

Abstract
To determine how acute ethanol intoxication may alter memory processing, we examined the effects of stepwise increases in ethanol on long-term potentiation (LTP) in hippocampal slices. LTP was inhibited by acute administration of 60 mM ethanol, but was readily induced if ethanol was increased gradually to 60 mM over 75 min. Administration of 2-amino-5 phosphonovalerate (APV), an N-methyl-D-aspartate receptor antagonist, during the stepwise increase in ethanol inhibited LTP, suggesting involvement of NMDA receptors in the development of tolerance. However, APV and nifedipine, an inhibitor of L-type calcium channels, failed to inhibit LTP when administered following the slow increase in ethanol. Ethanol-tolerant LTP was inhibited by thapsigargin, suggesting a major role for intracellular calcium release in this form of plasticity. The unique properties of ethanol-tolerant LTP suggest that memories formed during binge drinking are not acquired by standard synaptic mechanisms and that acute tolerance may involve the induction of novel mechanisms to maintain function.
Keywords: Alcohol (ethanol), tolerance , desensitization, allostasis
Acute alcohol intoxication is a significant public health problem. During the intoxicated state, problems with motor coordination and cognitive processing are common. Some individuals exhibit profound deficits in memory that may include a complete inability to learn new information, a condition referred to as a ‘blackout’ (White, 2003). Cognitive and behavioral manifestations associated with acute intoxication are often determined by how ethanol is consumed. Thus, it is possible that both the rate of alcohol consumption and the amount of ethanol consumed are key factors in determining overall effects on the CNS.
In hippocampal slices, numerous studies have shown that ethanol inhibits long-term potentiation (LTP), a cellular model of memory and learning (Sinclair and Lo, 1986; Morrisett and Swartzwelder, 1993; Schummers and Browning, 2001). The concentration of ethanol required for LTP inhibition, however, has varied widely among reports. While many studies have found that ethanol inhibits LTP induction at concentrations of 50 mM or more (Randall et al., 1995; Sugiura et al., 1995; Schummers et al., 1997), some reports have shown that concentrations as low as 5 mM also impair LTP induction (Blitzer et al., 1990). The inconsistencies in these studies may reflect experimental differences including slice preparation, recording and stimulation conditions, and the methods used for ethanol administration. We previously found that ethanol inhibits LTP induction when administered acutely at 60 mM for 15 min before and during delivery of a single 100 Hz x 1 sec high frequency stimulus (HFS). Similar 15 min applications of 60 mM ethanol also inhibit LTP when ethanol is removed for several hours prior to HFS (Izumi et al., 2005), suggesting that brief exposures to high concentrations of ethanol can have longer-lived adverse effects on synaptic function.
Although these observations suggest that the timing of ethanol administration is not critical for the inhibition of LTP, it remains unclear how the rate of ethanol administration and hence the rate of rise in ethanol concentration affects LTP induction. For example, 60 mM ethanol inhibits LTP in hippocampal slices when the perfusion rate of the media is 2 ml/min (Izumi et al., 2005). At this rate, it only takes ~2 min for the concentration in the slice chamber to reach 50 mM. Even heavy binge drinking does not raise blood ethanol levels to 50 mM in such a short time (Lange and Voas, 2001; Perkins et al., 2001). To examine the effects of more gradual increases in ethanol such as might occur during a bout of binge drinking, we used a protocol in which a final concentration of 60 mM was achieved by stepwise increases of 10 mM every 15 min. Under these conditions, we found that 60 mM ethanol failed to inhibit LTP, suggesting that an acute form of tolerance develops during slower stepwise increases in ethanol levels. We also examined whether this ethanol-tolerant LTP shares mechanisms with conventional LTP.
Materials and methods
Animal statement
All animal procedures were performed in accordance with the guidelines of the Washington University Animal Study Committee.
Hippocampal slices preparation
Transverse slices of hippocampus were prepared from Sprague-Dawley male rats at postnatal date (PND) 30 ± 2 using methods described previously (Zorumski et al., 1996). Albino rats were anesthetized with isoflurane and decapitated. The hippocampi were rapidly dissected and sliced at a thickness of 500 μm with a vibrating tissue slicer (World Precision Instruments Vibroslicer, Sarasota, FL, USA). Dissection and tissue slicing was done in a chamber filled with artificial cerebrospinal fluid (aCSF) maintained at an ice-cold temperature to avoid neurodegenerative changes. The aCSF contained (in mM): 124 NaCl, 5 KCl, 2 MgSO4, 2 CaCl2, 1.25 NaH2PO4, 22 NaHCO3, and 10 glucose, and was aerated with 95% O2 - 5% CO2 to ensure a pH of 7.4. The hippocampal slices were placed on stretched nylon in a beaker containing gassed aCSF for 1 h recovery at 30ºC. At the time of study, individual slices were transferred to the center of a submersion recording chamber, where they were allowed to stabilize for 15 min before recording.
Extracellular field potential recording
Extracellular recordings of field population excitatory postsynaptic potentials (EPSPs) and population spikes (PSs) were obtained from the dendritic layer and the pyramidal cell layer of CA1 using separate electrodes filled with 2 M NaCl (5-10 MΩ), using a microelectrode amplifier (Warner Instruments, Hamden, CT, USA). Schaffer collateral-commissural fibers were electrically stimulated with 0.1-0.2 ms constant current paired pulses at a 21 ms interval with a bipolar electrode (Rhodes Medical Instruments Inc., Summerland, CA, USA). The intensities for each recording were adjusted to produce an evoked response that was 50 -60 % of the maximum EPSP. PS amplitude was measured as the height from the apex of the first positive wave to the most negative point. The average of field EPSPs during an initial control period of the recording was used to determine the initial baseline response (represented as 100% in subsequent figures). A high-frequency stimulation (HFS) with a single 100 Hz × 1 s tetanus was applied for LTP induction. The degree of LTP was quantified based on analysis of input-output (IO) curves. After establishing a stable baseline, a control IO curve was obtained. A second IO curve was obtained 60 min after HFS. LTP was measured as a change in the EPSP slope at the 50% point on the respective IO curves sixty min after HFS and was defined as a 20% or greater increase in half maximal EPSP slope. The submersion-recording chamber (2.0 ml capacity) was continuously perfused with aCSF and regulated at 30ºC by a feedback circuit. The flow rate for all recordings was 2.0 ml/min with a gravity fed solution delivery system. In this system, the concentration of a drug is predicted to be C*(1-1/et) where C is the final concentration and t is perfusion time (min). Signals were digitized and analyzed using the PCLAMP software (Axon Instruments, Union city, CA, USA).
Chemicals
All chemicals were obtained from Sigma Chemical Company (St. Louis, MO, USA).
Statistical analysis
All quantitative results are expressed as mean ± standard error of the mean (SEM). Statistical differences between means were evaluated with Student’s t-test using commercial software (SigmaStat 3.1.1; Systat Software inc., Richmond, CA, USA). If the samples were not drawn from normally distributed population with the same variances, Mann-Whitney Rank Sum test was applied. P values of < 0.05 were considered significant.
