

Summary
The importance of neuropeptides in the hypothalamus has been experimentally established. Due to difficulties in assessing function in vivo, the roles of fast-acting neurotransmitters, glutamate and GABA, are largely unknown. Synaptic vesicular transporters (VGLUTs for glutamate and VGAT for GABA) are required for vesicular uptake and, consequently, synaptic release of neurotransmitters. Ventromedial hypothalamic (VMH) neurons are predominantly glutamatergic and express VGLUT2. To evaluate the role of glutamate release from VMH neurons, we generated mice lacking VGLUT2 selectively in SF1 neurons (major subset of VMH neurons). These mice have hypoglycemia during fasting secondary to impaired fasting-induced increases in the glucose-raising pancreatic hormone, glucagon, and impaired induction in liver of mRNAs encoding PGC1α and the gluconeogenic enzymes, PEPCK and G6Pase. Similarly, these mice have defective counter-regulatory responses to insulin-induced hypoglycemia and 2-deoxyglucose (anti-metabolite). Thus, glutamate release from VMH neurons is an important component of the neurocircuitry that functions to prevent hypoglycemia.
Introduction
Glutamate and GABA are the major excitatory and inhibitory neurotransmitters in the brain. They exert large effects on post-synaptic neurons and are widely distributed. With this in mind, it is remarkable how little is known about their roles in transducing specific behaviors and homeostatic responses. This is especially true when one considers the hypothalamus, a site where the majority of focus has been on neuropeptides. Indeed, critically important roles have been established for many hypothalamic neuropeptides and their receptors in regulating food intake, energy expenditure, insulin-glucose homeostasis, the sleep-wake cycle and neuroendocrine output of the pituitary gland (Cone, 2005; Guillemin, 2005; Saper et al., 2005; Williams et al., 2004). In striking contrast, the roles played by fast-acting neurotransmitters such as glutamate and GABA have been largely unexplored. This is not because they are thought to be unimportant, but instead because of the absence of methodological approaches for testing function in vivo (van den Pol, 2003). The underlying problem limiting investigations is the widespread distribution of glutamate and GABA, and their receptors. Because essentially all neurons have receptors for these neurotransmitters, site-specific injections of agonists or antagonists invoke responses in all neurons exposed, hence generating results of unclear physiologic meaning (van den Pol, 2003). Similarly, total gene knockouts aimed at disrupting neurotransmitter release or the ability to respond to neurotransmitters result in complex phenotypes, many of which are incompatible with life (Fremeau et al., 2004a; Moechars et al., 2006; Wallen-Mackenzie et al., 2006; Wojcik et al., 2006; Wojcik et al., 2004). Thus, there is tremendous need for an approach which allows one to study the roles of glutamate and GABA in selected sites within the brain, and hence in specific behaviors and homeostatic responses.
Towards these ends, we and others (Wallen-Mackenzie et al., 2006) have created lox-modified alleles of synaptic vesicle transporters. In order for a neuron to release glutamate or GABA, the neurotransmitter must first be packaged, at high concentrations, into synaptic vesicles. This is accomplished by specific transporters, VGLUT1, -2 and -3 in the case of glutamate (Fremeau et al., 2004b) and VGAT in the case of GABA (Gasnier, 2004). It has previously been demonstrated that gene knockout of VGLUT1 (Fremeau et al., 2004a; Wojcik et al., 2004), VGLUT2 (Moechars et al., 2006; Wallen-Mackenzie et al., 2006) and VGAT (Wojcik et al., 2004) creates neurons that are unable to release their respective neurotransmitter. By creating lox-modified alleles of these synaptic vesicle transporters, and by crossing these animals with transgenic mice expressing cre-recombinase in specific groups of neurons, trans-synaptic communication by these transmitters can be disrupted in a neuron-specific manner. Such studies make it possible to determine the role of fast-acting neurotransmitters in specific behaviors and homeostatic responses.
The ventromedial hypothalamus (VMH) integrates forebrain neuronal input with ascending information from the brainstem, and then sends output to regions involved in control of various behaviors and physiologic responses (Swanson, 1987). Indeed, VMH neurons send projections to a vast number of sites including numerous locations within the medial hypothalamus, the lateral hypothalamus, the zona incerta, parts of the midline thalamus, the bed nuclei of the stria terminalis, various parts of the amygdala, and the periaqueductal gray (Canteras et al., 1994; Krieger et al., 1979; Saper et al., 1976). The VMH also sends projections, at a lower density, to many other sites, including direct glutamatergic projection to nearby POMC neurons (Sternson et al., 2005). By means of these projections, the VMH is thought to control ingestive, sexual, and defensive behaviors (Canteras, 2002; Choi et al., 2005; King, 2006), the physiologic responses that accompany these behaviors, as well as counter-regulatory responses to hypoglycemia (Borg et al., 1997; Borg et al., 1994; Borg et al., 1995). Neurons in the VMH are largely glutamatergic as evidenced by high level expression of mRNA encoding the vesicular glutamate transporter, VGLUT2, and lower levels of expression of mRNA encoding the vesicular GABA transporter, VGAT, and the GABA synthesizing enzyme, glutamic acid decarboxylase (GAD) (Ovesjo et al., 2001; Ziegler et al., 2002). The importance of glutamate release from VMH neurons, versus other neurotransmitters and neuropeptides, in mediating the above mentioned behaviors and physiologic responses, however, is unknown.
Steroidogenic factor-1 (SF1, official gene name Nr5a1) is a member of the nuclear hormone receptor family and, in the CNS, is expressed exclusively in the VMH (Dhillon et al., 2006; Stallings et al., 2002). Gene knockout mice lacking SF1 have abnormal VMH development and are obese (Majdic et al., 2002). Recently, we generated Sf1-Cre transgenic mice that express cre-recombinase selectively in the VMH (Dhillon et al., 2006). These mice are useful for VMH-specific knockout of genes flanked by lox sites. We have used this approach to generate mice lacking leptin receptors (LEPRs) on SF1 neurons (Sf1-Cre, Leprflox/flox mice). These mice develop mild obesity when fed a chow diet and are sensitive to high fat diet-induced obesity (Dhillon et al., 2006). As mentioned above, the relevant mediator released by SF1 neurons is largely unknown. In the present study, we have evaluated the role of glutamate by generating mice that lack VGLUT2 selectively in SF1 neurons (Sf1-Cre, Vglut2flox/flox mice).
Results
Generation of Vglut2flox/flox mice
Gene targeting in ES cells was used to create mice with lox sites flanking exon 2 of the Vglut2 (Slc17a6) gene (Figure 1A). Cre-recombinase-mediated deletion of exon 2 is expected to generate a null Vglut2 allele as exon 1 contains only ˜5% of VGLUT2’s coding sequence. Furthermore, the reading frame is disrupted if exon 1 splices to exon 3. To confirm the “null” status of this allele, we generated mice homozygous for the deleted allele. The deleted allele was first created by crossing Vglut2+/flox mice with transgenic animals expressing cre in the female germ line (ZP3-Cre mice, Jackson Laboratory, stock #003651). Female ZP3-Cre, Vglut2+/flox mice were then used to generate offspring that were heterozygous for the deleted allele (Vglut2+/- mice). These heterozygotes (Vglut2+/-) were then bred to generate homozygous, presumably “null” animals (Vglut2-/-). Prior studies have demonstrated that mice lacking VGLUT2 are not viable (Moechars et al., 2006; Wallen-Mackenzie et al., 2006). Consistent with this, we were unable to detect any homozygous offspring at age 3 weeks (out of 31 offspring genotyped, 10 were Vglut2+/+, 21 were Vglut2+/- and 0 were Vglut2-/-). However, we were able to detect offspring homozygous for the deleted allele at embryonic day 19 (out of 10 embryos genotyped, 3 were Vglut2+/+, 5 were Vglut2+/- and 2 were Vglut2-/-). RT-PCR using mRNA isolated from brain tissue of Vglut2+/- mice confirmed that exon 2 was missing and that exon 1 was spliced to exon 3. These results demonstrate that the cre-mediated deletion of the Vglut2flox allele results in a null allele.
Figure 1. Generation of Mice lacking VGLUT2 in SF1 Neurons.
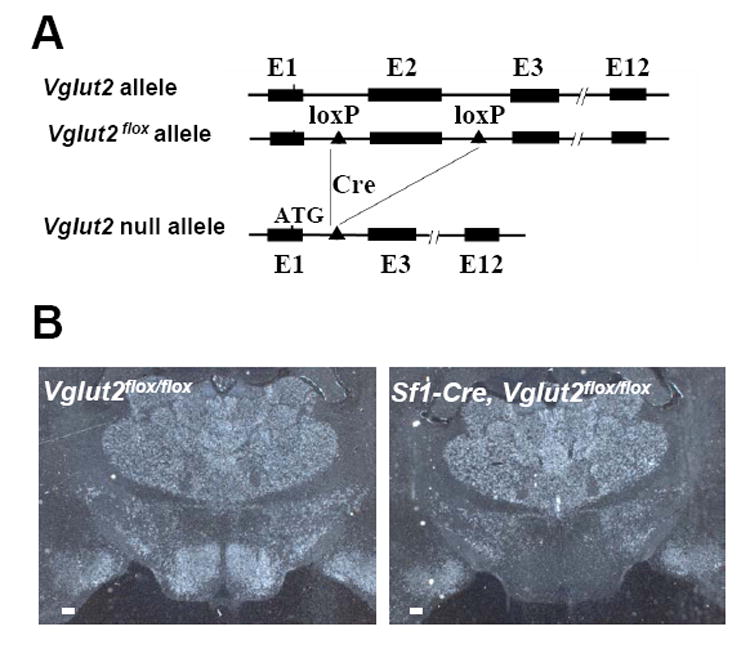
(A) The Vglut2 gene, the Vglut2flox allele and the cre-deleted Vglut2 null allele. E: exon; Cre: cre recombinase.
