

Abstract
Trisomy is a genetic abnormality of considerable medical importance. The most familiar example is trisomy 21, which causes Down Syndrome [Cummings, M. R. (1988) Human Heredity: Principles and Issues (West Publishing Company, New York)]. In a classic paper, Axelrod and Hamilton [Axelrod, R. & Hamilton, W. D. (1981) Science 211, 1390–1396] offered a chromosomal drive (CD) hypothesis based on the game iterated prisoner’s dilemma (IPD) to explain the evolution of an increased frequency of trisomic pregnancies with maternal age. In this paper we explore this hypothesis and its predictions in detail. On closer examination we find that IPD does not provide an adequate model for the CD hypothesis. Therefore, we develop a more suitable model and explore the conditions necessary for it to explain maternal age-dependent trisomy. Our results demonstrate that a relationship between the decay of a female’s reproductive potential and chromosomal drive must exist for the CD hypothesis to work. With appropriate parameter values, a comparison of model predictions with empirical estimates for the age-dependence of trisomy reveals a striking correspondence. We point out a close correspondence between other predictions made by the CD hypothesis and empirical observations, as well.
Meiotic nondisjunction is the failure of homologous chromosomes to segregate properly to opposite poles during meiosis resulting in the production of gametes that have an improper chromosome complement. When a normal gamete combines with a gamete that has an extra chromosome, the resulting zygote is trisomic. Trisomies have been identified for most chromosomes in humans (1–3), but the viability of most trisomic zygotes is low. A familiar example with a relatively high viability is trisomy 21, which results in Down Syndrome (4).
Trisomy has been the focus of extensive medical research, but the molecular mechanism by which nondisjunction occurs is still not understood (1, 5–8). One feature common to most autosomal trisomies is an increase in the frequency of trisomic pregnancies with increasing maternal age (1–3). A complete explanation for this is still lacking, but at least two possibilities have been proposed (also see refs. 9 and 10). One contends that the frequency of nondisjunction remains constant but that a screening mechanism becomes less effective at aborting trisomic zygotes in older females (11). The other maintains that the frequency of nondisjunction increases with maternal age (12). Past evidence has been indirect and inconclusive (11, 13), but recent techniques have allowed a direct examination of the relationship between maternal age and the frequency of nondisjunction in oocytes. Results demonstrate that there is an increase in the frequency of nondisjunction with maternal age (5, 6, 12, 14), and many researchers now argue for its importance in human trisomy, suggesting that a maternal screening mechanism is an unlikely or incomplete explanation (12, 15, 16). The issue, however, is not yet completely resolved (6–8, 12, 17–19).
Although these hypotheses center on the proximate cause of age-dependent trisomy, relatively little research addresses its evolutionary (ultimate) cause. Some evolutionary hypotheses have been suggested (20, 21), but none has been examined in any detail to determine its adequacy. A thorough examination is essential to understand the assumptions implicit in these hypotheses as well as to allow a rigorous comparison of predictions with empirical data. Here we examine one of these hypotheses, the chromosomal drive (CD) hypothesis (20), in detail. We demonstrate that an important and previously unrealized assumption needs to be made for this hypothesis to explain age-dependent trisomy. We consider the likelihood of this assumption being met in humans, and then compare the age-dependence predicted by the CD hypothesis with that observed for several autosomal trisomies provided this assumption is met.
Evolutionary Explanations for Trisomy
In light of recent evidence (5, 6, 12, 14–16) we proceed under the assumption that the proximate cause of age-dependent trisomy is an increase in the frequency of nondisjunction with maternal age. Although it is still unclear exactly how this might occur at the cellular level, our goal is to understand why it occurs, and thus we need only assume that some mechanism exists. The question we address is, why has this phenomenon evolved and/or why has not natural selection removed such a deleterious phenomenon? We emphasize that the hypotheses generated from such ultimate questions are generally complimentary to hypotheses of proximate mechanisms rather than alternatives.