Results
LTP induction in the presence of stepwise increases in ethanol
In slices from 30-day old rats, delivery of a single 100 Hz x 1 sec HFS consistently induced LTP (open circles in Fig. 1A; EPSP change: 186.1 ± 6.1% of baseline sixty minutes following the HFS, n = 5). As we previously reported (Izumi et al., 2005), acute administration of 60 mM ethanol for 15 min before and during the HFS completely inhibited LTP (closed circles in Fig. 1A; EPSP change: 102.2 ± 2.7% of baseline, n = 5; P < 0.01 vs. control LTP). Similarly, acute administration of 60 mM ethanol for 25 min before and during the HFS inhibited LTP (data not shown; 99.3 ± 0.6%, n = 5, P < 0.01 vs. control LTP).
Figure 1. LTP induction in the presence of stepwise increases in ethanol.

The graphs show the time course of change in population excitatory postsynaptic potential (EPSP) slope (mean ± SEM). (A) Acute administration of 60 mM ethanol for 15 min prior to a single train of high-frequency stimulation (HFS) (100Hz x 1s, designated by the arrow) blocked long-term potentiation (LTP) (filled circles, n = 5). In control slices, HFS successfully induced LTP that lasted at least 60 minutes (open circles, n = 5). (B) In contrast to panel A, LTP was induced when ethanol was increased stepwise by 10 mM every 15 min to a final concentration of 60 mM for 25 min prior to delivery of HFS (n = 5). (C) Similar to effects of 15 min administration, acute administration of 60 mM ethanol for 100 min prior to HFS blocked LTP induction (n = 5). Traces to the right of each graph are representative EPSPs obtained 10 min before (dashed) and 60 min after HFS (solid traces). Scale bar: 1 mV, 5 milliseconds.
Abrupt elevations of ethanol to 60 mM over a very short time (~ 2 min) are unlikely to occur during bouts of oral alcohol consumption. This led us to examine the effects of gradual stepwise increases in ethanol concentrations. Because acute 15-25 min exposure to 60 mM ethanol does not affect baseline EPSPs (Fig. 1A; Izumi et al., 2005), and we found that gradual increases in ethanol to 60 mM in 10 mM increments also do not affect basal EPSPs (n = 3, data not shown), in all subsequent studies, ethanol levels were increased to 60 mM using a stepwise protocol before we began synaptic stimulation and field potential recording. When ethanol levels were increased in 10 mM increments every 15 min to achieve a final level of 60 mM for 25 min prior to HFS (15 min for stabilization and 10 min for baseline recording) and when ethanol was washed out immediately after HFS, LTP was induced in all slices examined (Fig. 1B; EPSP change: 127.7 ± 1.6% of baseline, n = 5). In contrast, 60 mM ethanol acutely administered and maintained for 100 min before and during the HFS blocked LTP (Fig. 1C; EPSP change: 100.3 ± 2.4% of baseline, n = 5, P < 0.01 vs. stepwise increase in ethanol to 60 mM) just as it did in the 15-25 min exposures. This suggests that LTP induction following slow increases in ethanol results from changes occurring during the gradual rise in ethanol concentration rather than the prolonged presence of high levels of ethanol prior to HFS.
In the experiments in Figure 1, ethanol was abruptly discontinued immediately following delivery of the HFS. Thus, it is possible that the abrupt withdrawal of ethanol following HFS, as shown in Fig. 1B, alters LTP induction because this LTP is significantly smaller than conventional LTP in naïve slices (P < 0.01 vs. control LTP). Interestingly, we found that when ethanol levels were stepwise increased to 60 mM 25 min before HFS and when 60 mM ethanol was continuously administered after HFS, even more robust LTP was induced (Fig. 2A; EPSP change: 192.7 ± 17.3% of baseline, n = 5, P < 0.01 vs. LTP when ethanol is slowly increased and discontinued after HFS, but P = 0.646 vs. conventional LTP in naïve slices).
Figure 2. LTP in the presence of 60 mM ethanol and effects of preconditioning.
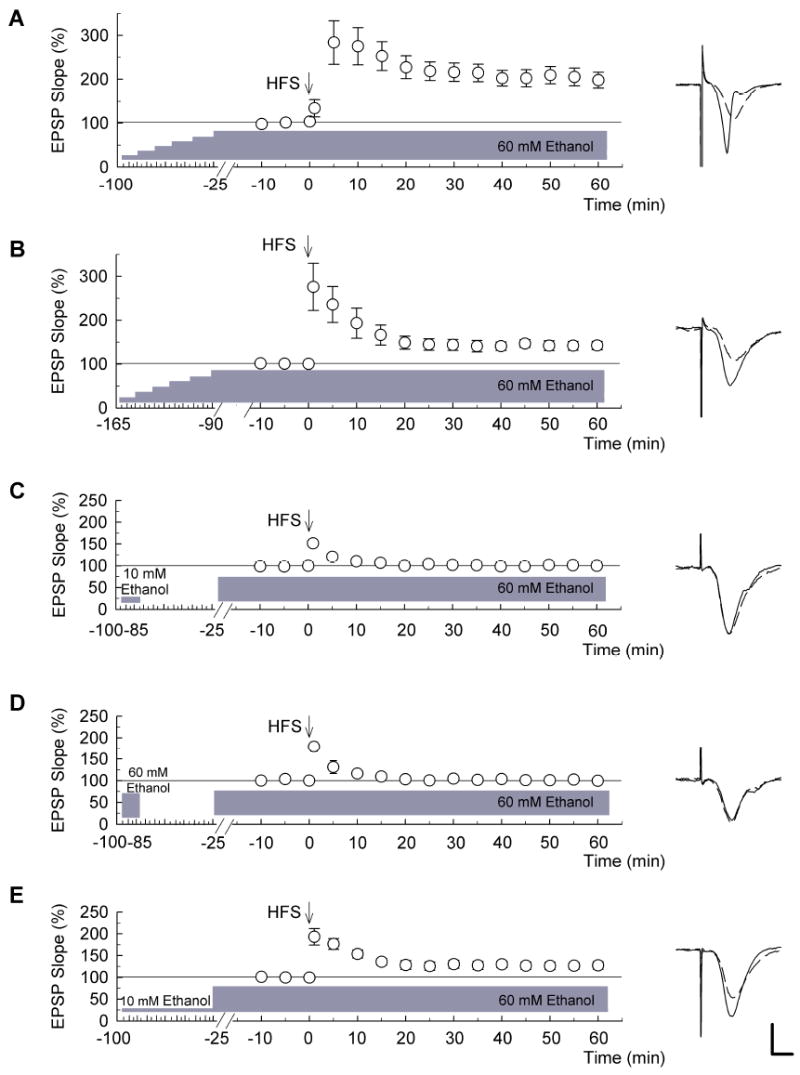
In these experiments, 60 mM ethanol was administered throughout the recording and continued for 60 min after HFS (n = 5). (A) When ethanol was increased stepwise by 10 mM every 15 min and 60 mM ethanol was continuously administered throughout the recording, robust long-term potentiation (LTP) was induced (n = 5). (B) After a stepwise increase of ethanol, 60 mM ethanol was continuously administered for 90 min before HFS. This prolonged administration of 60 mM ethanol administration did not inhibit induction of the LTP (n = 5). (C) Fifteen min preconditioning with 10 mM ethanol 60 min before continuous administration of 60 mM ethanol failed to allow LTP (n = 5). (D) Similarly, preconditioning with 60 mM ethanol for 15 min and acute administration of 60 mM ethanol at an interval of 60 min also blocked LTP induction (n = 5). (E) Administration of 10 mM ethanol for 75 min prior to a jump to 60 mM ethanol allowed LTP induction. Traces to the right of each graph are representative EPSPs obtained 10 min before (dashed) and 60 min after HFS (solid traces). Scale bar: 1 mV, 5 milliseconds.
