

Abstract
The expression of tumor suppressor gene DBC2 causes certain breast cancer cells to stop growing [1]. Recently, DBC2 was found to participate in diverse cellular functions such as protein transport, cytoskeleton regulation, apoptosis and cell cycle control [2]. Its tumor suppression mechanism, however, remains unclear. In this paper, we demonstrate that DBC2 suppresses breast cancer proliferation through down-regulation of Cyclin D1 (CCND1). Additionally, the constitutional overexpression of CCND1 prevented the negative impact of DBC2 expression on their growth. Under a CCND1 promoter, the expression of CCNE1 exhibited the same protective effect. Our results indicate that the down-regulation of CCND1 is an essential step for DBC2's growth suppression of cancer cells. We believe that this discovery contributes to a better understanding of DBC2's tumor suppressor function.
Keywords: DBC2, breast cancer, cyclin D1, growth suppression
Introduction
It is widely accepted that genetic alterations in cancer-related genes ultimately affect various cellular pathways and contribute to the rise of breast cancer. Studies on these cancer-related genes have not only contributed to a better understanding of various aspects of the cellular function but also laid the foundation for developing new strategies for cancer treatment. Although therapies for breast cancer have not been developed from studies of tumor suppressor genes, findings on their cellular function continue to shed light on the molecular mechanism of carcinogenesis.
DBC2, cloned from human chromosome 8p22, is a tumor suppressor gene involved in breast cancer development. Genetic analyses have demonstrated that the DBC2 locus is frequently deleted in breast cancer and that DBC2 expression is silenced in breast and lung cancers [1]. Biological studies have revealed that DBC2 reactivation can lead to growth arrest of breast cancer cells [1]. Further analyses of DBC2 have suggested its participation in cellular pathways related to ubiquitination, cell cycle control, protein transport, apoptosis and cytoskeleton regulation [2; 3; 4]. Its tumor suppression mechanism, however, remains unclear.
DBC2 belongs to the RHOBTB protein family. The first of its two BTB domains was demonstrated to interact with CUL3, suggesting the possibility that DBC2 serves as adapter to Cul3-based E3 ligase [5]. Indeed, BTB protein Mel-26 acts as an adaptor between the Cul-3 ubiquitin ligase and its substrate, MEI-1 [6]. All three BTB proteins in S. Pombe (Btb1, Btb2 and Btb3) were found in the E3 ubiquitin ligase complex with Pcu3 (CUL3 homolog) and Pip1 (RBX1 homolog) [7]. These findings elicit several questions: does DBC2 play a role in protein degradation through ubiquitination and, if so, what is the target substrate?
The cell cycle is driven by the activation of cyclin-dependent kinases (CDKs) whose activities are orchestrated by key regulators such as cyclins and CDK inhibitors (CDKIs). The concentrations of these regulators at critical points of the cell cycle are mostly controlled by ubiqutin-mediated proteolysis by 26S proteasome. Two previous observations imply that DBC2 may play a role in cell cycle control through ubiquitination: 1) manipulation of DBC2 functionality disturbs the expression level of many cell cycle regulators [2] and 2) DBC2 physically interacts with CUL3 ubiquitin ligase [5]. To test this hypothesis, the effects of DBC2 expression on cell cycle regulators were investigated.
Results and Discussions
We first studied the effects of exogenous DBC2 expression on cell cycle regulators such as CCNE, CCNB, CCND1, CCNH, p34 and p27. 293 cells were transformed with a mammalian expression vector containing DBC2 (pCMV-DBC2) or an empty vector (pCMV) and DBC2 expression was confirmed by western blot analysis (Figure 1). Western blot analysis of cell cycle regulators listed above revealed that CCND1 expression decreased in the pCMV-DBC2 transformants whereas CCNE, CCNB, CCNH, p34 and p27 expressions were unaffected (Figure 1).
Figure 1. Western blot analysis of 293 transformants.