(B) In situ hyrbridization for Vglut2 mRNA. VMH: ventromedial hypothalamus. Scale bar=100 μM.
Generation of Mice Lacking VGLUT2 in SF1 Neurons
To generate mice lacking VGLUT2 in SF1 neurons, as well as control mice, Vglut2flox/flox mice were crossed with Sf1-Cre, Vglut2flox/flox mice. To test for neuron-specific loss of Vglut2 mRNA, in situ hybridization was performed using a riboprobe corresponding to exon 2. As previously observed (Ziegler et al., 2002), Vglut2 mRNA is abundantly expressed in the VMH of control mice (Figure 1B, left panel). As expected, Vglut2 mRNA is selectively absent in the VMH of Sf1-Cre, Vglut2flox/flox mice (Figure 1B, right panel). In contrast, Vglut2 mRNA is undisturbed in all other brain regions of Sf1-Cre, Vglut2flox/flox mice (data not shown). We were unable to detect VMH expression of Vglut1 mRNA or Vglut3 mRNA in control (Vglut2flox/flox mice) or VMH-deleted (Sf1-Cre, Vglut2flox/flox) mice (see Supplemental Data, Figure S1 ). Thus, in Sf1-Cre, Vglut2flox/flox mice, Vglut2 mRNA is undetectable in the VMH and there is no compensatory upregulation of Vglut1 or Vglut3 mRNA.
Glutamate Release is Disrupted in SF1 Neurons of Sf1-Cre, Vglut2flox/flox Mice
To confirm that deletion of VGLUT2 in SF1 neurons leads to loss of glutamate release, we performed electrophysiologic recordings (whole-cell, voltage clamp mode) on SF1 neurons, plated at low density, that had formed autosynapses (autaptic cultures) (Hentges et al., 2004). To permit identification of SF1 neurons, Sf1-Cre transgenic mice were first crossed with animals bearing a cre-dependent, GFP reporter transgene (Z/EG, Jackson Laboratory) (Novak et al., 2000). Control neurons were derived from Sf1-Cre, Z/EG mice while VGLUT2 deleted neurons were derived from Sf1-Cre, Z/EG, Vglut2flox/flox mice. Excitatory (mediated by glutamate) and inhibitory (mediated by GABA) postsynaptic currents (EPSCs and IPSCs) were evoked every 20 seconds by depolarizing pulses (from a holding potential of -60 mV to +10 mV for 2 milliseconds). With the conditions utilized (see methods), both EPSCs and IPSCs were identified as inward current. Glutamatergic-mediated EPSCs versus GABAergic-mediated IPSCs were differentiated based upon decay time constants (0.8-3.0 ms for EPSCs and 10-15 ms for IPSCs) and effects of glutamate receptor and GABA receptor antagonists. Out of 12 control SF1 neurons, 8 neurons were found to have EPSCs mediated by glutamate (Figure 2A), while 2 neurons were found to have IPSCs mediated by GABA (Figure 2B, results summarized in 2C). Two neurons lacked EPSCs or IPSCs indicating that they release neither glutamate nor GABA, or that they failed to establish functional autosynapses. These results indicate that most SF1 neurons are glutamatergic while a smaller percentage are GABAergic. Of interest, out of 11 SF1 neurons from Sf1-Cre, Z/EG, Vglut2flox/flox mice, 0 neurons were found to have EPSCs, while 2 neurons were found to have IPSCs mediated by GABA (results summarized in Figure 2C). These findings indicate that, as expected, glutamate release is selectively disrupted in SF1 neurons of Sf1-Cre, Vglut2flox/flox mice.
Figure 2. Assessment of Neurotransmitter Release in Autaptic Cultures.
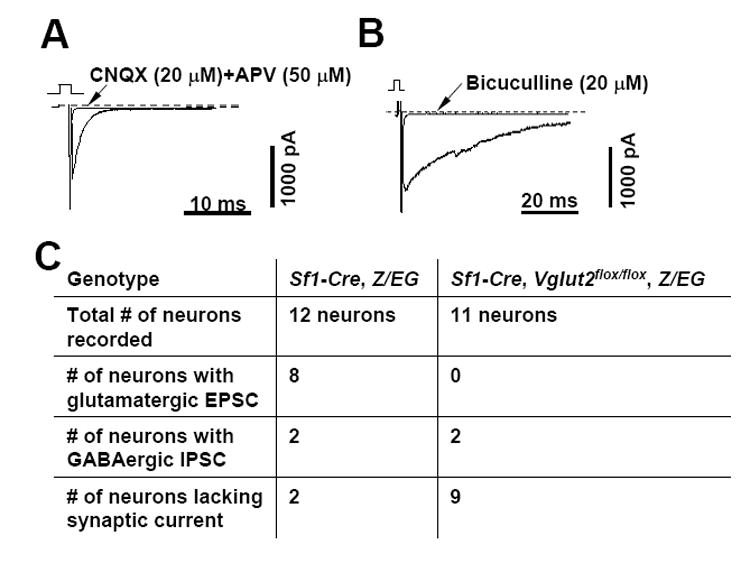
Control SF1 neurons were prepared from Sf1-Cre, Z/EG mice and VGLUT2 deficient SF1 neurons were prepared from Sf1-Cre, Z/EG, Vglut2flox/flox mice.
(A) Excitatory Postsynaptic Current (EPSC) from a control SF1 neuron recorded in the absence and then in the presence of glutamate receptor antagonists (CNQX and APV).
(B) Inhibitory Postsynaptic Current (IPSC) from a control SF1 neuron recorded in the absence and then in the presence of a GABA receptor antagonist (bicuculline).
(C) Summary of recordings from control (Sf1-Cre, Z/EG) and VGLUT2 deficient (Sf1- Cre, Z/EG, Vglut2flox/flox) SF1 neurons.
Cell Bodies and Projections of SF Neurons in Sf1-Cre, Vglut2flox/flox Mice
It is conceivable that disruption of glutamatergic transmission could alter the survival and/or projections of SF1 neurons. To address this, we compared the density of cell bodies and projections of control (Sf1-Cre, Z/EG) and VGLUT2-deleted (Sf1-Cre, Z/EG, Vglut2flox/flox) SF1 neurons. The SF1 neurons and their projections were identified by immunohistochemical detection of GFP in brain sections of 10 week old mice. These studies indicated that the density of SF1 cell bodies in the VMH was unchanged in Sf1-Cre, Z/EG, Vglut2flox/flox mice ( Supplemental Data, Figure S 2A ). To check for projections, we examined the bed nuclei of stria terminalis (BST) and the periaqueductal gray (PAG) area, two sites that are densely innervated by VMH neurons. These analyses indicated that projections were not grossly disturbed in Sf1-Cre, Z/EG, Vglut2flox/flox mice ( Supplemental Data, Figure S 2B ). In total, these studies indicate that glutamatergic transmission is not required for the survival of SF1 neurons or for the development of their projections.
Body Weight in Sf1-Cre, Vglut2flox/flox mice
Sf1-Cre, Vglut2flox/flox mice fed a standard chow diet had normal body weights (data not shown). However, when fed a high fat, high sucrose diet, Sf1-Cre, Vglut2flox/flox mice developed a modest increase in body weight compared with controls (Vglut2flox/flox mice) (Figure 3A). This was associated with a small increase in food intake (Figure 3B) and fat stores (Figure 3C) but not with an obvious effect on energy expenditure (Figure 3D). These results suggest that release of glutamate from SF1 neurons plays little or no role, in the case of chow-fed mice, and a small role, in the case of fat-fed mice, in regulating energy balance.
Figure 3. Effect of High Fat, High Sucrose Diet (HFD) on Energy Balance.