The effects of slow increases in ethanol concentration on LTP induction last for more than 90 min. When ethanol levels were stepwise increased to 60 mM in 10 mM increments and 60 mM ethanol was continuously administered starting 90 min before HFS and throughout the recording, HFS could still induce LTP though the magnitude was diminished (Fig. 2B; EPSP change: 142.4 ± 9.5% of baseline, n = 5, P < 0.05 vs. LTP when HFS was delivered 25 min after reaching 60 mM ethanol).
To examine whether slowly increased ethanol at a concentration above 60 mM inhibited LTP, ethanol levels were stepwise elevated in 10 mM increments to 100 mM over 100 min. Twenty-five min after this slow introduction of 100 mM ethanol, HFS could still induce LTP (EPSP change: 158.6 ± 14.6%, n = 5, data not shown).
If LTP following stepwise increases in ethanol reflects a form of acute tolerance, then preconditioning with ethanol may also allow LTP induction in the presence of high ethanol concentrations. However, 15 min preconditioning of slices with 10 mM ethanol 60 min prior to acute administration of 60 mM ethanol failed to allow LTP (Fig. 2C; EPSP change: 100.1 ± 1.2% of baseline, n = 5, P < 0.01 vs. stepwise increase in ethanol to 60 mM). Similarly, 15 min preconditioning with 60 mM ethanol 60 min before acute administration of 60 mM ethanol also failed to allow LTP (Fig. 2D; EPSP change: 99.5 ± 2.6% of baseline, n = 5). In contrast, preconditioning slices with 10 mM ethanol continuously for 60 min prior to acute administration of 60 mM ethanol promoted LTP induction (Fig.2E; EPSP change: 127.3 ± 8.6%, n = 5), although the magnitude was less than that observed in the stepwise protocol of Fig. 2A (P < 0.01 vs. stepwise increase in ethanol to 60 mM). These results suggest that persistent rather than brief preconditioning may be important for allowing LTP in the presence of high ethanol concentrations.
We have previously shown that the inhibition of LTP by acute administration of 60 mM ethanol is overcome by picrotoxin, a non-competitive gamma-aminobutyric acid A type (GABAA) receptor antagonist that also reduces paired pulse depression of PSs evoked at an interpulse interval of 21 ms (Izumi et al., 2005). Thus, it is possible that LTP in the presence of stepwise increases in ethanol results from similar GABAergic disinhibition. If so, stepwise increases in ethanol may mimic the effects of picrotoxin in diminishing local network inhibition by also diminishing PS paired pulse depression. However, we found that paired pulse depression of PS amplitude (16.6 ± 5.7%; ratio of 2nd / 1st PS at baseline, n = 3) was not reduced, but rather augmented by stepwise increases in ethanol (2.7 ± 1.5%; ratio of 2nd / 1st after stepwise increase in ethanol to 60 mM, data not shown, P < 0.05, Paired t-test). This suggests that changes in GABAA receptor-mediated inhibition, at least as manifest by paired pulse PS depression, are unlikely to account for LTP induction in the presence of stepwise increases in ethanol.
Effects of APV, nifedipine and MCPG on LTP with stepwise increases in ethanol
The effects we observe on LTP in the presence of stepwise increases in ethanol could result from either a form of desensitization to ethanol or an alternative biological adaptation of hippocampal synapses to the effects of ethanol over the period of administration. In the case of ethanol desensitization, one would expect that the mechanisms contributing to LTP in the presence of ethanol would be the same as those observed under baseline conditions. Under baseline conditions, LTP induced by a single 100 Hz x 1 sec HFS is completely blocked by 100 μM D, L-2-amino-5-phosphonovalerate (D, L-APV), an antagonist of N-methyl-D-aspartate receptors (NMDARs) (Fig. 3A; EPSP change: 99.2 ± 0.9% of baseline, n = 5) (see also Bliss and Collingridge, 1993; Malenka, 1994, Lo and Mize, 2002). CA1 synapses are also known to exhibit another form of LTP that is independent of NMDARs. This form of LTP requires voltage-dependent Ca2+ channels (VDCCs) for its induction (Grover and Teyler, 1990) and is blocked by the L-type VDCC inhibitor, nifedipine (Cavus and Teyler, 1996; Izumi et al., 1998). VDCC dependent LTP is typically induced by 200 Hz x 1 sec HFS in the presence of 100 μM APV (open circles, Fig. 3B; EPSP change: 157.8 ± 5.5%, n = 5), and is blocked by administration of 20 μM nifedipine during the 200 Hz HFS (filled circles, Fig. 3B; EPSP change: 99.9 ± 0.9%, n = 5, P < 0.01 vs. APV alone).
Figure 3. Effects of APV and nifedipine on induction of the LTP with stepwise elevated ethanol.