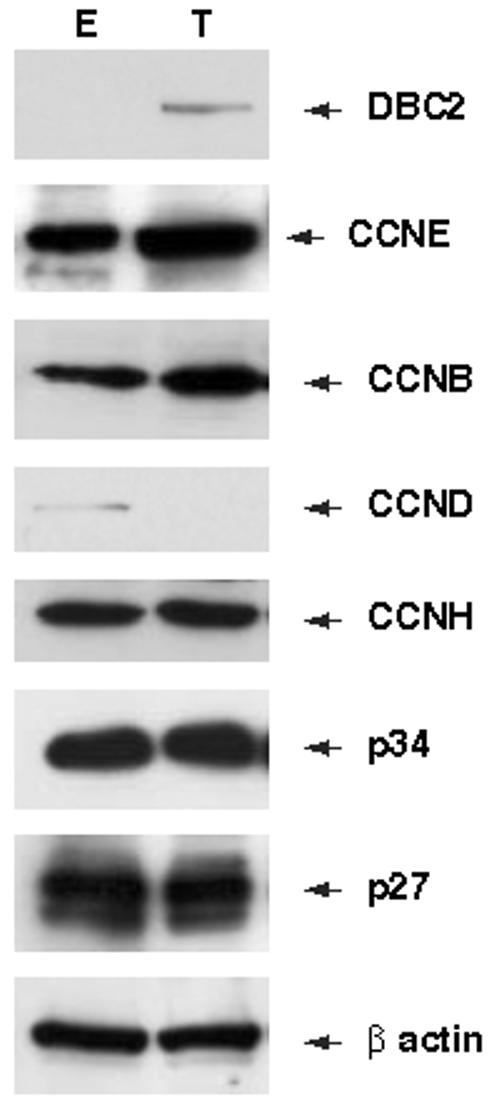
293 cells were transformed with pCMV or pCMV-DBC2 using Lipofectamine Transfection Reagent (Invitrogen, Carlsbad, CA USA) and harvested with CelLytic MT, 48 hours after transformation. T and E in the figure represent pCMV-DBC2 and pCMV transformants, respectively. Screening of cell cycle regulators. Expression levels of CCNE, CCNB, CCND1, CCNH, p34 and p27 were analyzed with corresponding antibodies: H-145, H-433, M-20, C-18, H-297 and C-19 respectively (Santa Cruz Biotechnology).
To confirm the down-regulation of CCND1 by DBC2, we utilized an inducible gene expression system [8]. We established two host cell lines harboring inducible DBC2 (T-47D39, 293A8) in which DBC2 expression was induced by administrating muristerone A. DBC2 induction was then verified by western blot analysis, revealing that the amount of induced DBC2 was dependent on the dose of administered muristerone A (Figure 2A).
Figure 2. DBC2 induction and CCND1 expression.
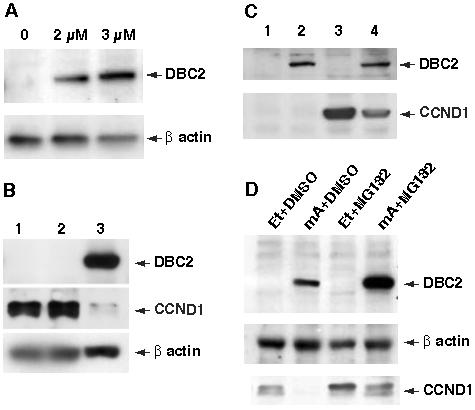
A. Induction of DBC2 by muristerone A (mA). 293A8 cells were cultured in a medium containing various concentrations of mA and examined for DBC2 expression by western blot analysis with anti-DBC2 antibody (N15). The concentration of mA is indicated at the top of each lane. B. DBC2 induction and endogenous CCND1. T-47D cells were cultured in DMEM (1), DMEM with ethanol (2) or DMEM with 5 μM muristerone A (3). The cells were harvested 6 hours after induction and analyzed by western blot analysis with anti-flag M2 antibody and anti-CCND1 antibody (M-20) (Santa Cruz Biotech, CA, USA). Drastic reduction of CCND1 protein in lane 3 was observed. C. 293A8 cells were transformed with either pCMV-CCND1 (lanes 3 and 4) or an empty vector (lanes 1 and 2). The transformants were cultured in DMEM with ethanol (lanes 1 and 3) or DMEM with 2 μM muristerone A (lanes 2 and 4) and then harvested 6 hours after induction. The cell lysates were analyzed by western blot analysis with anti-CCND1 antibody (M-20). Reduced CCND1 expression was noted when DBC2 was induced by muristerone A administration. D. DBC2 was induced in T-47D39 cells by culturing the cells in DMEM containing 5 μM muristerone A for 5 hours. The same amount of ethanol was added to the medium for the control. To inhibit 26S proteasome, the cells were incubated in a medium containing 50 μM MG132 for an hour until harvest. DMSO was added as a control. The cell lysates were analyzed by western blot analysis. Et and mA in the figure stand for ethanol and muristerone A, respectively.