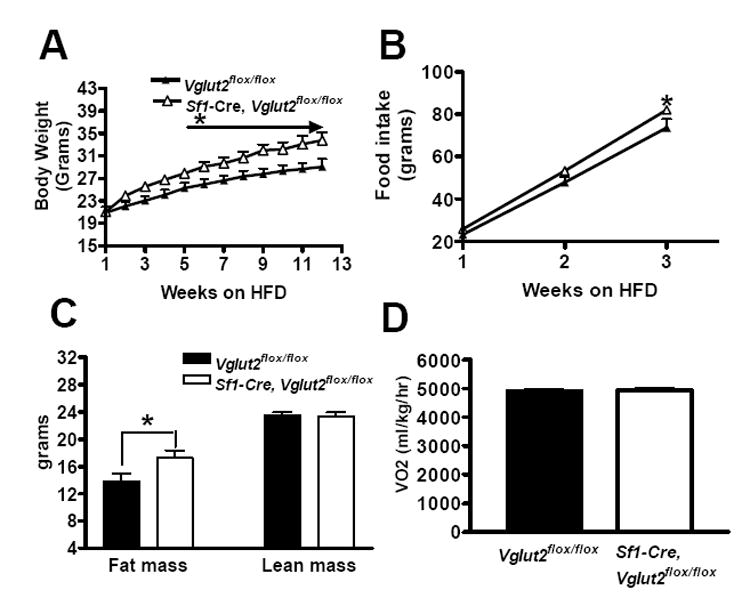
Male mice were studied. The HFD was started at the age of 8-10 weeks. All results are expressed as mean +/- SE (n = 8-10 mice per group). * = P < 0.05.
(A) Body weight.
(B) Cumulative food intake.
(C) Body composition.
(D) Energy expenditure as assessed by oxygen consumption.
Impaired Glucose Homeostasis in the Fasted State
Sf1-Cre, Vglut2flox/flox mice had lower blood glucose levels, compared with Vglut2flox/flox controls, after fasting for 24 hours (mg/dl: 55 ± 3 versus 68 ± 5 in males, Figure 4A; 53 ± 2 sersus 67 ± 5 in females, Figure 4B). Insulin values fell with fasting in both groups, and were similar in Sf1-Cre, Vglut2flox/flox mice versus Vglut2flox/flox mice in both the fed or fasted states (data not shown). In contrast, Sf1-Cre, Vglut2flox/flox mice had an impaired fasting-mediated increase in the pancreatic hormone glucagon (Figure 4C). Glucagon works on the liver where it stimulates glycogenolysis (breakdown of glycogen to glucose) and gluconeogenesis (synthesis of glucose from pyruvate, lactate, glycerol and amino acids) (Vidal-Puig and O’Rahilly, 2001). The effect on gluconeogenesis is mediated, in part, by augmenting expression of PGC1α, a transcription co-activator that stimulates expression of genes encoding gluconeogenic enzymes such as glucose-6-phosphatase (G6Pase) and phosphoenolpyruvate carboxykinase (PEPCK) (Yoon et al., 2001). As expected, in livers of control Vglut2flox/flox mice, fasting increased expression of Ppargc1α (PGC1α), Pck1 (PEPCK) and G6pc (G6Pase) mRNAs (Figure 4D, 4E and 4F). Of note, these fasting-induced increases in gene expression were absent in livers of Sf1-Cre, Vglut2flox/flox mice. These findings indicate that hypoglycemia during fasting in Sf1-Cre, Vglut2flox/flox mice is mediated, at least in part, by failure to increase blood glucagon levels, and subsequent failure to induce hepatic expression of PGC1α, and its gluconeogenic targets, G6Pase and PEPCK.
Figure 4. The Effects of 24 hrs of Fasting on Blood Glucose, Glucagon and Liver Gene Expression.
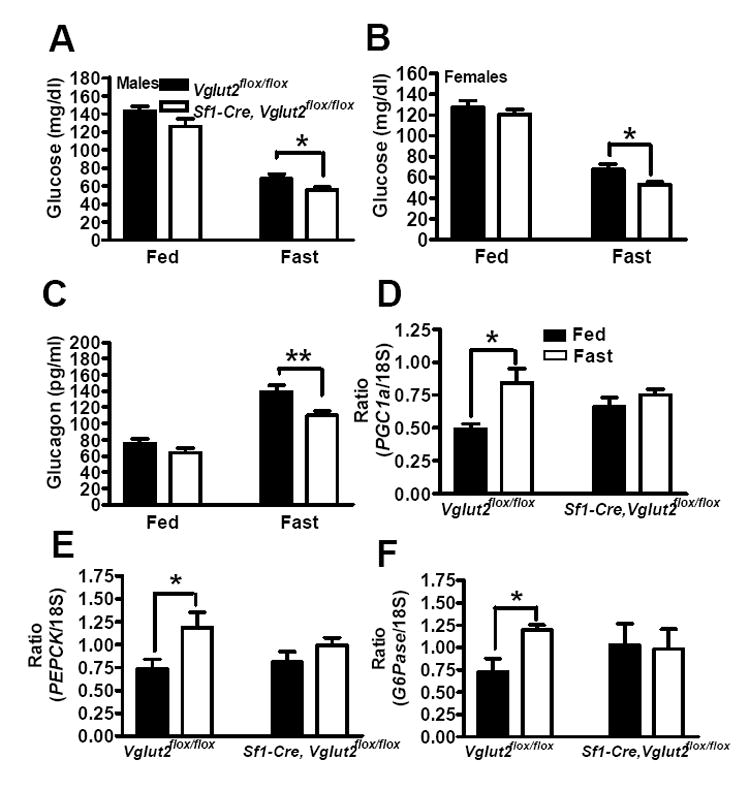
Male and female mice, age 8-10 weeks, were studied. All results are expressed as the mean +/- SE (n = 13-15 per group). * = p < 0.05, ** = p < 0.01.
(A) Blood glucose in male mice.
(B) Blood glucose in female mice.
(C) Plasma glucagon in female mice..
(D, E and F) PGC1α, PEPCK and G6Pase mRNA levels in livers from female mice.
Impaired Counter-Regulation to Insulin-Induced Hypoglycemia
Acute administration of insulin induces hypoglycemia. Counter-regulatory measures, initiated in part by the brain, operate to limit the degree of hypoglycemia. As shown in Figure 5A, a single dose of insulin (1.5 U/kg) at 60 mins caused a greater fall in blood glucose in Sf1-Cre, Vglut2flox/flox mice. This greater degree of hypoglycemia was associated with an impaired glucagon response (Figure 5B).
Figure 5. Effects of Insulin-Induced Hypoglycemia on Counter-Regulatory Response.
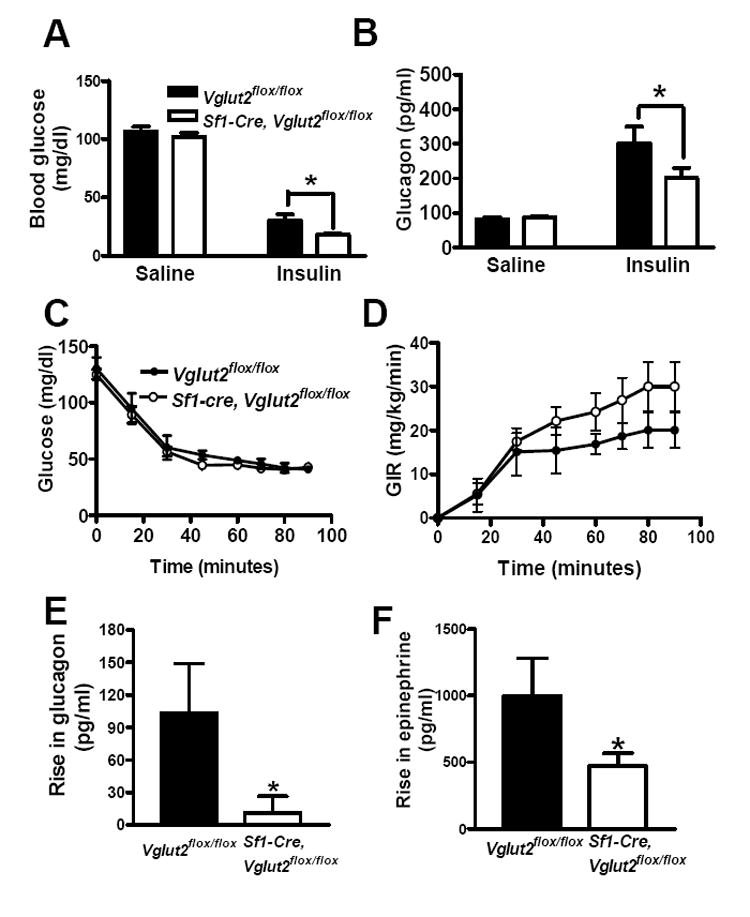
For the single injection study (A and B), male mice (n = 12-15), age 10-12 weeks, were fasted overnight and then given a single dose of insulin (1.5 U/kg). For the hyperinsulinemic hypoglycemic clamp studies (C, D, E and F), male mice (n=5-10), age 12-14 weeks old, were used. Insulin infusion started at time 0 and continued during the entire 90 minute period. Glucose was infused at a variable rate with the goal of maintaining glucose at approximately 45 mg/dl. All results are expressed as the mean +/- SE. * = P < 0.05.
(A) Blood glucose at 60 mins.
(B) Plasma glucagon at 60 mins.
(C) Blood glucose levels during clamp.
(D) Glucose infusion rate (GIR) during the clamp.
(E) Rise in glucagon (increase at 60-90 minute interval over level observed at time 0).