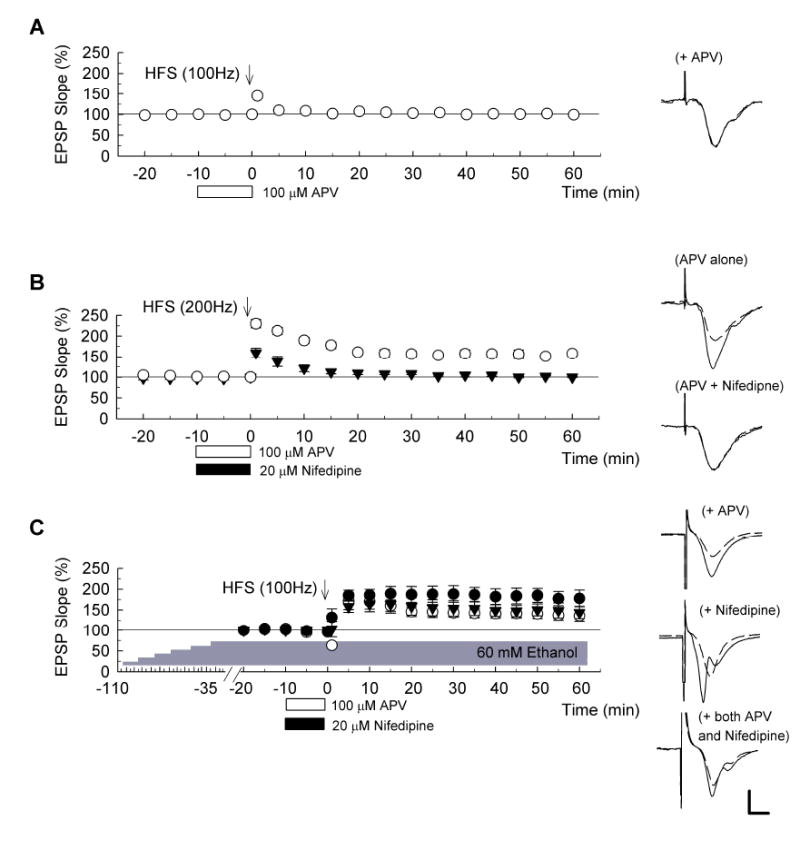
(A) In control slices, a 100Hz x 1s HFS in the presence of 100 μM D, L-APV, open bar) failed to induce LTP (n = 5). (B) In contrast, 200Hz x 1s HFS successfully induced LTP in the presence of 100 μM APV (open bar) as denoted with open circles (n = 5). However, induction of this form of LTP was blocked by co-administration of APV plus 20 μM nifedipine (filled bar) as denoted with filled triangles (n = 5). (C) Slices were treated with ethanol stepwise increased by 10 mM every 15 min and 60 mM, the final concentration of ethanol was continuously administrated throughout the recording. When HFS (arrow) was delivered in the presence of 20 μM nifedipine (filled bar), robust long-term potentiation (LTP) was induced (filled circles, n = 5). Administration of 100 μM APV (open bar) also failed to block LTP induction (open circles) (n = 5). Similarly, coadministration of APV and nifedipine did not block LTP induction (triangles, n = 5). Traces to the right of each graph are representative EPSPs obtained 10 min before (dashed) and 60 min after HFS (solid traces). Scale bar: 1 mV, 5 milliseconds.
Surprisingly, we found that a single HFS administered following stepwise increases in ethanol still generated LTP in the presence of 100 μM APV administered for 10 min prior to and during HFS (filled circles, Fig. 3C; EPSP change: 136.0 ± 13.1%, n = 5, P < 0.01 vs. APV alone), though the magnitude of LTP was smaller than that without APV (P < 0.01 vs. stepwise increase in ethanol without APV). This suggests that LTP in the presence of gradually increasing ethanol differs mechanistically from conventional CA1 LTP and likely reflects an alternative synaptic adaptation and not simply recovery of baseline function.
Next, we tested whether LTP following slow increases in ethanol requires VDCCs. Following stepwise increases in ethanol, however, a single 100 Hz x 1 sec HFS successfully induced LTP in the presence of 20 μM nifedipine (open circles in Fig. 3C; changes in EPSPs: 177.8 ± 20.0%, n= 5). Furthermore, co-administration of 100 μM APV plus 20 μM nifedipine failed to block LTP induction following slow increases in ethanol (triangles in Fig. 3C; changes in EPSPs: 143.7 ± 14.8%, n = 5) though the magnitude of LTP was statistically smaller than that without two agents (P < 0.05). These results again suggest that mechanisms contributing to LTP following stepwise increases in ethanol differ from typical forms of LTP.
Induction of the conventional form of LTP in hippocampal slices can also involve activation of metabotropic glutamate receptors (mGluRs) (Izumi et al., 1991; Frenguelli et al., 1993). As previously reported (Izumi et al., 2000), we found that conventional LTP is blocked by 500 μM α-methyl-4-carboxyphenylglycine (MCPG), a broad spectrum mGluR antagonist, administrated during 100 Hz x 1 sec HFS (Fig. 4A; EPSP change: 101.5 ± 1.5% of baseline, n = 5). However, administration of 500 μM MCPG during HFS failed to inhibit LTP following stepwise increases in ethanol (Fig. 4B; changes in EPSPs: 166.9. ± 17.0 %, n = 5, P < 0.01 vs. MCPG alone). This again supports the idea that the LTP in slowly elevated ethanol has unique properties.
Figure 4. Effects of MCPG on induction of the LTP with stepwise elevated ethanol.
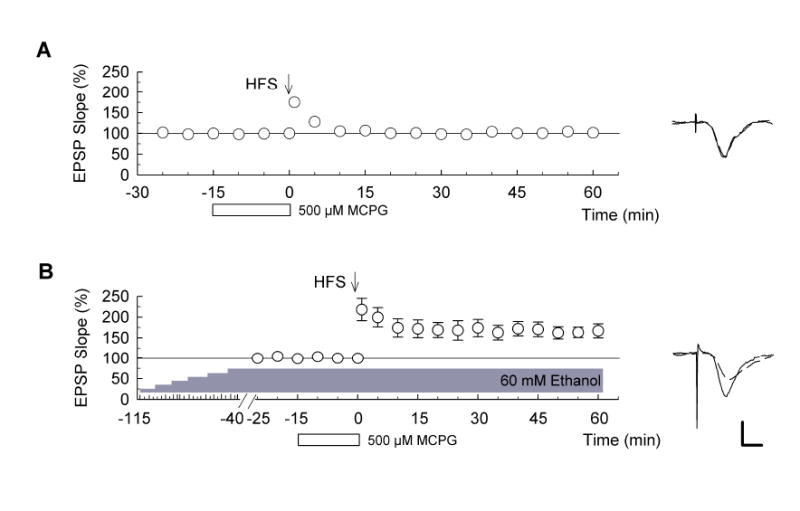
(A) In control slices, administration of 500 μM MCPG (open bar) 15 min before high-frequency stimulation (HFS) (arrow) blocked LTP (n = 5). (B) In slices treated with ethanol stepwise increased by 10 mM every 15 min, however, HFS delivered 40 min after reaching 60 mM induced LTP in the presence of 500 μM MCPG (n = 5). Traces to the right of the graphs show representative EPSPs obtained 15 min before (dashed lines) and 60 min after (solid) HFS. Scale bar: 1 mV, 5 milliseconds.
Involvement of calcium in LTP with stepwise increases in ethanol
Although the LTP following slowly elevated ethanol does not require NMDARs, VDCCs or mGluRs, CA1 LTP is known to be critically dependent upon calcium. In previous studies, we found that a reduction of extracellular calcium from 2 mM to 1 mM for 5 min before and during HFS blocks conventional CA1 LTP (Izumi et al., 1987). Similarly, this change in extracellular calcium blocked the LTP observed following stepwise increases in ethanol (Fig. 5A; EPSP change: 86.8 ± 7.3%, n = 5, P < 0.01 vs. stepwise increase in ethanol alone). To determine whether intracellular calcium stores participate in ethanol-tolerant LTP, we examined 5 μM thapsigargin, a store-operated Ca2+ channel inhibitor that also blocks conventional LTP (Izumi et al., 2000). In contrast to APV, nifedipine and MCPG, thapsigargin also blocked LTP in the presence of slowly increased ethanol (Fig. 5B; EPSP change: 100.7 ± 2.7%, n = 5, P < 0.01 vs. stepwise increase in ethanol alone).