T-47D39 cells express a significant amount of endogenous CCND1. We investigated the effects of DBC2 induction in T-47D39 cells on the endogenous CCND1. Western blot analysis confirmed successful induction of DBC2 and revealed that CCND1 protein is substantially less in T-47D39 cells cultured with muristerone A than in those with just ethanol (Figure 2B). A previous study demonstrated that DBC2 expression did not affect the amount of CCND1 transcript [2]. These findings indicate that down-regulation of CCND1 by DBC2 is likely to be through posttranscriptional regulation. To test this hypothesis, we studied DBC2's influence over exogenous CCND1 under a constitutive promoter. A mammalian expression vector containing CCND1 under a CMV promoter (pCMV-CCND1) was transfected into 293A8 cells and DBC2 was induced. When DBC2 was induced, the amount of CCND1 protein markedly decreased although still existed considerablly (Figure 2C). These findings corroborate with our hypothesis that DBC2 negatively regulates CCND1 expression posttranscriptionally.
DBC2 belongs to a BTB protein family and also binds to CUL3 ubiquitin ligase. In order to clarify the role of DBC2 in the ubiquitin pathway, we studied the influence of 26S proteasome inhibition by MG132 on protein levels of DBC2 and CCND1. We found that DBC2 protein significantly increased in MG132 treated cells in comparison to those without MG132. (Figure 2D) This indicates that DBC2 is, in fact, an active target of 26S-mediated protein degradation. As we expected, CCND1 protein increased when cells were cultured with MG132 (Figure 2D). CCND1 protein, however, still decreased by DBC2 expression even in the cells treated with MG132, which implies that CCND1 down-regulation by DBC2 can be at least partially attributed to a pathway that is independent of 26S proteasome.
DBC2 expression hinders the growth of T-47D cells through an unknown mechanism [1]. Since T-47D is driven by a CCND1 overexpression that can be depleted by reactivation of DBC2, we hypothesized that the growth suppression of T-47D by DBC2 reactivation requires the down-regulation of CCND1. We tested this hypothesis through a series of rescue experiments. First, we determined whether constitutive overexpression of CCND1 in T-47D cells could negate the growth arrest caused by DBC2. Even after DBC2 was induced, CCND1 protein remained at a detectable level by western blot analysis (Figure 3A). Such manipulation of T-47D cells prevented growth arrest by DBC2, indicating that negative regulation of CCND1 is essential for DBC2 to halt T-47D proliferation (Figure 3B). 293 cells, on the other hand, continued to grow even when DBC2 was expressed. Transformation with the CCND1 construct did not have a significant impact on the growth of 293 cells, suggesting growth of 293 cells were driven by other cellular pathways than those involving CCND1.
Figure 3. Growth curves of T47D transformants.
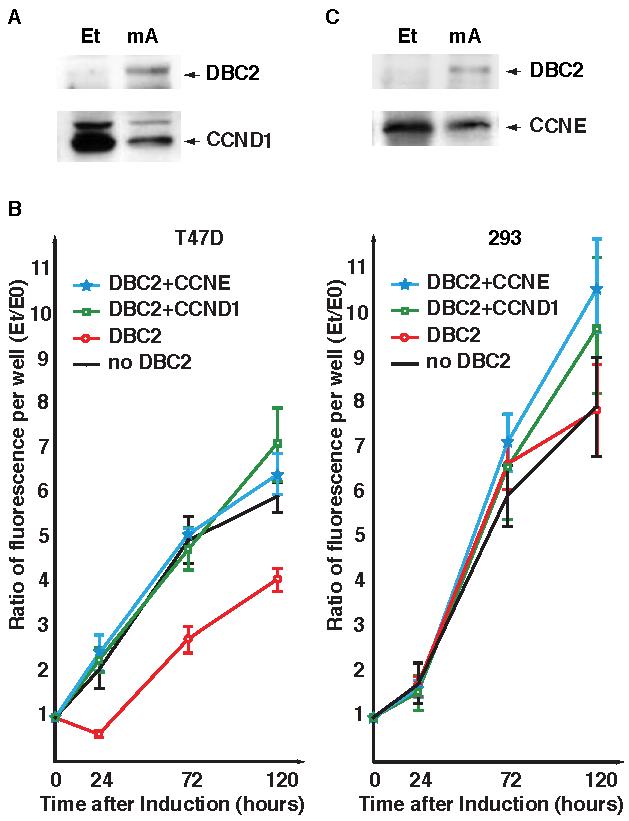
A. Western blot analysis of T47D39 transfected with pCMV-CCND1. Et and mA in the figure stand for ethanol and muristerone A, respectively. DBC2 was detected by anti-DBC2 antibody (N15) and CCND1 was detected by anti-CCND1 antibody (M-20). CCND1 protein decreased after DBC2 expression, but still remained detectable. B. Growth curves of T47D and 293 cells. The left pane shows growth curves of T-47D transformants and the right pane, those of 293. The average ratios over time point 0 are plotted in the graph. The error bars represent 2SD. In both panes, solid circles with black lines represent transformants with the empty vector without DBC2; open circles (red), those with the empty vector and DBC2 expression; open squares (green), those with pCMV-CCND and DBC2 expression; stars (blue), those with pCCND-CCNE and DBC2 expression. T47-D cells exhibited slow growth when DBC2 was expressed. DBC2's influence was offset by either CCNE or CCND1 expression. None of the manipulations disturbed the growth of 293 cells. C. Western blot analysis of T47D39 transfected with pCCND-CCNE. DBC2 was detected by anti-DBC2 antibody (N15) and CCNE was detected by anti-CCNE antibody (H-145). A significant amount of CCNE was observed in T47D39 cells before and after DBC2 expression.