One evolutionary explanation for the age dependence of nondisjunction is provided by the evolutionary theory of senescence (22). This theory asserts that natural selection on characters expressed late in life is weaker than that on characters expressed early in life. Therefore, we might expect an increase in malfunction of all physiological processes with advancing age simply because such deterioration has a relatively small fitness cost. We consider this to be a null hypothesis because it asserts that age-dependent nondisjunction is essentially unavoidable. The alternative evolutionary hypothesis examined here is based on a suggestion by Axelrod and Hamilton (20). They proposed that nondisjunction and trisomy are the side effects of adaptive evolution at the chromosome level. Kloss and Nesse (21) have previously considered the validity of this hypothesis and rejected it because there was no strong evidence at that time of the frequency of nondisjunction increasing with maternal age. As already mentioned, however, new techniques have allowed a more direct examination of this question and have revealed that nondisjunction does become more frequent as females age. Therefore, it is worthwhile to reexamine this hypothesis in detail.
Axelrod and Hamilton (20) suggested that nondisjunction results from chromosomal drive [see Haig (23) for discussion of a similar phenomenon in angiosperms]. Their article focused on the game known as iterated prisoner’s dilemma (IPD), and they noted that when two opponents play the game, cooperation can be favored if the probability of a future encounter between them is high. They then suggested that IPD might provide a suitable model for nondisjunction in humans. During oogenesis one chromosome of each homologous pair is discarded into the polar body (4), and, as a result, if a chromosome can enhance its likelihood of going to the ovum rather than the polar body, it will be at an advantage. They suggested viewing such a drive strategy as defection in IPD, and a passive strategy as cooperation. As a female nears menopause, the probability of a future encounter between chromosomes decreases to zero; thus, according to their IPD results each chromosome should defect, and when both defect simultaneously, “an extra chromosome in the offspring could be the occasional result” (20).
Although their verbal argument seems plausible, there are some difficulties with using IPD as a model for this phenomenon. The general IPD results are based on models with a constant probability of future encounter and assume that the number of encounters is potentially infinite (20). It is not clear that these results carry over to instances where the probability of future encounters changes. For example, it can be shown that when there are a finite number of interactions in IPD (e.g., because of menopause), the only stable strategy is constant defection. This result is enough to demonstrate that IPD cannot explain the age dependence of trisomy. Furthermore, the IPD model assumes that a chromosome in a developing egg is able to detect a defection during the development of previous eggs (20). This assumption seems quite restrictive. These considerations rule out IPD as an adequate model of trisomy, but the CD hypothesis might still be tenable. Presumably, however, if the CD hypothesis is to provide an explanation, conditions other than those specified in IPD models must be met. We now construct a model specific to the CD hypothesis to explore what these conditions are.
A Model of Chromosomal Drive
Consider a pair of homologous chromosomes in a female and suppose that each chromosome has a drive strategy, z(t), that determines its aggressiveness at getting into the ovum (z = 0 is passive). z depends on t, the time since female sexual maturity, because we want to consider the possibility of this strategy changing as the female ages. The total reproductive output (fitness), W, of any chromosome is the sum from sexual maturity (t = 0) to menopause (t = T) of its rate of reproductive output, m, at each time multiplied by the probability that the female is still reproductively active at that time, l(t);
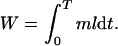 |
1 |
First, consider m. For each chromosome a higher drive results in a higher probability of getting into the ovum, but it is also likely that the probability of nondisjunction, N(z̄), increases as the average level of drive within the female, z̄, increases. Thus, m is the product of two factors: (i) the probability that the chromosome gets into the ovum given only one chromosome does, and (ii) the probability that only one chromosome gets into the ovum (i.e., 1 − Prob[nondisjunction]). This formulation assumes that trisomic zygotes have zero fitness and no nullosomic zygotes are formed (the CD hypothesis predicts that nullosomics should not occur; see below). We model (i) as a lottery; if a chromosome uses strategy z against a chromosome using ẑ then this probability is (z + α)/(z + ẑ + 2α). Here, each chromosome has α “free tickets” in the lottery and z (and ẑ) is the additional number that each “purchases.” A large α means that a chromosome’s drive must be very large to affect (i) substantially. Thus, expressing everything in terms of z and z̄, the average drive within a female,