(F) Rise in epinephrine (increase at 60-90 minute interval over level observed at time 0).
To confirm these findings, we performed hypoglycemic clamp studies in which hypoglycemia is induced and sustained at similar levels in a controlled fashion (Figure 5C). Sf1-Cre, Vglut2flox/flox mice tended to require higher rates of glucose infusion (GIR) during the clamp which is consistent with decreased rates of endogenous glucose production (Figure 5D). Of note, hypoglycemia induced a large increase in plasma glucagon levels in control mice and, importantly, this response was undetectable in Sf1-Cre, Vglut2flox/flox mice (Figure 5E). Similarly, hypoglycemia induced a large increase in blood levels of epinephrine in control mice and this response was blunted in Sf1-Cre, Vglut2flox/flox mice (Figure 5F). In total, these results demonstrate that Sf1-Cre, Vglut2flox/flox mice have an impaired counter-regulatory response to insulin-induced hypoglycemia.
Impaired Counter-Regulation to Central 2-Deoxyglucose
2-Deoxyglucose (2-DG) is a non-metabolizable glucose analogue that inhibits glucolysis in cells. Injection of 2-DG into the third ventricle of the brain is frequently used as a means of mimicking central glucopenia (Borg et al., 1995; Miki et al., 2001). This manipulation is useful because “glucopenia” is limited to the brain, and also because it is not confounded by the presence of hyperinsulinemia. In control mice, central 2-DG treatment for 30 minutes increased blood glucose levels (Figure 6A), plasma glucagon levels (p=0.06) (Figure 6B) and PEPCK mRNA levels in the liver (Figure 6C). There was also a trend for G6Pase mRNA to increase (Figure 6D). All of these responses to central 2-DG were absent in Sf1-Cre, Vglut2flox/flox mice. These results demonstrate that counter-regulatory responses to central glucopenia are defective in Sf1-Cre, Vglut2flox/flox mice.
Figure 6. Effect of Centrally Administered 2-Deoxyglucose (2-DG) on Blood Glucose, Glucagon and Liver Gene Expression.
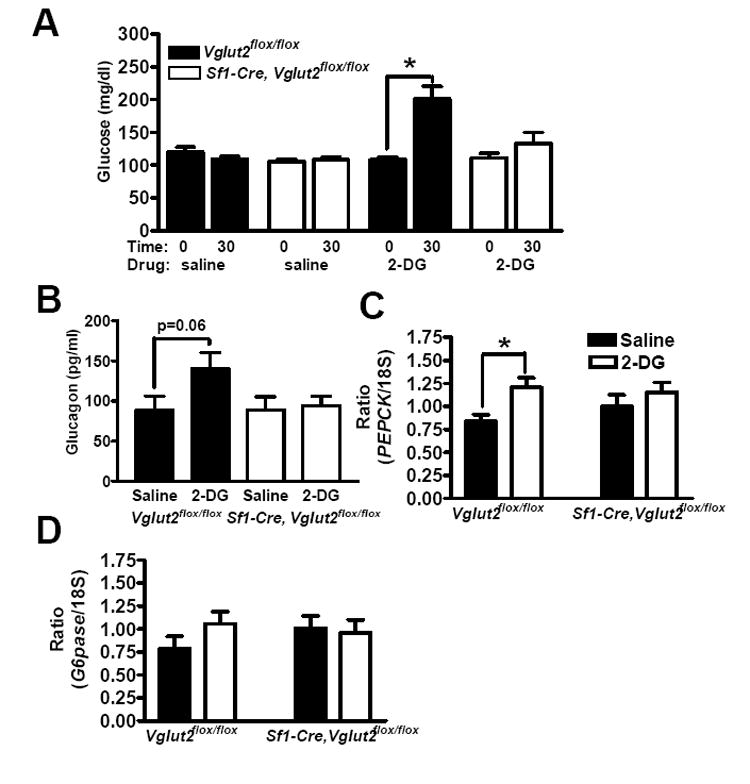
Female mice, age 10 weeks old, were studied. 2-DG or saline was administered i.c.v.. All results are expressed as the mean +/- SE (n = 9-10 per group). * = P < 0.05.
(A) Blood glucose was assessed at 0 and 30 minutes after saline or 2-DG treatment.
(B) Glucagon was assessed 30 minutes after saline or 2-DG treatment.
(C and D) PEPCK and G6Pase mRNA levels in the liver were assessed 30 minutes after saline or 2-DG treatment.
However, some counter-regulatory responses may be normal in Sf1-Cre, Vglut2flox/flox mice. Peripheral 2-DG administration is also known to stimulate food intake (Ritter et al., 2006; Sindelar et al., 2004), which functions to raise blood glucose levels. The hyperphagic response to fasting or 2-DG was preserved in Sf1-Cre,Vglut2flox/flox mice (see Supplemental Data, Figure S3 ). These results suggest that glutamate release from SF1 neurons is not involved in the pathway by which central glucopenia stimulates food intake.
Induction of c-Fos in the Dorsal Motor Nucleus of the Vagus (DMV)
Induction of c-Fos is a well established indicator of neuronal activation (Herrera and Robertson, 1996). SF1 neurons release glutamate, which is an excitatory neurotransmitter, and this release is required for the glucagon response to hypoglycemia. Thus, there is an excitatory efferent pathway from SF1 neurons to pancreatic α cells. Neurons situated in this pathway activated (directly or indirectly) by glutamate released from SF1 neurons could be identified by comparing hypoglycemic-mediated c-Fos induction in Vglut2flox/flox mice versus Sf1-Cre, Vglut2flox/flox mice. Indeed, in insulin treated hypoglycemic Vglut2flox/flox mice, significantly more neurons express c-Fos (18 ± 2.0/DMV/section) in the DMV of 3 consecutive sections immediately rostral to area postrema ( Figure 7B1, B2, B3 and D ), compared to that of saline treated mice (1.4 ± 0.1/DMV/section; Figure 7A1, A2, A3 and D ). In contrast, in insulin treated Sf1-Cre, Vglut2flox/flox mice, the induction of c-Fos is significantly reduced (11 ± 1.7/DMV/section; Figure 7C1, C2, C3 and D ). Thus, these DMV neurons likely act as a link between SF1 neurons and pancreatic α cells.
Figure 7. Induction of c-Fos in Dorsal Motor Nucleus of the Vagus (DMV).
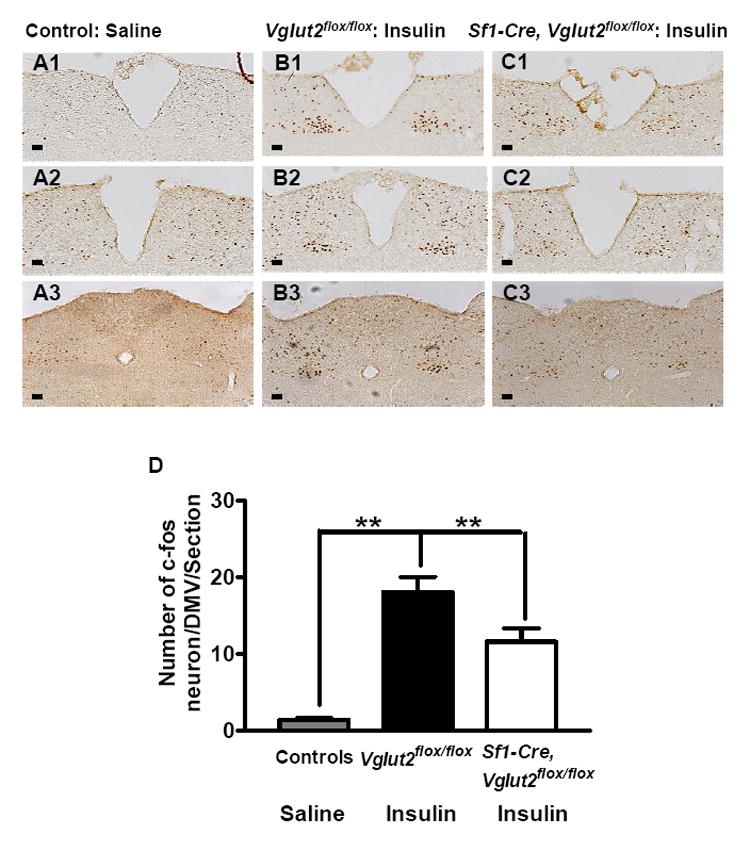
60 mins after treatment with saline or insulin (1.5 U/kg), brains from Vglut2flox/flox and Sf1-Cre, Vglut2flox/flox mice were removed and immunostained for c-Fos. The saline treated Vglut2flox/flox and Sf1-Cre,Vglut2flox/flox mice were pooled together as controls since c-Fos induction was not different between these mice. 3 consecutive sections immediately rostral to the area postrema were analyzed. Data was presented as mean ± SEM (n=4). Scale bar = 100 μM.
(A1, A2 and A3) c-Fos expression in saline treated mice.
(B1, B2 and B3) c-Fos expression in insulin treated Vglut2flox/flox mice.
