth B, Cersosimo E, Devlin JT, Horton E, Abumrad N. alpha-Ketoisocaproate is superior to leucine in sparing glucose utilization in humans. Am J Physiol Endocrinol Metab. 1986;251:E648–53. doi: 10.1152/ajpendo.1986.251.6.E648. [DOI] [PubMed] [Google Scholar]
- 49.Boden G, Tappy L. Effects of amino acids on glucose disposal. Diabetes. 1990;39:1079–84. doi: 10.2337/diab.39.9.1079. [DOI] [PubMed] [Google Scholar]
- 50.Roden M, Price TB, Perseghin G, Petersen KF, Rothman DL, Cline GW, Shulman GI. Mechanism of free fatty acid-induced insulin resistance in humans. J Clin Invest. 1997;97:2859–65. doi: 10.1172/JCI118742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Frias JP, Macaraeg GB, Ofrecio J, Yu JG, Olefsky JM, Kruszynska YT. Decreased susceptability to fatty acid-induced peripheral tissue insulin resistance in women. Diabetes. 2001;50:1344–50. doi: 10.2337/diabetes.50.6.1344. [DOI] [PubMed] [Google Scholar]
- 52.Perseghin G, Scifo P, Pagliato E, Battezzati A, Benedini S, Soldini L, Testolin G, Del Maschio A, Luzi L. Gender factors affect fatty acids-induced insulin resistance in non-obese humans: effects of oral steroidal contraception. J Clin Endocrinol Metab. 2001;86:3188–96. doi: 10.1210/jcem.86.7.7666. [DOI] [PubMed] [Google Scholar]