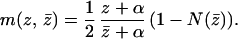 |
2 |
Now consider l(t). In general, we have l(t) = exp(−∫0tμdτ), where μ is mortality rate. Before menopause, a female’s reproduction may cease through her mortality or through the failure of her reproductive system (e.g., ovarian cancer). In the absence of extended parental care, these two scenarios are evolutionarily equivalent (see below). Therefore, we refer to μ as a generalized mortality rate; it can be thought of as the female’s probability of mortality per unit time or the probability of failure of her reproductive system per unit time. How will generalized mortality rate be affected by chromosomal drive? At present there is no direct evidence available to answer this question decisively, but it seems likely that a high average level of drive might cause a high generalized mortality rate. This is particularly true if strong meiotic drive has pleiotropic effects in mitosis. For example, a high average level of chromosomal drive could increase the likelihood of failure of a female’s reproductive system through an increased likelihood of cysts and/or cancer (24). Some support for such effects is found in the high frequency of nondisjunction observed in cancerous cells (25). Additionally, it is conceivable that a high average level of drive might affect all cells of a female’s body adversely, thereby increasing her mortality rate. In either case, to capture such effects we simply assume that generalized mortality rate, μ(z̄), increases with z̄. As discussed below, this assumption turns out to be central to the CD hypothesis.
We now characterize the evolutionarily stable strategy (26) strategy z*(t) by using a combination of inclusive fitness and control theory (27). In control theoretic terminology, z(t) is the control variable and l(t) is the state variable. The inclusive fitness increment of a chromosome increasing its drive during oogenesis at time t, given the female is still reproductively active, is,
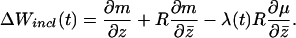 |
3 |
Here, λ(t) is the expected future fitness at time t, and R is the coefficient of consanguinity of two chromosomes within a female [i.e., the relatedness between two homologous chromosomes selected randomly with replacement (27)]. Eq. 3 provides insight into the form of the evolutionarily stable strategy because ΔWincl(t) must equal zero at all times along z*(t) (27).
Rather than presenting a full analysis here, we simply note that this model is a special case of a general altruism model in which, if ∂2N/∂z̄2 ≥ 0 and ∂2μ/∂z̄2 ≥ 0, then z*(t) is monotonically increasing with time (28). These conditions imply that both the probability of nondisjunction and generalized mortality rate increase linearly or faster with the average level of drive. Note that these are sufficient conditions and therefore we expect the strength of chromosomal drive and thus the frequency of nondisjunction and trisomy to increase with maternal age under quite general circumstances.
To understand why this occurs, consider expression 3. The first two terms of Eq. 3 comprise the inclusive fitness increment through current reproductive output. The first term gives the amount that a chromosome’s current reproductive output will increase with a unit increase in its level of drive, when the average level of drive is held constant. If a chromosome increases its drive, however, this increases the average level of drive as well. This affects the current reproductive output of both chromosomes in two ways. First, it increases the probability of nondisjunction, and second, it decreases the value of each individual “ticket” in the lottery by increasing the total number of tickets held. The derivative ∂m/∂z̄ encompasses both of these effects, and, therefore, the coefficient of consanguinity, R, in the second term is calculated by sampling with replacement to include these effects for both chromosomes (27). A higher drive, however, also jeopardizes the future reproduction of both chromosomes through an increased generalized mortality rate (the third term in Eq. 3). This effect is large when the female is far from menopause because λ(t) is large. As the female approaches menopause, however, λ(t) decreases to zero (at menopause there is no future reproduction), and thus the effect of drive on mortality rate becomes unimportant; therefore, z*(t) increases. At menopause, λ(T) = 0, Eq. 3 becomes
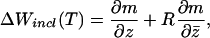 |
4 |
and drive reaches its maximum. Note however, that we do not expect unlimited drive because there is still a balance being struck between the two terms of Eq. 4.