Figure 5. Involvement of calcium in LTP with stepwise increased ethanol.
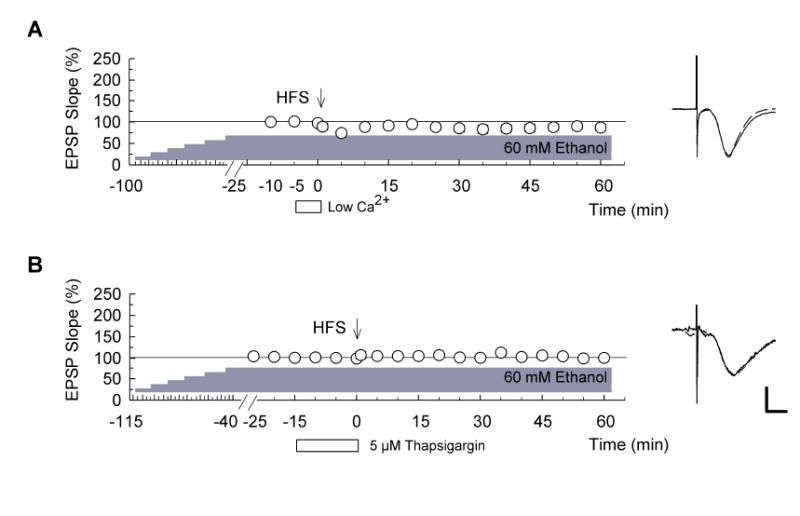
(A) Reduction of extracellular calcium from 2 mM to 1 mM (open bar) for 5 min before high-frequency stimulation (HFS) (100Hz x 1s duration, arrow) inhibited long-term potentiation (LTP) induction in the presence of 60 mM ethanol (n = 5). (B) Similarly, administration of 5 μM thapsigargin (open bar) for 15 min before HFS blocked LTP induction in the presence of 60 mM ethanol (n = 5). In both graphs ethanol levels were stepwise increased by 10 mM every 15 min and HFS was delivered 25 (panel A) or 40 min (panel B) after the concentration of ethanol reached 60 mM. Traces to the right of the graphs are representative EPSPs. Dashed traces show EPSP at baseline. Scale bar: 1 mV, 5 milliseconds.
Involvement of NMDARs in the induction of acute ethanol tolerance
Because other forms of CNS tolerance can involve activation of NMDARs (Kato et al., 1992; Wong et al., 1996; Grabb and Choi, 1999), we examined whether a broad spectrum NMDAR antagonist modifies the effects of slow increases in ethanol. When 100 μM APV was administered during the stepwise increase in ethanol concentration and maintained until just prior to delivery of HFS, LTP could not be induced (Fig. 6A; EPSP changes: 94.0 ± 3.6%, n = 5, P < 0.01 vs. stepwise increase in ethanol in the absence of APV). This effect of APV appears to reflect effects occurring during the period of increase in ethanol concentration and not simply an effect at the time of the HFS because administration of APV only during the stepwise increase in ethanol with discontinuation for 15 min before HFS still blocked LTP induction (Fig. 6B; EPSP change: 97.2 ± 2.3%, n = 5, P < 0.01 vs. stepwise increase in ethanol in the absence of APV). Because the induction of LTP in slowly elevated ethanol is not blocked by APV acutely administered for 10 min just before HFS (Fig. 3C), this suggests that the inhibition of LTP in the presence of APV plus gradually increasing ethanol results from block of NMDARs during the development of this form of ethanol tolerance.
Figure 6. Involvement of NMDA receptors in the induction of acute ethanol tolerance.
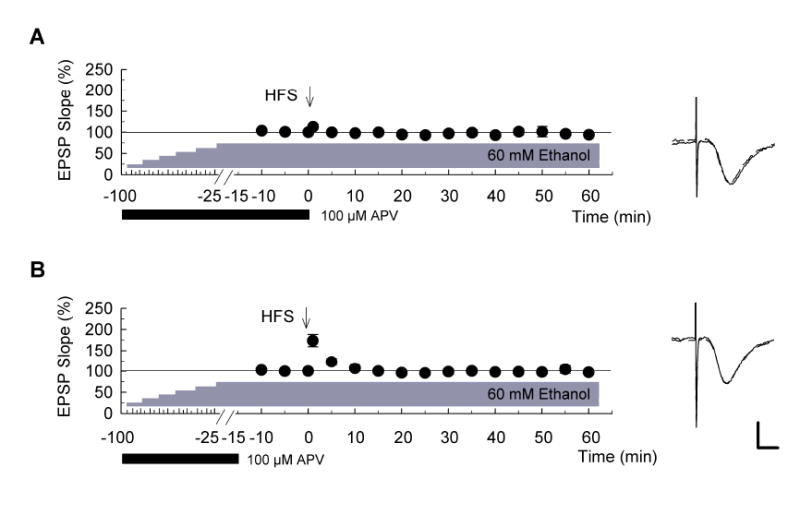
Graphs show the time course of change in EPSP slopes when concentrations of ethanol were stepwise increased by 10 mM every 15 minutes up to 60 mM in the presence of 100 μM D, L-APV, filled bar). Administration of APV was terminated just prior to delivery of high-frequency stimulation (HFS) (A) or 15 min before HFS (B) (n = 5 for each). 60 mM ethanol was continuously administered throughout the recording period. LTP was not induced under these conditions (filled circles). Traces to the right of each graph show representative EPSPs recorded 10 min before (dashed line) and 60 min after HFS (solid). Scale bar: 1 mV, 5 milliseconds.
Discussion
Although acute administration of 60 mM ethanol for 15 min prior to and during HFS inhibits LTP, it is unclear how such rapid and large increases in ethanol relate to effects occurring during bouts of binge drinking. To examine synaptic effects of ethanol under conditions that are more likely to reflect oral ethanol intake, we used a protocol in which ethanol was increased in a stepwise fashion from 10 mM to 60 mM in 10 mM increments every 15 minutes. Under these conditions, we observed a form of tolerance that was manifest by an inability of 60 mM ethanol to block LTP induction. We also found that 60 mM ethanol failed to inhibit LTP when administered acutely following longer preconditioning with 10 mM ethanol. Interestingly, the magnitude of LTP following stepwise increases in ethanol was significantly smaller when 60 mM ethanol was abruptly discontinued following HFS than when ethanol was continuously present before and following HFS, suggesting that abrupt withdrawal may dampen processes contributing to synaptic plasticity. It is also possible that the continued presence of ethanol is required during the expression of ethanol-tolerant LTP. The ability to induce LTP in the setting of acute ethanol tolerance suggests a mechanism by which synaptic plasticity, and perhaps memory formation, can occur even during severe binge drinking.