(C1, C2 and C3) c-Fos expression in insulin treated Sf1-Cre, Vglut2flox/flox mice.
(D) Summary of c-Fos counting in DMV neurons.
Discussion
Neuropeptides versus Fast-Acting Neurotransmitters
Important questions exist regarding roles of neuropeptides versus the fast-acting neurotransmitters, glutamate and GABA, in regulating hypothalamic responses (van den Pol, 2003). Neuropeptides and neurotransmitters may both function as independent transmitters, or alternatively, neuropeptides may work by modulating the actions of glutamate and GABA. Key insight into the function of neuropeptides has been gained through the administration of agonists and antagonists, as well as the targeted disruption of genes encoding neuropeptides and their receptors. In contrast, with regards to assessing the roles of glutamate and GABA in hypothalamic function, these approaches have not been (or would not be) particularly informative. Thus, the role of fasting-acting neurotransmitters in regulating hypothalamic function, in vivo, is largely unknown. Based upon the results of brain slice electrophysiology studies, it is likely that fast-acting neurotransmitters play important roles (Cowley et al., 1999; Cowley et al., 2001; van den Pol, 2003). A major goal of the present study was to develop an approach for studying, in vivo, their roles in regulating behaviors and physiologic processes.
Towards these ends, we have used neuron-specific knockout of the vesicular glutamate transporter, VGLUT2, to assess the role of glutamate. VGLUT2, but not VGLUT1 or 3, is expressed in the hypothalamus (Gras et al., 2002; Ziegler et al., 2002). The present study focused on the VMH, a site that is rich in glutamatergic neurons and is thought to play important roles in controlling an array of behaviors and physiologic responses (Swanson, 1987). Specifically, we used cre / lox technology to generate mice that lack VGLUT2 in SF1 neurons (Sf1-Cre, Vglut2flox/flox mice). SF1 neurons constitute the majority of VMH neurons (Stallings et al., 2002). Electrophysiologic studies confirmed that glutamatergic transmission by SF1 neurons was completely disrupted by this approach (Figure 2C). Thus, Sf1-Cre, Vglut2flox/flox mice should be extremely useful for determining the function of glutamate release by VMH neurons. This general approach (neuron-specific deletion of vesicular transporters) and the generated reagents (for example, Vglut2flox/flox mice) should have broad applicability to studies where the goal is to link neurotransmission by selected neurons with specific behaviors and physiologic responses.
Counter-Regulation to Hypoglycemia and the Role of the Brain
The brain requires a continuous supply of glucose for proper function and for survival. To prevent hypoglycemia, low blood glucose elicits the following adaptive responses (Cryer, 2005): 1) insulin secretion by pancreatic β-cells decreases, 2) glucagon secretion by pancreatic α-cells increases, 3) if needed, epinephrine secretion by the adrenal medulla gland increases, and 4) food intake is stimulated. Reduced insulin secretion is thought to be a direct response by β-cells to low blood glucose. The other responses, however, are driven primarily by the brain. These critically important counter-measures are initiated by fasting, and iatrogenically, when hypoglycemia develops following insulin treatment. Indeed, insulin-induced hypoglycemia complicates and severely limits the widespread application of intensive insulin therapy in type 1 diabetes, which has been shown to markedly reduce microvascular complications of chronic hyperglycemia (Cryer, 2005). This issue is further exacerbated by that fact that counter-regulatory responses to hypoglycemia are defective in individuals with diabetes (Cryer, 2005). For this reason, understanding the mechanisms responsible for counter-regulatory responses, and the reasons why they are inoperative in diabetics, is an extremely important area for investigation.
The CNS detects hypoglycemia and then initiates counter-regulatory responses. The brain contains “glucose-excited” neurons (firing rate increases as glucose levels rise) and “glucose-inhibited” neurons (firing rate increases as glucose levels fall) (Anand et al., 1962; Burdakov et al., 2005b; Oomura et al., 1974; Routh, 2002). Glucose depolarizes glucose-excited neurons by closing ATP-sensitive K+ channels (Ashford et al., 1990; Miki et al., 2001; Routh, 2002). The mechanism by which glucose hyperpolarizes glucose-inhibited neurons, on the other hand, is less well understood, but may involve altered activity of chloride channels (Routh, 2002), Na/K pumps (Silver and Erecinska, 1998), or could also involve tandem-pore K+ (K2p) channels, as was recently shown for glucose-inhibited orexin neurons (Burdakov et al., 2006). Neurons capable of sensing glucose are abundant in various parts of the hypothalamus (including the VMH, the lateral hypothalamus and the arcuate nucleus) and in the hindbrain (Burdakov et al., 2005a; Ibrahim et al., 2003; Marty et al., 2005; Muroya et al., 1999; Ritter et al., 2006; Routh, 2002). In principle, the method employed by the brain to detect hypoglycemia could involve decreased activity of glucose-excited neurons, increased activity of glucose-inhibited neurons, input from peripheral blood glucose sensors (Burcelin et al., 2000), or possibly some combination of these three processes.
The Role of the VMH
The neurocircuitry underlying detection of hypoglycemia and subsequent counter-regulatory responses is not well known. The VMH is believed to play a role for the following reasons: 1) the VMH contains neurons capable of sensing glucose (Ashford et al., 1990; Routh, 2002), 2) local glucopenia in the VMH induced by infusion of 2-deoxyglucose produces counter-regulatory responses (Borg et al., 1995), 3) lesions of the VMH impair counter-regulatory responses (Borg et al., 1994), and 4) infusion of glucose directly into the VMH during systemic hypoglycemia attenuates counter-regulatory responses (Borg et al., 1997). While these findings implicate the VMH, they are limited by uncertainty regarding the range of diffusion of infused molecules (glucose and 2-deoxyglucose) and the extent of ablative lesions. Of note, glucose-excited (POMC neurons, MCH neurons) and glucose-inhibited (NPY/AGRP neurons, orexin neurons) neurons are found nearby in the arcuate nucleus and the lateral hypothalamus (Burdakov et al., 2005a; Ibrahim et al., 2003; Muroya et al., 1999).
In the present study, we have shown that Sf1-Cre, Vglut2flox/flox mice have clear defects in counter-regulatory responses. This manifests itself as hypoglycemia during fasting and during insulin treatment, and as a defective hyperglycemic response to neuroglycopenia (induced by central infusion of 2-deoxyglucose). The greatest counter-regulatory defect was seen in hypoglycemia-induced secretion of glucagon. A modest impairment was seen in hypoglycemia-induced secretion of epinephrine. Of note, no impairment was seen in the hyperphagic response to neuroglycopenia. This latter finding is in agreement with studies implicating hindbrain catecholaminergic cell groups with projection to the PVH (Ritter et al., 2006) and neuropeptide Y (Sindelar et al., 2004), which is not found in the VMH, in mediating neuroglycopenia-induced hyperphagia. In total, the present study definitively establishes a role for SF1 neurons, and glutamate release by these neurons, in the homeostatic neurocircuitry that prevents hypoglycemia.
Afferents and Efferents of SF1 Neurons
The “first-order neurons” responsible for detecting hypoglycemia are unknown. It could be the SF1 neurons themselves. A subset of VMH neurons are known to sense glucose (Song and Routh, 2005), some of which could be SF1 neurons. Alternatively, it is possible that the primary “glucose-sensing” neurons are upstream of SF1 neurons. One possibility are the hindbrain catecholaminergic neurons. These neurons are responsive to hypoglycemia and send projections to the VMH (Ritter et al., 2006). Consistent with this, the norepinephrine level is increased in the VMH area in response to hypoglycemia (Beverly et al., 2001) and VMH neurons express adrenergic receptors (Boundy and Cincotta, 2000). Another possibility are the orexin neurons in the lateral hypothalamus, which are excited by low glucose (Burdakov et al., 2005a; Burdakov et al., 2006). Orexin receptors 1 (OX1R) and 2 (OX2R) are both expressed at high levels in the VMH (Marcus et al., 2001). Other neurons, including glucose-excited POMC neurons and MCH neurons or glucose-inhibited NPY/AGRP neurons, could also be upstream of SF1 neurons (Burdakov et al., 2005a; Ibrahim et al., 2003; Muroya et al., 1999; Ritter et al., 2006). Likewise, it is possible that peripheral glucose sensors (Burcelin et al., 2000) could provide input to VMH. Identifying the primary site responsible for detecting hypoglycemia, and determining how this site connects with SF1 neurons, is an important area of investigation.
Similarly, the efferent pathways connecting SF1 neurons with glucagon secretion are also of interest. The autonomic nervous system controls glucagon secretion and, importantly, blockade of parasympathetic and sympathetic input to pancreatic α-cells prevents hypoglycemia-induced and neuroglycopenia-induced increases in glucagon (Rossi et al., 2005; Taborsky et al., 1998). Given this, parasympathetic preganglionic neurons in the dosal motor nucleus of the vagus (DMV) and sympathetic preganglionic neurons in the spinal cord could be downstream of SF1 neurons. Consistent with an important role for parasympathetic preganlionic neurons, we have found that hypoglycemia-mediated induction of c-Fos in the DMV, at the level of area postrema, is significantly attenuated in Sf1-Cre, Vglut2flox/flox mice (Figure 7). This finding is likely to be relevant since DMV neurons at this level send direct projections to pancreas (Rinaman and Miselis, 1987). How SF1 neurons connect with DMV neurons, however, remains to be determined. The connection may be indirect since direct projection from the VMH to the DMV have yet to be described (Canteras et al., 1994).