CCNE knockin rescues CCND1 deficiency in vivo [9]. This raises the possibility that CCNE expression under the CCND1 promoter may prevent growth arrest by DBC2. To examine this possibility, CCNE ORF was cloned downstream of the CCND1 promoter (pCCND-CCNE) and transfected into T-47D cells. Exogenous CCNE expression was slightly decreased but still at a high level when DBC2 was induced (Figure 3C). T-47D39 cells containing the pCCND-CCNE did not succumb to DBC2 induction and kept growing as steadily as the control with no induction (Figure 3B). These results clearly demonstrate that DBC2 stops growth of T-47D through negative regulation of the CCND1 pathway.
RHOBTB proteins have been found in diverse organisms such as slime molds, arthropods, tunicates and vertebrates. Among three mammalian RHOBTBs, only RHOBTB1 is evolutionarily conserved, suggesting that RHOBTB1 is likely to carry out fundamental cellular activities. In contrast, orthologous genes of DBC2 have been discovered only in mammals, which implies that DBC2 function may be related to subtle orchestration of cellular pathways, but not vital functions. In fact, many cells including breast cancer cells proliferate better without DBC2 protein.
It was demonstrated that RAS and c-erb-B2 absolutely depend on CCND1 to trigger malignant transformations, but other genes do not [10]. This suggests that certain tumor cells are resistant to DBC2 expression, since DBC2 achieves growth arrest through the negative regulation of CCND1. For example, oncogenic pathways driven by c-myc and Wint-1 are not disturbed by CCND1 depletion and therefore are likely to be unaffected by reactivation of DBC2. This would explain why many breast cancer cells retain DBC2 expression.
Materials and Methods
Cell lines and cell culture
Cell culture media and reagents were purchased from Invitrogen. All cell lines were obtained from American Tissue Type Collection (ATCC) and maintained in Dulbecco's Modified Eagle Medium (ICN Biomedicals Inc., Costa Mesa, CA) supplemented with 10 % FBS and antibiotics (50 units/ml penicillin and 50 μg/ml streptomycin) at 37 °C in a 5 % CO2 incubator, except for T-47D cells which were cultured with 15 % FBS. The cell lines with the inducible DBC2 gene were established by previously described methods [8]. For induction, the host cells were cultured for 4 hours or longer in the medium containing muristerone A at a final concentration of 2 to 5 μM. Proteasome inhibition was achieved by adding MG132 (Sigma-Aldrich) at 50 μM 6 hours prior to the harvest.
Reagents
All restriction enzymes were purchased from New England Biolabs. LR Clonase was purchased from Invitrogen.
Vector constructs
The CMV promoter of pcDNA6.2 was removed by digestion with SacI and SpeI and replaced with the promoter region of CCND1 (sequence between positions 69,162,057 and 69,165,110 of NC_000011, gi: 51511727). The CCNE coding region was cloned by recombination between the modified vector and pENTR221-CCNE (IOH27850) procured from Invitrogen with LR clonase (Invitrogen). The pCMV-CCND1 vector was constructed by cloning the coding region of CCND1 into pcDNA6.2-v5-DEST (Invitrogen) with LR clonase. For constitutive DBC2 expression, pCMV-DBC2 was constructed by cloning the coding region of DBC2 into pCMV-tag2B (Stratagene). Transformation was performed with Lipofectamine (Invitrogen).