The development of this acute ethanol tolerance appears to involve the activation of NMDARs during the period when ethanol concentrations are increasing. These observations are consistent with prior studies of other forms of brain tolerance in which NMDARs participate in the development and/or expression of the tolerance. For example, a form of tolerance to the neurotoxic effects of ischemia can result from preconditioning with brief bouts of ischemia. This ischemic tolerance requires NMDAR activation during preconditioning (Kato et al., 1992; Grabb and Choi, 1999). Similarly, tolerance following chronic exposure to opioids can be reversed by ketamine, an NMDAR antagonist (Wong et al., 1996), suggesting that NMDARs play a key role in the expression of this form of tolerance.
In our studies, NMDAR activation appears to play a critical role in the development of acute ethanol tolerance (Khanna et al., 1991), but not in the expression of the tolerance. That is, APV administered during stepwise increases in ethanol prevented LTP even when APV was removed prior to HFS. However, APV administered only after the period of stepwise increase failed to completely block LTP, the expression of this acute tolerance. Because induction of conventional LTP requires NMDAR activation, LTP in the presence of APV in the setting of acute ethanol tolerance suggests the generation of a non-traditional form of LTP. Nonetheless, because the degree of LTP in the presence of APV is significantly less than the degree of LTP observed in the absence of APV, it seems likely that NMDARs still contribute to ethanol-tolerant LTP even though NMDARs are not absolutely required for its induction. It is also known that delivery of very high frequency stimulation (200 Hz) or 100 Hz HFS in the presence of high extracellular calcium also induces LTP in the presence of NMDAR antagonists. This form of LTP is inhibited by nifedipine, an L-type VDCC blocker (Shankar et al., 1998). In the presence of stepwise increased ethanol, however, nifedipine also failed to inhibit LTP, suggesting that this form of LTP does not require VDCCs. A combination of APV and nifedipine similarly failed to block ethanol-tolerant LTP.
Conventional LTP induction depends upon calcium and a decrease in extracellular calcium at the time of tetanic stimulation impairs LTP (Dunwiddie and Lynch, 1979; Izumi et al., 1987). In the present study, low calcium administered during HFS also inhibited LTP in the presence of stepwise increased ethanol, suggesting that even though NMDARs and L-type VDCCs are not required for this LTP, calcium still plays an essential role. Because the two major ion channels through which extracellular calcium enters neurons to induce CA1 LTP are not critical for ethanol-tolerant LTP, we examined a role for calcium release from intracellular stores. Thapsigargin, a store-operated calcium channel inhibitor, is known to modulate conventional LTP (Harvey and Collingridge, 1992), and we found that thapsigargin completely blocked ethanol-tolerant LTP. Although the effects of thapsigargin are complex (Mengesdorf et al. 2001), this finding suggests that calcium release from intracellular stores is pivotal for inducing this form of plasticity. The inability of NMDAR and VDCC antagonists to completely block ethanol-tolerant LTP suggests that stepwise increases in ethanol may alter the properties of intracellular calcium storage systems such that LTP can be induced without significant calcium influx from these channels. Prior studies have shown that ethanol alters calcium homeostasis in cultured hippocampal neurons (Webb et al., 1997), cultured human lymphoblast cells (Nagy, 2000) and myotubes (Nicolas, 1998). It remains possible, however, that entry of extracellular calcium from an unknown channel participates in the induction of LTP following stepwise increases in ethanol. Given the importance of extracellular calcium in ethanol-tolerant LTP, it is also possible that mechanisms involved in calcium-induced intracellular calcium release (CICR) are upregulated during acute ethanol tolerance. Such a finding would be consistent with the greater LTP observed in the absence of APV. In the absence of NMDAR or VDCC input, however, intracellular calcium release, likely supported by extracellular calcium, appears to be sufficient to generate at least some degree of lasting synaptic enhancement. In control slices, intracellular calcium release is insufficient to drive LTP induction when NMDARs are blocked (Fig. 3A).
Stepwise increases in ethanol might also modulate mGluRs because ethanoltolerant LTP was insensitive to MCPG, an mGluR antagonist that inhibits LTP in control slices (Izumi and Zorumski, 1994). Whether acute ethanol tolerance alters mGluR function is unclear, although prenatal ethanol exposure decreases mGluR5 expression in the dentate gyrus (Galindo et al., 2004) and reduces mGluR-activated phosphoinositide (PI) hydrolysis (Queen et al., 1993). PI hydrolysis is involved in the formation of inositol triphosphate (IP3), an intracellular messenger that activates IP3 receptors on calcium stores, leading to calcium release. While changes in mGluR function could occur during acute ethanol tolerance, the inability of MCPG to inhibit ethanol-tolerant LTP indicates that mGluRs do not drive this form of plasticity.
In addition to LTP, post-tetanic potentiation (PTP) is also sensitive to acute administration of 60 mM ethanol (Fig. 1A, C and Fig. 2C, D). The partial depression of PTP may result from inactivation of NMDA receptors because PTP is also partially depressed in the presence of APV (Fig. 3A). It is also possible that calcium influx through VDCCs is involved in PTP because PTP induced by 200 Hz × 1 s HFS is attenuated by nifedipine (Fig. 3B). The large degree of PTP in the presence of stepwise increases in ethanol (Fig. 1B and Fig. 2A, B and E) suggests that calcium entry through NMDAR channels or VDCCs is altered by slow increases in ethanol or sustained preconditioning with low levels of ethanol. Supporting this, PTP was depressed by APV or nifedipine in the presence of 60 mM ethanol following stepwise increases in concentration (Fig. 3C). Of note is that LTP in the presence of 60 mM ethanol following stepwise increases is not blocked by APV and/or nifedipine (Fig. 3C), indicating that the degree of PTP does not predict success or failure of LTP.
LTP can be induced not only when ethanol is increased in a stepwise fashion but also when a low concentration is administered prior to delivery of a high concentration. This strongly suggests that the ability to generate LTP in the presence of 60 mM ethanol represents a form of acute tolerance. As mentioned above, however, LTP in the presence of ethanol differs from control LTP based on lack of APV, nifedipine and MCPG sensitivity. Although tolerance reflects the ability of a biological system to maintain or restore function in the continued presence of a stimulus that alters its function, how this adaptation occurs varies among systems and stimuli. In some cases, the system may simply up- or down-regulate its basal function to accommodate. For example, when receptors are continuously exposed to direct agonists or antagonists, tolerance is often manifest by changes in receptor expression or receptor function (Chandler et al., 1998; Kumar et al., 2004; Bailey and Connor, 2005). This form of tolerance seems unlikely to explain our results. Rather, we observe an adaptation in which the usual driving forces for LTP (NMDAR, mGluR or VDCC activation) are no longer required to initiate the process but rather calcium-dependent intracellular calcium release becomes the primary trigger. It remains unclear how intracellular calcium release is augmented in response to HFS following ethanol tolerance and whether the changes represent increases in sensitivity to activating stimuli (e.g. small influxes of extracellular calcium), enhanced ion release mechanisms, changes in intracellular ion homeostasis and clearance, or a combination of mechanisms.