Body Weight and release of glutamate from SF1 Neurons
Mice homozygous for a disrupted SF1 allele and mice lacking leptin receptors on SF1 neurons (Sf1-Cre, Leprflox/flox mice) are modestly obese when fed a chow diet and develop marked obesity when fed a high fat diet (Dhillon et al., 2006; Majdic et al., 2002). With this in mind, it is surprising that Sf1-Cre, Vglut2flox/flox mice are not obese when fed a chow diet and had very limited obesity when fed a high fat diet. These findings raise the possibility that some other effector released from SF1 neurons, for example BDNF (Xu et al., 2003) or GABA (as shown in Figure 2, some SF1 neurons are GABAergic), may play a role in regulating body weight. We suspect that BDNF release from SF1 neurons is not involved since mice lacking BDNF in SF1 neurons (Sf1-Cre, Bdnfflox/flox mice) failed to develop obesity (data not shown). We are presently testing the role of GABA by assessing energy homeostasis in Sf1-Cre, Vgatflox/flox mice.
Summary
In summary, this study demonstrates that neuron-specific manipulation of vesicle neurotransmitter transporters, such as VGLUT2, is a useful means of linking fast-acting neurotransmitter release with specific functions. In addition, we have established that SF1 neurons in the VMH, and glutamate release by these neurons, is an important component of the neurocircuitry that functions to prevent hypoglycemia. Using SF1 neurons as a starting point, it will be important to identify the afferent and efferent components of this circuitry. Knowledge of these pathways should provide a framework for studies aimed at understanding the clinically significant problem of defective counter-regulation to hypoglycemia in diabetes.
Methods
Animal Care
Care of all animals and procedures were approved by the Beth Israel Deaconess Medical Center Institutional animal care and use committee. Mice were housed at 22°C–24°C with a 14 hr light/10 hr dark cycle with standard mouse chow (Teklad F6 Rodent Diet 8664, 4.05 kcal/g, 3.3 kcal/g metabolizable energy, 12.5% kcal from fat, Harlan Teklad, Madison, WI) and water provided ad libitum.
Generation of Vglut2flox/flox mice and Sf1-Cre, Vglut2flox/flox mice
The lox modified Vglut2 (Slc17a6) targeting construct was constructed using a mouse 129 BAC genomic clone and recombineering technology (Lee et al., 2001). The 2 lox P sites were inserted in the first intron and the second intron, respectively. Sf1-Cre, Vglut2flox/+ mice were generated by mating Sf1-Cre transgenic mice (FVB/N background) (Dhillon et al., 2006) with Vglut2flox/+ mice (129, C57Bl/6 background). Study subjects were generated by mating Sf1-Cre, Vglut2flox/flox mice with Vglut2flox/flox mice. See Supplemental Text for details.
High Fat, High Sucrose Diet Study
Groups of 8-10 week old male controls (Vglut2flox/flox mice) and Sf1-Cre, Vglut2flox/flox mice were switched to the high fat, high sucrose diet (HFD) and maintained on HFD for 12 weeks. See Supplemental Text for study details.
Electrophysiologic Recordings on Autaptic Cultured Neurons
Micro-punched VMH tissues were digested using a papain dissociation system (Worthington, Lakewood, NJ). Digested neurons were seeded on poly-L-lysine-coated glass coverslips in Neurobasal A medium (Invitrogen, Grand Island, NY). The neurons were planted at low density to encourage the formation of autosynapses
Synaptic currents were recorded in whole-cell voltage clamp mode and release of synaptic vesicles was evoked by a depolarizing pulse to +10 mV from the holding potential of –60 mV, applied every 20 sec. The solution used in the recording pipette permits identification of both glutamatergic EPSCs and GABAergic IPSCs as inward current. EPSCs and IPSCs were identified by their deferent decay time constants and by using specific glutamate receptor antagonists (QBNX and D-APV) and GABA receptor antagonist (bicuculline). See Supplemental Text for details.
In situ Hybridization Studies
The in situ hybridizations were performed as previously described (Kishi et al., 2003). See Supplemental Text for details.
Glucose Homeostasis Studies
Fed blood glucose levels were measured between 9am-10am. Fasted blood glucose, liver gluconeogenic gene expression and glucagon levels were measured after 24 hrs fasting.
For the insulin-induced hypoglycemia study, overnight fasted males were administrated i.p. with saline or insulin at 1.5 U/kg. At 1 hour post administration, blood was collected for determination of various parameters.
For i.c.v. injections, a stainless steel cannula (30 gauge) was implanted in the right lateral ventricle. On the test day, 2-deoxyglucose (1mg/3μl/mouse) or saline was injected i.c.v. into fed mice. At 30 minutes after injection, liver samples were collected, and glucose, insulin and glucagon were determined. See Supplemental Text for details.
Quantitative PCR Assay
Liver RNA was reverse transcribed with RETROscript (Ambion, Inc., Austin, TX) and amplified using Assay on Demand Tagman probes and primers (Applied Biosystems, Foster City, CA). Quantitative PCR was performed on an Mx4000 instrument (Stratagene). See Supplemental Text for details.
Hyperinsulinemia Hypoglycemia Clamp Studies
The hyperinsulinemia hypoglycemia protocol was a modification of a procedure previously described for rats (McCrimmon et al., 2006). Briefly, at t = 0, a 90-min 10 mU ∙ kg-1 ∙ min-1 infusion of human regular insulin (Eli Lilly) was begun. The glucose was allowed to fall to ~45 mg/dl and was then maintained at this level at 60- 90 min. Samples for measurement of the hormones epinephrine, norepinephrine (HPLC with electrochemical detection) and glucagon (RIA, Linco Research Inc.) were taken at 0, 60 and 90 min. See Supplemental Text for details.
Hypoglycemia-Medicated c-Fos Induction
Vglut2flox/flox and Sf1-Cre, Vglut2flox/flox mice were acclimated for 7 days before i.p. injection of either saline (4 mice from each genotype) or insulin (1.5U/kg, 4 mice from each genotype). Blood glucose was then monitored and the mice were transcardially perfused at 60 mins. The brains were cut in 5 series with 25 μM thickness and immunostained for c-Fos (α-c-Fos with Cat# PC38 raised in rabbit at 1:25,000).
In the DMV, the greatest induction of c-Fos was observed in 3 consecutive sections immediately rostral to the appearance of area postrema in Vglut2flox/flox mice. Therefore, c-Fos positive neurons were counted in the DMV of these sections. The saline treated Vglut2flox/flox mice and Sf1-Cre, Vglut2flox/flox mice were pooled together as a saline control as there was no difference in c-Fos induction in these mice. Only neurons showing clear, round c-Fos stained nuclei were counted as positive for c-Fos.
Statistical Methods
Data sets were analyzed for statistical significance using PRISM (GraphPad, San Diego, CA) for a two-tailed unpaired Student’s t test.