If generalized mortality rate is unaffected by the average level of drive in a female (i.e., is independent of z̄), then Eq. 3 becomes Eq. 4 at all times. This implies that z*(t) is then constant because nothing in Eq. 4 is explicitly time-dependent (28). A chromosome’s fitness is then completely determined by its rate of reproductive output per unit time, m, regardless of the amount of time remaining, and, therefore, there is no “reason” for it to alter its level of drive. Note that this is true even if generalized mortality rate changes with time, as would be the case if the female senesced, for example. Thus, an important conclusion is that a positive relationship between generalized mortality rate and the strength of chromosomal drive is necessary for the CD hypothesis to predict an age dependence in the frequency of trisomy; otherwise, the model predicts maximal drive (given by Eq. 4) just as does IPD.
Assuming that drive does increase generalized mortality (by hastening the decay of a female’s reproductive system, for example), a comparison of the predicted age dependence of trisomy with data reveals remarkable concordance (Fig. 1). It is difficult to know which functions to choose for N(z̄) and μ(z̄) or which parameter values to use, but most choices yield predictions qualitatively similar to observations. In particular, the data reveal a pattern whereby the frequency of trisomic pregnancies increases slowly with age between sexual maturity and age 35 and then there is a dramatic increase in frequency from age 35 onward (Fig. 1). This sharp increase in frequency after age 35 was predicted for all functions tried and for all parameter values that place the predicted frequency within the correct range. It appears to be a generic property of having a finite reproductive time horizon (i.e., menopause). Thus, one of the main features of the data on trisomy in humans is predicted by the CD hypothesis.
Figure 1.

Maternal age dependence of trisomy. Vertical axis (ρ) is percentage of clinically recognized pregnancies that are trisomic. Horizontal axis (t) is year of reproductive life (female has a 30-year reproductive life span). Thin lines are data from ref. 3. Solid, C group chromosomes; dash, D group chromosomes; dot, chromosomes 17–20; dot–dash, chromosome 16; dot–dot–dash, chromosome 21. Thick lines are model predictions using N(z̄) = φz̄2 and μ(z̄) = μ0 + βz̄2. Solid, r = 0.5; α = 5; μ0 = 0.05; φ = 1/300; β = 0.005. Dotted, the same values except r = 0.75. Model predictions were calculated by using scientific workplace 2.5.
The model also predicts that, as R increases, the frequency of trisomy at all ages decreases because the genetic conflict between homologous chromosomes is reduced (Fig. 1). Reducing the effectiveness of drive at getting into the ovum (increasing α) or increasing the effect of drive on generalized mortality rate also decreases the frequency of trisomy at all ages. The frequency of trisomy nevertheless is always predicted to increase with maternal age provided generalized mortality increases with z̄. Unfortunately, no data are yet available to test these additional predictions.
If extended parental care is important, then it becomes necessary to distinguish between the two types of generalized mortality. The reason is that the death of a female clearly affects the parental care given to existing offspring whereas the failure of her reproductive system need not. All else equal, if z̄ does affect mortality rate per se, then this will likely select for lower levels of nondisjunction and hence trisomy.
Last, we note that, in deriving expression (Eq. 2) for the rate of reproductive output, we neglected a benefit of higher chromosomal drive that may select for higher levels of nondisjunction and trisomy. Although we defined m as the rate of reproductive output per unit time, our expression (Eq. 2) really represents reproductive output per pregnancy. If most trisomic zygotes are aborted early in pregnancy, however, then a higher average level of drive will result in a higher average number of pregnancies per female lifetime, and this reduces the cost of drive (we thank D. Haig, Harvard University, for pointing this out). Including this effect in the model makes it more difficult to calculate the ESS schedule of drive, but it is not difficult to show that, qualitatively, all of the above results still hold. Thus, all else equal, we expect that this effect will increase the frequency of nondisjunction and trisomy slightly; however, the age dependence of nondisjunction and trisomy still remains.