If ethanol-tolerant LTP indeed differs from conventional LTP and if LTP serves as a cellular mechanism for memory formation, then these findings suggest that memory processing during binge drinking differs from conventional learning. It is unclear whether memories formed during alcohol intoxication have similar properties to conventional memories and whether they are similarly persistent. It is also possible that ethanol, even if administered by stepwise increases, still inhibits conventional LTP because the LTP we observe in the setting of tolerance differs from conventional LTP in its proximal induction mechanisms. However, induction of robust LTP in stepwise increased ethanol and its partial depression by APV may hint that two distinct forms of LTP, conventional and ethanol-tolerant LTP, coexist in the setting of acute tolerance. This raises the possibility that in other forms of CNS tolerance, novel mechanisms may coexist with preexisting mechanisms to restore function.
In addition to considering these two forms of LTP, the tolerance seen in stepwise increased ethanol should also be evaluated as having two components - development and expression. Development of acute tolerance occurs during the time that ethanol levels are increasing and the ability of a single HFS to generate LTP represents the expression of the tolerance. While the expression of acute ethanol tolerance, as manifest by LTP induction in the presence of ethanol, is not blocked completely by APV, the development of this acute ethanol tolerance is clearly prevented by APV, indicating that NMDAR activation is crucial for driving the changes underlying this form of tolerance. Understanding other mechanisms contributing to the LTP-sparing effects of acute ethanol tolerance may point to ways to preserve and restore function in a variety of pathological conditions.
Acknowledgments
This work was supported by AA12951, AG18434, DK56341 and the Bantly Foundation. The authors thank Terutomo Sato for technical advice.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Bailey CP, Connor M. Opioids: cellular mechanisms of tolerance and physical dependence. Curr Opin Pharmacol. 2005;5:60–68. doi: 10.1016/j.coph.2004.08.012. [DOI] [PubMed] [Google Scholar]
- Baker SP, Braver ER, Chen LH, Li G, Williams AF. Drinking histories of fatally injured drivers. Inj Prev. 2002;8:221–226. doi: 10.1136/ip.8.3.221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bliss TV, Collingridge GL. A synaptic model of memory: long-term potentiation in the hippocampus. Nature. 1993;361:31–39. doi: 10.1038/361031a0. [DOI] [PubMed] [Google Scholar]
- Blitzer RD, Gil O, Landau EM. Long-term potentiation in rat hippocampus is inhibited by low concentrations of ethanol. Brain Res. 1990;537:203–208. doi: 10.1016/0006-8993(90)90359-j. [DOI] [PubMed] [Google Scholar]
- Cavus I, Teyler T. Two forms of long-term potentiation in area CA1 activate different signal transduction cascades. J Neurophysiol. 1996;76:3038–3047. doi: 10.1152/jn.1996.76.5.3038. [DOI] [PubMed] [Google Scholar]
- Chandler LJ, Harris RA, Crews FT. Ethanol tolerance and synaptic plasticity. Trends Pharmacol Sci. 1998;19:491–495. doi: 10.1016/s0165-6147(98)01268-1. [DOI] [PubMed] [Google Scholar]
- Dunwiddie TV, Lynch G. The relationship between extracellular calcium concentrations and the induction of hippocampal long-term potentiation. Brain Res. 1979;169:103–110. doi: 10.1016/0006-8993(79)90377-9. [DOI] [PubMed] [Google Scholar]
- Frenguelli BG, Potier B, Slater NT, Alford S, Collingridge GL. Metabotropic glutamate receptors and calcium signalling in dendrites of hippocampal CA1 neurones. Neuropharmacology. 1993;32:1229–1237. doi: 10.1016/0028-3908(93)90017-w. [DOI] [PubMed] [Google Scholar]
- Galindo R, Frausto S, Wolff C, Caldwell KK, Perrone-Bizzozero NI, Savage DD. Prenatal ethanol exposure reduces mGluR5 receptor number and function in the dentate gyrus of adult offspring. Alcohol Clin Exp Res. 2004;28:1587–1597. doi: 10.1097/01.alc.0000141815.21602.82. [DOI] [PubMed] [Google Scholar]
- Grabb MC, Choi DW. Ischemic tolerance in murine cortical cell culture: critical role for NMDA receptors. J Neurosci. 1999;19:1657–1662. doi: 10.1523/JNEUROSCI.19-05-01657.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grover LM, Teyler TJ. Two components of long-term potentiation induced by different patterns of afferent activation. Nature. 1990;347:477–479. doi: 10.1038/347477a0. [DOI] [PubMed] [Google Scholar]
- Harvey J, Collingridge GL. Thapsigargin blocks the induction of long-term potentiation in rat hippocampal slices. Neurosci Lett. 1992;139:197–200. doi: 10.1016/0304-3940(92)90551-h. [DOI] [PubMed] [Google Scholar]
- Izumi Y, Ito K, Miyakawa H, Kato H. Requirement of extracellular Ca2+ after HFS for induction of long-term potentiation in guinea pig hippocampal slices. Neurosci Lett. 1987;77:176–180. doi: 10.1016/0304-3940(87)90582-9. [DOI] [PubMed] [Google Scholar]
- Izumi Y, Clifford DB, Zorumski CF. 2-Amino-3-phosphonopropionate blocks the induction and maintenance of long-term potentiation in rat hippocampal slices. Neurosci Lett. 1991;122:187–190. doi: 10.1016/0304-3940(91)90854-m. [DOI] [PubMed] [Google Scholar]
- Izumi Y, Zorumski CF. Developmental changes in the effects of metabotropic glutamate receptor antagonists on CA1 long-term potentiation in rat hippocampal slices. Neurosci Lett. 1994;176:89–92. doi: 10.1016/0304-3940(94)90878-8. [DOI] [PubMed] [Google Scholar]
- Izumi Y, Zorumski CF. LTP in CA1 of the adult rat hippocampus and voltage-activated calcium channels. Neuroreport. 1998;9:3689–3691. doi: 10.1097/00001756-199811160-00022. [DOI] [PubMed] [Google Scholar]
- Izumi Y, Zarrin AR, Zorumski CF. Arachidonic acid rescues hippocampal longterm potentiation blocked by group I metabotropic glutamate receptor antagonists. Neuroscience. 2000;100:485–491. doi: 10.1016/s0306-4522(00)00301-8. [DOI] [PubMed] [Google Scholar]