Supplementary Material
Acknowledgments
The authors acknowledge Bruce Bean and Jun Lu for helpful discussions. This work was supported by the National Institutes of Health (RO1 DK071051 and PO1 DK56116 to BBL, R01 DK072409 and R37 DK020495 to RSS, R01 DK069831 to RJM, DK071320 to JKE), the NIH-funded Boston Obesity Nutrition Research Center (P30 DK046200, the Transgenic Core, Directed by BBL, helped generate the SF1-Cre transgenic mice and the Vglut2flox/flox mice, also HD was the recipient of a Pilot and Feasibility Award), the NIH-funded Boston Area Diabetes Endocrinology Research Center (P30 DK057521, the Transgenic Core, directed by BBL, helped generate mice as above), the NIH-funded Program Project Energy Expenditure Core (PO1 DK56116), the Smith Family Foundation Pinnacle Program Project Award from American Diabetes Association (JKE), the JDRF (JDRF Center for the Study of Hypoglycemia, RSS) and the American Heart Association (Fellowships to QT and HD).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Anand BK, Chhina GS, Singh B. Effect of glucose on the activity of hypothalamic “feeding centers”. Science. 1962;138:597–598. doi: 10.1126/science.138.3540.597. [DOI] [PubMed] [Google Scholar]
- Ashford ML, Boden PR, Treherne JM. Glucose-induced excitation of hypothalamic neurones is mediated by ATP-sensitive K+ channels. Pflugers Arch. 1990;415:479–483. doi: 10.1007/BF00373626. [DOI] [PubMed] [Google Scholar]
- Beverly JL, De Vries MG, Bouman SD, Arseneau LM. Noradrenergic and GABAergic systems in the medial hypothalamus are activated during hypoglycemia. Am J Physiol Regul Integr Comp Physiol. 2001;280:R563–569. doi: 10.1152/ajpregu.2001.280.2.R563. [DOI] [PubMed] [Google Scholar]
- Borg MA, Sherwin RS, Borg WP, Tamborlane WV, Shulman GI. Local ventromedial hypothalamus glucose perfusion blocks counterregulation during systemic hypoglycemia in awake rats. J Clin Invest. 1997;99:361–365. doi: 10.1172/JCI119165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borg WP, During MJ, Sherwin RS, Borg MA, Brines ML, Shulman GI. Ventromedial hypothalamic lesions in rats suppress counterregulatory responses to hypoglycemia. J Clin Invest. 1994;93:1677–1682. doi: 10.1172/JCI117150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borg WP, Sherwin RS, During MJ, Borg MA, Shulman GI. Local ventromedial hypothalamus glucopenia triggers counterregulatory hormone release. Diabetes. 1995;44:180–184. doi: 10.2337/diab.44.2.180. [DOI] [PubMed] [Google Scholar]
- Boundy VA, Cincotta AH. Hypothalamic adrenergic receptor changes in the metabolic syndrome of genetically obese (ob/ob) mice. Am J Physiol Regul Integr Comp Physiol. 2000;279:R505–514. doi: 10.1152/ajpregu.2000.279.2.R505. [DOI] [PubMed] [Google Scholar]
- Burcelin R, Dolci W, Thorens B. Glucose sensing by the hepatoportal sensor is GLUT2-dependent: in vivo analysis in GLUT2-null mice. Diabetes. 2000;49:1643–1648. doi: 10.2337/diabetes.49.10.1643. [DOI] [PubMed] [Google Scholar]
- Burdakov D, Gerasimenko O, Verkhratsky A. Physiological changes in glucose differentially modulate the excitability of hypothalamic melanin-concentrating hormone and orexin neurons in situ. J Neurosci. 2005a;25:2429–2433. doi: 10.1523/JNEUROSCI.4925-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burdakov D, Jensen LT, Alexopoulos H, Williams RH, Fearon IM, O’Kelly I, Gerasimenko O, Fugger L, Verkhratsky A. Tandem-pore K+ channels mediate inhibition of orexin neurons by glucose. Neuron. 2006;50:711–722. doi: 10.1016/j.neuron.2006.04.032. [DOI] [PubMed] [Google Scholar]
- Burdakov D, Luckman SM, Verkhratsky A. Glucose-sensing neurons of the hypothalamus. Philos Trans R Soc Lond B Biol Sci. 2005b;360:2227–2235. doi: 10.1098/rstb.2005.1763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canteras NS. The medial hypothalamic defensive system: hodological organization and functional implications. Pharmacol Biochem Behav. 2002;71:481–491. doi: 10.1016/s0091-3057(01)00685-2. [DOI] [PubMed] [Google Scholar]
- Canteras NS, Simerly RB, Swanson LW. Organization of projections from the ventromedial nucleus of the hypothalamus: a Phaseolus vulgaris-leucoagglutinin study in the rat. J Comp Neurol. 1994;348:41–79. doi: 10.1002/cne.903480103. [DOI] [PubMed] [Google Scholar]
- Choi GB, Dong HW, Murphy AJ, Valenzuela DM, Yancopoulos GD, Swanson LW, Anderson DJ. Lhx6 delineates a pathway mediating innate reproductive behaviors from the amygdala to the hypothalamus. Neuron. 2005;46:647–660. doi: 10.1016/j.neuron.2005.04.011. [DOI] [PubMed] [Google Scholar]
- Cone RD. Anatomy and regulation of the central melanocortin system. Nat Neurosci. 2005;8:571–578. doi: 10.1038/nn1455. [DOI] [PubMed] [Google Scholar]
- Cowley MA, Pronchuk N, Fan W, Dinulescu DM, Colmers WF, Cone RD. Integration of NPY, AGRP, and melanocortin signals in the hypothalamic paraventricular nucleus: evidence of a cellular basis for the adipostat. Neuron. 1999;24:155–163. doi: 10.1016/s0896-6273(00)80829-6. [DOI] [PubMed] [Google Scholar]
- Cowley MA, Smart JL, Rubinstein M, Cerdan MG, Diano S, Horvath TL, Cone RD, Low MJ. Leptin activates anorexigenic POMC neurons through a neural network in the arcuate nucleus. Nature. 2001;411:480–484. doi: 10.1038/35078085. [DOI] [PubMed] [Google Scholar]
- Cryer PE. Mechanisms of hypoglycemia-associated autonomic failure and its component syndromes in diabetes. Diabetes. 2005;54:3592–3601. doi: 10.2337/diabetes.54.12.3592. [DOI] [PubMed] [Google Scholar]
- Dhillon H, Zigman JM, Ye C, Lee CE, McGovern RA, Tang V, Kenny CD, Christiansen LM, White RD, Edelstein EA, et al. Leptin directly activates SF1 neurons in the VMH, and this action by leptin is required for normal body-weight homeostasis. Neuron. 2006;49:191–203. doi: 10.1016/j.neuron.2005.12.021. [DOI] [PubMed] [Google Scholar]
- Fremeau RT, Jr, Kam K, Qureshi T, Johnson J, Copenhagen DR, Storm-Mathisen J, Chaudhry FA, Nicoll RA, Edwards RH. Vesicular glutamate transporters 1 and 2 target to functionally distinct synaptic release sites. Science. 2004a;304:1815–1819. doi: 10.1126/science.1097468. [DOI] [PubMed] [Google Scholar]
- Fremeau RT, Jr, Voglmaier S, Seal RP, Edwards RH. VGLUTs define subsets of excitatory neurons and suggest novel roles for glutamate. Trends Neurosci. 2004b;27:98–103. doi: 10.1016/j.tins.2003.11.005. [DOI] [PubMed] [Google Scholar]
- Gasnier B. The SLC32 transporter, a key protein for the synaptic release of inhibitory amino acids. Pflugers Arch. 2004;447:756–759. doi: 10.1007/s00424-003-1091-2. [DOI] [PubMed] [Google Scholar]
- Gras C, Herzog E, Bellenchi GC, Bernard V, Ravassard P, Pohl M, Gasnier B, Giros B, El Mestikawy S. A third vesicular glutamate transporter expressed by cholinergic and serotoninergic neurons. J Neurosci. 2002;22:5442–5451. doi: 10.1523/JNEUROSCI.22-13-05442.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guillemin R. Hypothalamic hormones a.k.a. hypothalamic releasing factors. J Endocrinol. 2005;184:11–28. doi: 10.1677/joe.1.05883. [DOI] [PubMed] [Google Scholar]
- Hentges ST, Nishiyama M, Overstreet LS, Stenzel-Poore M, Williams JT, Low MJ. GABA release from proopiomelanocortin neurons. J Neurosci. 2004;24:1578–1583. doi: 10.1523/JNEUROSCI.3952-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrera DG, Robertson HA. Activation of c-fos in the brain. Prog Neurobiol. 1996;50:83–107. doi: 10.1016/s0301-0082(96)00021-4. [DOI] [PubMed] [Google Scholar]
- Ibrahim N, Bosch MA, Smart JL, Qiu J, Rubinstein M, Ronnekleiv OK, Low MJ, Kelly MJ. Hypothalamic proopiomelanocortin neurons are glucose responsive and express K(ATP) channels. Endocrinology. 2003;144:1331–1340. doi: 10.1210/en.2002-221033. [DOI] [PubMed] [Google Scholar]
- King BM. The rise, fall, and resurrection of the ventromedial hypothalamus in the regulation of feeding behavior and body weight. Physiol Behav. 2006;87:221–244. doi: 10.1016/j.physbeh.2005.10.007. [DOI] [PubMed] [Google Scholar]
- Kishi T, Aschkenasi CJ, Lee CE, Mountjoy KG, Saper CB, Elmquist JK. Expression of melanocortin 4 receptor mRNA in the central nervous system of the rat. J Comp Neurol. 2003;457:213–235. doi: 10.1002/cne.10454. [DOI] [PubMed] [Google Scholar]