Additional Predictions of the CD Hypothesis
Aside from age dependence, the CD hypothesis also makes at least four other predictions that can be tentatively evaluated with available data. The first is that all trisomy should result from nondisjunction in females because there is no genetic conflict in males. This pattern is witnessed in the majority of autosomal trisomies recorded (1). Although paternal nondisjunction plays a role with some chromosomes (1), it is recognized that the vast majority of trisomic conceptions arise through maternal nondisjunction. If the CD hypothesis is true, examples involving paternal nondisjunction may represent instances where chromosomal drive has not (yet) evolved to be completely sex-limited in its expression.
A second, related prediction of the CD hypothesis is that most trisomy should result from nondisjunction at meiosis I because, neglecting recombination (23), there is no genetic conflict at meiosis II. Although there can be significant levels of recombination at loci distal to the centromere, it is probably loci near the centromere that have the greatest potential for influencing the pattern of segregation, and these will have very low recombination rates [see Haig and Grafen (29)]. Again, this pattern is witnessed in the vast majority of trisomies recorded. There are some examples in which it appears that nondisjunction at meiosis II plays an important role [e.g., chromosome 18 (23)], but it has recently been suggested that nondisjunction in these instances might actually be precipitated by events that occur during meiosis I (1, 6–8).
Together the above two predictions of the CD hypothesis state that all trisomic conceptions should arise through meiosis I nondisjunction in females. The data are consistent with this prediction, but it might be argued that most trisomic conceptions arise in this way because of the peculiarities of oogenesis. During oogenesis, oocytes remain in arrested meiosis I throughout the life of the female until they are ovulated, whereas no similar process occurs during spermatogenesis. Therefore, it is conceivable that nondisjunction is directly related to this phenomenon and that nondisjunction is more likely the longer that an oocyte remains in arrested meiosis. Although this is quite plausible, we wish to stress that it provides a proximate explanation whereas the CD hypothesis provides an ultimate (evolutionary) explanation. The two are not competing hypotheses because they address different questions. For example, it is conceivable that the peculiarities of oogenesis are involved in the occurrence of nondisjunction and this provides a convenient proximate mechanism for the evolution of chromosomal drive that can be limited to female meiosis I.
A third prediction of the CD hypothesis is that organisms lacking a definite menopause should display a lower frequency of trisomy and/or a weaker relationship of trisomy with maternal age. It is still unclear whether menopause is a largely human phenomenon, or whether it occurs in many other species but is more obvious in humans because of the extension of the human life span as a result of modern living conditions. Assuming that some species do not have a definite menopause, however, our model suggests that chromosomes in such species should exhibit a weaker level of drive, and drive should not increase as sharply with female age. The reason is that it is the rapid decline in future reproductive potential as the time horizon is approached that causes the sharp increase in nondisjunction and trisomy. The data on the occurrence of trisomy in other organisms are sparse, but there is some suggestion that trisomy in humans is far more prevalent and its age dependency is far stronger than in other organisms (1, 5, 30, 31). Also notice that, even if humans in ancestral environments rarely lived long enough to experience menopause, the sharp increase in trisomy at the age of menopause is still predicted. Although very few females would ever reach this age, the optimal temporal schedule of chromosomal drive would still involve this sharp increase. The strength of selection on that portion of the temporal schedule, however, would be very small such that other evolutionary forces not included in the model might come to predominate (e.g., mutation pressure).
The fourth prediction of the CD hypothesis is that all nondisjunction should result in trisomy rather than nullosomies (offspring with missing chromosomes). This prediction is unique to the CD hypothesis. If nondisjunction results from random errors, one would expect an equal chance of too many or too few chromosomes in offspring. If nondisjunction results from CD, however, then chromosomes should never make the “mistake” of going to the polar body. Thus, we would expect only trisomies. The data on this are limited but tend to exhibit this pattern (4, 31). It has been suggested, however, that the absence of nullosomies is a reflection of their extremely low viability (4, 31). Further empirical research on this question would be very useful for assessing the validity of the CD hypothesis.