Western blot analysis
Cell lysates were prepared with CelLytic MT (Sigma-Aldrich). The primary antibodies used in this study were purchased from Santa Cruz Biotech unless otherwise stated: anti-CCND1 (M-20 and DCS-6, 1:3500), anti-CCNE (H-145, 1:3000), anti-β actin (I-19, 1:7000), anti-p34 (H-297, 1:3000), anti-p27 (C-19, 1:2000), anti-cyclin B (H-433, 1:3000) and anti-cyclin H (C-18, 1:3000) antibodies. Anti-DBC2 antibody (N-15, 1:1000) was obtained from Delta Biolabs. Anti-flag M2 antibody (1:2000) was purchased from Sigma-Aldrich. The secondary antibody was goat anti-rabbit IgG conjugated with HRP (1:25000) obtained from Santa Cruz Biotech. The antibodies were detected with ECL Plus Blotting Reagent (Amersham).
Cell Proliferation Assay
The cells were plated into 96-well plates and harvested at 4 time points. The harvested cells were treated with Cyquant Cell Proliferation Assay kit (Invitrogen). The fluorescence per well was quantitated twice with Thermo Fluoroskan Ascent.
Acknowledgment
This work was supported by a grant from the National Institute of Health (R01-CA-100006-01) to MH. We are indebted to Carissa Bob Meyer and Carina Sorenson for their help in the construction of the expression vectors. We thank Faith Chang and Noriko Simorowski for their technical assistance.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Hamaguchi M, Meth JL, Von Klitzing C, Wei W, Esposito D, Rodgers L, Walsh T, Welcsh P, King MC, Wigler MH. DBC2, a candidate for a tumor suppressor gene involved in breast cancer. Proc Natl Acad Sci U S A. 2002;99:13647–13652. doi: 10.1073/pnas.212516099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Siripurapu V, Meth JL, Kobayashi N, Hamaguchi M. DBC2 Significantly Influences Cell Cycle, Apoptosis, Cytoskeleton and Membrane Trafficking Pathways. J Molecular Biology. 2004 doi: 10.1016/j.jmb.2004.11.043. in press. [DOI] [PubMed] [Google Scholar]
- 3.Chang FK, Sato N, Kobayashi-Simorowski N, Yoshihara T, Meth JL, Hamaguchi M. DBC2 is essential for transporting vesicular stomatitis virus glycoprotein. J Mol Biol. 2006;364:302–8. doi: 10.1016/j.jmb.2006.09.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Xu L, Wei Y, Reboul J, Vaglio P, Shin TH, Vidal M, Elledge SJ, Harper JW. BTB proteins are substrate-specific adaptors in an SCF-like modular ubiquitin ligase containing CUL-3. Nature. 2003;425:316–21. doi: 10.1038/nature01985. [DOI] [PubMed] [Google Scholar]
- 5.Wilkins A, Ping Q, Carpenter CL. RhoBTB2 is a substrate of the mammalian Cul3 ubiquitin ligase complex. Genes Dev. 2004;18:856–61. doi: 10.1101/gad.1177904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pintard L, Willis JH, Willems A, Johnson JL, Srayko M, Kurz T, Glaser S, Mains PE, Tyers M, Bowerman B, Peter M. The BTB protein MEL-26 is a substrate-specific adaptor of the CUL-3 ubiquitin-ligase. Nature. 2003;425:311–6. doi: 10.1038/nature01959. [DOI] [PubMed] [Google Scholar]
- 7.Geyer R, Wee S, Anderson S, Yates J, Wolf DA. BTB/POZ domain proteins are putative substrate adaptors for cullin 3 ubiquitin ligases. Mol Cell. 2003;12:783–90. doi: 10.1016/s1097-2765(03)00341-1. [DOI] [PubMed] [Google Scholar]
- 8.Stolarov J, Chang K, Reiner A, Rodgers L, Hannon GJ, Wigler MH, Mittal V. Design of a retroviral-mediated ecdysone-inducible system and its application to the expression profiling of the PTEN tumor suppressor. Proc Natl Acad Sci U S A. 2001;98:13043–8. doi: 10.1073/pnas.221450598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Geng Y, Whoriskey W, Park MY, Bronson RT, Medema RH, Li T, Weinberg RA, Sicinski P. Rescue of cyclin D1 deficiency by knockin cyclin E. Cell. 1999;97:767–77. doi: 10.1016/s0092-8674(00)80788-6. [DOI] [PubMed] [Google Scholar]
- 10.Yu Q, Geng Y, Sicinski P. Specific protection against breast cancers by cyclin D1 ablation. Nature. 2001;411:1017–21. doi: 10.1038/35082500. [DOI] [PubMed] [Google Scholar]