- Krieger MS, Conrad LC, Pfaff DW. An autoradiographic study of the efferent connections of the ventromedial nucleus of the hypothalamus. J Comp Neurol. 1979;183:785–815. doi: 10.1002/cne.901830408. [DOI] [PubMed] [Google Scholar]
- Lee EC, Yu D, Martinez de Velasco J, Tessarollo L, Swing DA, Court DL, Jenkins NA, Copeland NG. A highly efficient Escherichia coli-based chromosome engineering system adapted for recombinogenic targeting and subcloning of BAC DNA. Genomics. 2001;73:56–65. doi: 10.1006/geno.2000.6451. [DOI] [PubMed] [Google Scholar]
- Majdic G, Young M, Gomez-Sanchez E, Anderson P, Szczepaniak LS, Dobbins RL, McGarry JD, Parker KL. Knockout mice lacking steroidogenic factor 1 are a novel genetic model of hypothalamic obesity. Endocrinology. 2002;143:607–614. doi: 10.1210/endo.143.2.8652. [DOI] [PubMed] [Google Scholar]
- Marcus JN, Aschkenasi CJ, Lee CE, Chemelli RM, Saper CB, Yanagisawa M, Elmquist JK. Differential expression of orexin receptors 1 and 2 in the rat brain. J Comp Neurol. 2001;435:6–25. doi: 10.1002/cne.1190. [DOI] [PubMed] [Google Scholar]
- Marty N, Dallaporta M, Foretz M, Emery M, Tarussio D, Bady I, Binnert C, Beermann F, Thorens B. Regulation of glucagon secretion by glucose transporter type 2 (glut2) and astrocyte-dependent glucose sensors. J Clin Invest. 2005;115:3545–3553. doi: 10.1172/JCI26309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCrimmon RJ, Song Z, Cheng H, McNay EC, Weikart-Yeckel C, Fan X, Routh VH, Sherwin RS. Corticotrophin-releasing factor receptors within the ventromedial hypothalamus regulate hypoglycemia-induced hormonal counterregulation. J Clin Invest. 2006;116:1723–1730. doi: 10.1172/JCI27775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miki T, Liss B, Minami K, Shiuchi T, Saraya A, Kashima Y, Horiuchi M, Ashcroft F, Minokoshi Y, Roeper J, Seino S. ATP-sensitive K+ channels in the hypothalamus are essential for the maintenance of glucose homeostasis. Nat Neurosci. 2001;4:507–512. doi: 10.1038/87455. [DOI] [PubMed] [Google Scholar]
- Moechars D, Weston MC, Leo S, Callaerts-Vegh Z, Goris I, Daneels G, Buist A, Cik M, van der Spek P, Kass S, et al. Vesicular glutamate transporter VGLUT2 expression levels control quantal size and neuropathic pain. J Neurosci. 2006;26:12055–12066. doi: 10.1523/JNEUROSCI.2556-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muroya S, Yada T, Shioda S, Takigawa M. Glucose-sensitive neurons in the rat arcuate nucleus contain neuropeptide Y. Neurosci Lett. 1999;264:113–116. doi: 10.1016/s0304-3940(99)00185-8. [DOI] [PubMed] [Google Scholar]
- Novak A, Guo C, Yang W, Nagy A, Lobe CG. Z/EG, a double reporter mouse line that expresses enhanced green fluorescent protein upon Cre-mediated excision. Genesis. 2000;28:147–155. [PubMed] [Google Scholar]
- Oomura Y, Ooyama H, Sugimori M, Nakamura T, Yamada Y. Glucose inhibition of the glucose-sensitive neurone in the rat lateral hypothalamus. Nature. 1974;247:284–286. doi: 10.1038/247284a0. [DOI] [PubMed] [Google Scholar]
- Ovesjo ML, Gamstedt M, Collin M, Meister B. GABAergic nature of hypothalamic leptin target neurones in the ventromedial arcuate nucleus. J Neuroendocrinol. 2001;13:505–516. doi: 10.1046/j.1365-2826.2001.00662.x. [DOI] [PubMed] [Google Scholar]
- Rinaman L, Miselis RR. The organization of vagal innervation of rat pancreas using cholera toxin-horseradish peroxidase conjugate. J Auton Nerv Syst. 1987;21:109–125. doi: 10.1016/0165-1838(87)90014-2. [DOI] [PubMed] [Google Scholar]
- Ritter S, Dinh TT, Li AJ. Hindbrain catecholamine neurons control multiple glucoregulatory responses. Physiol Behav. 2006;89:490–500. doi: 10.1016/j.physbeh.2006.05.036. [DOI] [PubMed] [Google Scholar]
- Rossi J, Santamaki P, Airaksinen MS, Herzig KH. Parasympathetic innervation and function of endocrine pancreas requires the glial cell line-derived factor family receptor alpha2 (GFRalpha2) Diabetes. 2005;54:1324–1330. doi: 10.2337/diabetes.54.5.1324. [DOI] [PubMed] [Google Scholar]
- Routh VH. Glucose-sensing neurons: are they physiologically relevant? Physiol Behav. 2002;76:403–413. doi: 10.1016/s0031-9384(02)00761-8. [DOI] [PubMed] [Google Scholar]
- Saper CB, Scammell TE, Lu J. Hypothalamic regulation of sleep and circadian rhythms. Nature. 2005;437:1257–1263. doi: 10.1038/nature04284. [DOI] [PubMed] [Google Scholar]
- Saper CB, Swanson LW, Cowan WM. The efferent connections of the ventromedial nucleus of the hypothalamus of the rat. J Comp Neurol. 1976;169:409–442. doi: 10.1002/cne.901690403. [DOI] [PubMed] [Google Scholar]
- Silver IA, Erecinska M. Glucose-induced intracellular ion changes in sugar-sensitive hypothalamic neurons. J Neurophysiol. 1998;79:1733–1745. doi: 10.1152/jn.1998.79.4.1733. [DOI] [PubMed] [Google Scholar]
- Sindelar DK, Ste Marie L, Miura GI, Palmiter RD, McMinn JE, Morton GJ, Schwartz MW. Neuropeptide Y is required for hyperphagic feeding in response to neuroglucopenia. Endocrinology. 2004;145:3363–3368. doi: 10.1210/en.2003-1727. [DOI] [PubMed] [Google Scholar]
- Song Z, Routh VH. Differential effects of glucose and lactate on glucosensing neurons in the ventromedial hypothalamic nucleus. Diabetes. 2005;54:15–22. doi: 10.2337/diabetes.54.1.15. [DOI] [PubMed] [Google Scholar]
- Stallings NR, Hanley NA, Majdic G, Zhao L, Bakke M, Parker KL. Development of a transgenic green fluorescent protein lineage marker for steroidogenic factor 1. Endocr Res. 2002;28:497–504. doi: 10.1081/erc-120016829. [DOI] [PubMed] [Google Scholar]
- Sternson SM, Shepherd GM, Friedman JM. Topographic mapping of VMH --> arcuate nucleus microcircuits and their reorganization by fasting. Nat Neurosci. 2005;8:1356–1363. doi: 10.1038/nn1550. [DOI] [PubMed] [Google Scholar]
- Swanson LW. The Hypothalamus. Vol. 5. Elservier; Amsterdam: 1987. [Google Scholar]
- Taborsky GJ, Jr, Ahren B, Havel PJ. Autonomic mediation of glucagon secretion during hypoglycemia: implications for impaired alpha-cell responses in type 1 diabetes. Diabetes. 1998;47:995–1005. doi: 10.2337/diabetes.47.7.995. [DOI] [PubMed] [Google Scholar]
- van den Pol AN. Weighing the role of hypothalamic feeding neurotransmitters. Neuron. 2003;40:1059–1061. doi: 10.1016/s0896-6273(03)00809-2. [DOI] [PubMed] [Google Scholar]
- Vidal-Puig A, O’Rahilly S. Metabolism. Controlling the glucose factory. Nature. 2001;413:125–126. doi: 10.1038/35093198. [DOI] [PubMed] [Google Scholar]
- Wallen-Mackenzie A, Gezelius H, Thoby-Brisson M, Nygard A, Enjin A, Fujiyama F, Fortin G, Kullander K. Vesicular glutamate transporter 2 is required for central respiratory rhythm generation but not for locomotor central pattern generation. J Neurosci. 2006;26:12294–12307. doi: 10.1523/JNEUROSCI.3855-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams G, Cai XJ, Elliott JC, Harrold JA. Anabolic neuropeptides. Physiol Behav. 2004;81:211–222. doi: 10.1016/j.physbeh.2004.02.005. [DOI] [PubMed] [Google Scholar]
- Wojcik SM, Katsurabayashi S, Guillemin I, Friauf E, Rosenmund C, Brose N, Rhee JS. A shared vesicular carrier allows synaptic corelease of GABA and glycine. Neuron. 2006;50:575–587. doi: 10.1016/j.neuron.2006.04.016. [DOI] [PubMed] [Google Scholar]
- Wojcik SM, Rhee JS, Herzog E, Sigler A, Jahn R, Takamori S, Brose N, Rosenmund C. An essential role for vesicular glutamate transporter 1 (VGLUT1) in postnatal development and control of quantal size. Proc Natl Acad Sci U S A. 2004;101:7158–7163. doi: 10.1073/pnas.0401764101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu B, Goulding EH, Zang K, Cepoi D, Cone RD, Jones KR, Tecott LH, Reichardt LF. Brain-derived neurotrophic factor regulates energy balance downstream of melanocortin-4 receptor. Nat Neurosci. 2003;6:736–742. doi: 10.1038/nn1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon JC, Puigserver P, Chen G, Donovan J, Wu Z, Rhee J, Adelmant G, Stafford J, Kahn CR, Granner DK, et al. Control of hepatic gluconeogenesis through the transcriptional coactivator PGC-1. Nature. 2001;413:131–138. doi: 10.1038/35093050. [DOI] [PubMed] [Google Scholar]
- Ziegler DR, Cullinan WE, Herman JP. Distribution of vesicular glutamate transporter mRNA in rat hypothalamus. J Comp Neurol. 2002;448:217–229. doi: 10.1002/cne.10257. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.