Conclusions
The CD hypothesis asserts that homologous chromosomes are involved in an evolutionary game and that adaptive evolution of chromosomal drive strategies results in the evolution of age-dependent nondisjunction and trisomy as a side effect. The model presented above reveals, however, that an important assumption of this hypothesis is that generalized mortality rate increases with an increase in the average level of drive in a female. This means that either a high level of drive increases the likelihood of a female’s mortality per se or that it increases the likelihood of her reproductive system failing (e.g., through ovarian cancer and/or cysts). Although data on this are still lacking, it does not seem to be an unreasonable assumption.
Supposing that this assumption is valid, our results illustrate that predictions from the CD hypothesis match empirical data very well. In particular, a robust prediction of the model is a gradual increase in the frequency of trisomic conceptions from sexual maturity until around age 35, and then a very rapid increase in frequency. The frequency of trisomy observed for most human chromosomes tends to exhibit this pattern (Fig. 1). As discussed in the preceding section, the CD hypothesis makes at least four other predictions aside from age dependence that are largely consistent with observations as well.
Most of the above discussion suggests that a chromosomal drive mechanism indeed may be the underlying cause of nondisjunction and trisomy in humans. If correct, however, it may be difficult to detect directly, particularly in a monomorphic population in which all chromosomes exhibit drive (32). Ironically, this will increase the probability of nondisjunction and trisomy, but each chromosome will still have a 50:50 chance of getting into the ovum.
Acknowledgments
We thank G. C. Williams for his ideas on this subject and D. S. Wilson, G. C. Williams, D. Schluter, S. P. Otto, L. Nagel, and D. Haig for several insightful comments on the manuscript. T.D. also thanks S. P. Otto for her amazing hospitality during his stay at the University of British Columbia. Funding was provided by the Natural Sciences and Engineering Research Council of Canada.
ABBREVIATIONS
- CD
chromosomal drive
- IPD
iterated prisoner’s dilemma
References
- 1.Koehler K E, Hawley R S, Sherman S, Hassold T. Hum Mol Genet. 1996;5:1495–1504. doi: 10.1093/hmg/5.supplement_1.1495. [DOI] [PubMed] [Google Scholar]
- 2.Risch N, Stein Z, Kline J, Warburton D. Am J Hum Genet. 1986;39:68–78. [PMC free article] [PubMed] [Google Scholar]
- 3.Hassold T, Chiu D. Hum Genet. 1985;70:11–17. doi: 10.1007/BF00389450. [DOI] [PubMed] [Google Scholar]
- 4.Cummings M R. Human Heredity: Principles and Issues. New York: West Publishing Company; 1988. [Google Scholar]
- 5.Angell R R, Xian J, Keith J, Ledger W, Baird D T. Cytogenet Cell Genet. 1994;65:194–202. doi: 10.1159/000133631. [DOI] [PubMed] [Google Scholar]
- 6.Orr-Weaver T. Nat Genet. 1996;14:374–376. doi: 10.1038/ng1296-374. [DOI] [PubMed] [Google Scholar]