- Izumi Y, Nagashima K, Murayama K, Zorumski CF. Acute effects of ethanol on hippocampal long-term potentiation and long-term depression are mediated by different mechanisms. Neuroscience. 2005;136:509–517. doi: 10.1016/j.neuroscience.2005.08.002. [DOI] [PubMed] [Google Scholar]
- Kato H, Liu Y, Araki T, Kogure K. MK-801, but not anisomycin, inhibits the induction of tolerance to ischemia in the gerbil hippocampus. Neurosci Lett. 1992;139:118–121. doi: 10.1016/0304-3940(92)90871-4. [DOI] [PubMed] [Google Scholar]
- Khanna JM, Wu PH, Weiner J, Kalant H. NMDA antagonist inhibits rapid tolerance to ethanol. Brain Res Bull. 1991;26:643–645. doi: 10.1016/0361-9230(91)90109-w. [DOI] [PubMed] [Google Scholar]
- Kumar S, Fleming RL, Morrow AL. Ethanol regulation of gamma-aminobutyric acid A receptors: genomic and nongenomic mechanisms. Pharmacol Ther. 2004;101:211–226. doi: 10.1016/j.pharmthera.2003.12.001. [DOI] [PubMed] [Google Scholar]
- Lange JE, Voas RB. Defining binge drinking quantities through resulting blood alcohol concentrations. Psychol Addict Behav. 2001;15:310–316. doi: 10.1037//0893-164x.15.4.310. [DOI] [PubMed] [Google Scholar]
- Lo FS, Mize RR. Properties of LTD and LTP of retinocollicular synaptic transmission in the developing rat superior colliculus. Eur J Neurosci. 2002;15:1421–1432. doi: 10.1046/j.1460-9568.2002.01979.x. [DOI] [PubMed] [Google Scholar]
- Malenka RC. Synaptic plasticity in the hippocampus: LTP and LTD. Cell. 1994;78:535–538. doi: 10.1016/0092-8674(94)90517-7. [DOI] [PubMed] [Google Scholar]
- Mengesdorf T, Althausen S, Oberndorfer I, Paschen W. Response of neurons to an irreversible inhibition of endoplasmic reticulum Ca(2+)-ATPase: relationship between global protein synthesis and expression and translation of individual genes. Biochem J. 2001;356:805–812. doi: 10.1042/0264-6021:3560805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrisett RA, Swartzwelder HS. Attenuation of hippocampal long-term potentiation by ethanol: a patch-clamp analysis of glutamatergic and GABAergic mechanisms. J Neurosci. 1993;13:2264–2272. doi: 10.1523/JNEUROSCI.13-05-02264.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagy J. Alcohol dependence at the cellular level: effects of ethanol on calcium homeostasis of IM-9 human lymphoblast cells. J Stud Alcohol. 2000;61:225–231. doi: 10.15288/jsa.2000.61.225. [DOI] [PubMed] [Google Scholar]
- Nicolas JM, Antunez E, Thomas AP, Fernandez-Sola J, Tobias E, Estruch R, Urbano-Marquez A. Ethanol acutely decreases calcium transients in cultured human myotubes. Alcohol Clin Exp Res. 1998;22:1086–1092. doi: 10.1111/j.1530-0277.1998.tb03705.x. [DOI] [PubMed] [Google Scholar]
- Perkins HW, Linkenbach J, Dejong W. Estimated blood alcohol levels reached by “binge” and “nonbinge” drinkers: a survey of young adults in Montana. Psychol Addict Behav. 2001;15:317–320. [PubMed] [Google Scholar]
- Queen SA, Sanchez CF, Lopez SR, Paxton LL, Savage DD. Dose- and age-dependent effects of prenatal ethanol exposure on hippocampal metabotropic-glutamate receptor-stimulated phosphoinositide hydrolysis. Alcohol Clin Exp Res. 1993;17:887–893. doi: 10.1111/j.1530-0277.1993.tb00859.x. [DOI] [PubMed] [Google Scholar]
- Quinlan KP, Brewer RD, Siegel P, Sleet DA, Mokdad AH, Shults RA, Flowers N. Alcohol-impaired driving among U.S. adults, 1993-2002. Am J Prev Med. 2005;28:346–350. doi: 10.1016/j.amepre.2005.01.006. [DOI] [PubMed] [Google Scholar]
- Randall RD, Lee SY, Meyer JH, Wittenberg GF, Gruol DL. Acute alcohol blocks neurosteroid modulation of synaptic transmission and long-term potentiation in the rat hippocampal slice. Brain Res. 1995;701:238–248. doi: 10.1016/0006-8993(95)01007-9. [DOI] [PubMed] [Google Scholar]
- Schummers J, Bentz S, Browning MD. Ethanol’s inhibition of LTP may not be mediated solely via direct effects on the NMDA receptor. Alcohol Clin Exp Res. 1997;21:404–408. doi: 10.1111/j.1530-0277.1997.tb03783.x. [DOI] [PubMed] [Google Scholar]
- Schummers J, Browning MD. Evidence for a role for GABA(A) and NMDA receptors in ethanol inhibition of long-term potentiation. Brain Res Mol Brain Res. 2001;94:9–14. doi: 10.1016/s0169-328x(01)00161-9. [DOI] [PubMed] [Google Scholar]
- Shankar S, Teyler TJ, Robbins N. Aging differentially alters forms of long-term potentiation in rat hippocampal area CA1. J Neurophysiol. 1998;79:334–341. doi: 10.1152/jn.1998.79.1.334. [DOI] [PubMed] [Google Scholar]
- Sinclair JG, Lo GF. Ethanol blocks tetanic and calcium-induced long-term potentiation in the hippocampal slice. Gen Pharmacol. 1986;17:231–233. doi: 10.1016/0306-3623(86)90144-8. [DOI] [PubMed] [Google Scholar]
- Sugiura M, Shoyama Y, Saito H, Abe K. The effects of ethanol and crocin on the induction of long-term potentiation in the CA1 region of rat hippocampal slices. Jpn J Pharmacol. 1995;67:395–397. doi: 10.1254/jjp.67.395. [DOI] [PubMed] [Google Scholar]
- Webb B, Heaton MB, Walker DW. Ethanol effects on cultured embryonic hippocampal neuronal calcium homeostasis are altered by nerve growth factor. Alcohol Clin Exp Res. 1997;21:1643–1652. [PubMed] [Google Scholar]
- White AM. What happened? Alcohol, memory blackouts, and the brain. Alcohol Res Health. 2003;27:186–196. [PMC free article] [PubMed] [Google Scholar]
- Wong CS, Liaw WJ, Tung CS, Su YF, Ho ST. Ketamine potentiates analgesic effect of morphine in postoperative epidural pain control. Reg Anesth. 1996;21:534–541. [PubMed] [Google Scholar]
- Zorumski CF, Mennerick S, Izumi Y. Assessment of synaptic effects of nitric oxide in hippocampal neurons. In: Maines MD, editor. Methods in neurosciences: From Nitric oxide synthase: characterization and functional analysis. Vol. 31. San Diego: Academic Press; 1996. pp. 282–299. [Google Scholar]