- 7.Lamb N E, Freeman S B, Savage-Austin A, Pettay D, Taft L, Hersey J, Gu Y, Shen J, Saker D, May K M, et al. Nat Genet. 1996;14:400–405. doi: 10.1038/ng1296-400. [DOI] [PubMed] [Google Scholar]
- 8.Koehler K E, Boulton C L, Collins H E, French R L, Herman K C, Lacefield S M, Madden L D, Schuetz C D, Hawley R S. Nat Genet. 1996;14:406–413. doi: 10.1038/ng1296-406. [DOI] [PubMed] [Google Scholar]
- 9.Zheng C, Byers B. Hum Genet. 1992;90:1–6. doi: 10.1007/BF00210736. [DOI] [PubMed] [Google Scholar]
- 10.Henderson S A, Edwards R G. Nature (London) 1968;217:22–28. doi: 10.1038/218022a0. [DOI] [PubMed] [Google Scholar]
- 11.Aymes S, Lippman-Hand A. Am J Hum Genet. 1982;34:558. [PMC free article] [PubMed] [Google Scholar]
- 12.Angell R R. Hum Reproduct. 1994;9:1199–1200. doi: 10.1093/oxfordjournals.humrep.a138675. [DOI] [PubMed] [Google Scholar]
- 13.Stein Z, Stein W, Susser M. Lancet. 1986;8487:944–947. doi: 10.1016/s0140-6736(86)91046-9. [DOI] [PubMed] [Google Scholar]
- 14.Dailey T, Dale B, Cohen J, Munn S. Am J Hum Genet. 1996;59:176–184. [PMC free article] [PubMed] [Google Scholar]
- 15.Petersen M B, Antonarakis S E, Hassold T J, Freeman S B, Sherman S L, Avramopoulos D, Mikkelsen M. Hum Mol Genet. 1993;2:1691–1695. doi: 10.1093/hmg/2.10.1691. [DOI] [PubMed] [Google Scholar]
- 16.Hawley R S, Frazier J A, Rasooly R. Hum Mol Genet. 1994;3:1521–5128. doi: 10.1093/hmg/3.9.1521. [DOI] [PubMed] [Google Scholar]
- 17.Antonarakis S E. Am J Hum Genet. 1996;59:1395–1397. [PMC free article] [PubMed] [Google Scholar]
- 18.Zheng C, Byers B. Am J Hum Genet. 1996;59:1397–1398. [PMC free article] [PubMed] [Google Scholar]
- 19.Bowman M C, Saunders D M. Hum Reproduct. 1994;9:1200–1201. doi: 10.1093/oxfordjournals.humrep.a138676. [DOI] [PubMed] [Google Scholar]
- 20.Axelrod R, Hamilton W D. Science. 1981;211:1390–1396. doi: 10.1126/science.7466396. [DOI] [PubMed] [Google Scholar]
- 21.Kloss R J, Nesse R M. Ethol Sociobiol. 1992;13:283–288. [Google Scholar]
- 22.Rose M R. Evolutionary Biology of Aging. Oxford: Oxford Univ. Press; 1991. [Google Scholar]
- 23.Haig D. J Theor Biol. 1986;123:471–480. [Google Scholar]
- 24.Eaton S B, Pike M C, Short R V, Lee N C, Trussell J, Hatcher R A, Wood J W, Worthman C M, Jones N G B, Konner M J, et al. Q Rev Biol. 1994;69:353–367. doi: 10.1086/418650. [DOI] [PubMed] [Google Scholar]
- 25.Holliday R. Trends Genet. 1989;5:42–45. doi: 10.1016/0168-9525(89)90020-6. [DOI] [PubMed] [Google Scholar]
- 26.Maynard Smith J. Evolution and the Theory of Games. Cambridge, U.K.: Cambridge Univ. Press; 1982. [Google Scholar]
- 27.Day T, Taylor P D. Proc Roy Soc London B. 1997;264:639–644. [Google Scholar]
- 28.Day, T. & Taylor, P. D. (1998) American Naturalist, in press. [DOI] [PubMed]
- 29.Haig D, Grafen A. J Theor Biol. 1991;153:531–558. doi: 10.1016/s0022-5193(05)80155-9. [DOI] [PubMed] [Google Scholar]
- 30.Angell R R. Human Genetics. 1991;86:383–387. doi: 10.1007/BF00201839. [DOI] [PubMed] [Google Scholar]
- 31.Vig B K, Sandberg A A, editors. Progress and Topics in Cytogenetics. 7A. New York: Liss; 1987. [Google Scholar]
- 32.Hurst L D, Atlan A, Bengtsson B O. Q Rev Biol. 1996;71:317–364. doi: 10.1086/419442. [DOI] [PubMed] [Google Scholar]