

Abstract
Background
Human pulmonary arterial hypertension (PAH) is characterized by proliferation of vascular smooth muscle and, in its more severe form, by the development of occlusive neointimal lesions. However, few animal models of pulmonary neointimal proliferation exist, thereby limiting a complete understanding of the pathobiology of PAH. Recent studies of the endothelin (ET) system demonstrate that deficiency of the ETB receptor predisposes adult rats to acute and chronic hypoxic PAH, yet these animals fail to develop neointimal lesions. Herein, we determined and thereafter showed that exposure of ETB receptor–deficient rats to the endothelial toxin monocrotaline (MCT) leads to the development of neointimal lesions that share hallmarks of human PAH.
Methods and Results
The pulmonary hemodynamic and morphometric effects of 60 mg/kg MCT in control (MCT+/+) and ETB receptor–deficient (MCTsl/sl) rats at 6 weeks of age were assessed. MCTsl/sl rats developed more severe PAH, characterized by elevated pulmonary artery pressure, diminished cardiac output, and right ventricular hypertrophy. In MCTsl/sl rats, morphometric evaluation revealed the presence of neointimal lesions within small distal pulmonary arteries, increased medial wall thickness, and decreased arterial-to-alveolar ratio. In keeping with this, barium angiography revealed diminished distal pulmonary vasculature of MCTsl/sl rat lungs. Cells within neointimal lesions expressed smooth muscle and endothelial cell markers. Moreover, cells within neointimal lesions exhibited increased levels of proliferation and were located in a tissue microenvironment enriched with vascular endothelial growth factor, tenascin-C, and activated matrix metalloproteinase-9, factors already implicated in human PAH. Finally, assessment of steady state mRNA showed that whereas expression of ETB receptors was decreased in MCTsl/sl rat lungs, ETA receptor expression increased.
Conclusions
Deficiency of the ETB receptor markedly accelerates the progression of PAH in rats treated with MCT and enhances the appearance of cellular and molecular markers associated with the pathobiology of PAH. Collectively, these results suggest an overall antiproliferative effect of the ETB receptor in pulmonary vascular homeostasis.
Keywords: arteriosclerosis, endothelin, metalloproteinases, nitric oxide, pediatrics
Pulmonary arterial hypertension (PAH) is characterized by progressive increases in pulmonary artery pressure (PAP) and resistance. Although treatments for PAH have improved over the last decade, there is still no cure for PAH. In PAH disorders, the pulmonary vasculature initially undergoes constriction and remodeling with muscularization of peripheral arteries followed by medial hypertrophy. A decrease in peripheral arteries is then followed by the formation of neointimal lesions within small pulmonary arteries, which are composed of multiple cell types, including vascular smooth muscle (VSMCs) and endothelial cells (ECs). In addition, cells within neointimal lesions are contained within a tissue microenvironment that is enriched with a variety of growth factors and specific extracellular matrix proteins, including vascular endothelial growth factor (VEGF) and tenascin-C (TN-C). Collectively, these and other factors collaborate to actively promote cellular proliferation, survival, and migration,1–5 which may lead to pulmonary vascular occlusion.
To date, only a few animal models of neointimal proliferation of the pulmonary vasculature have been described. For example, Botney6,7 showed that rat intra-acinar pulmonary arteries developed a neointima when exposed to both monocrotaline (MCT) and increased blood flow generated from a systemic to pulmonary artery shunt, whereas MCT or a shunt alone caused only medial hypertrophy. Neointimal lesions also developed after pneumonectomy and MCT treatment.8 In addition, Tuder and Voelkel9 demonstrated that perturbing VEGF receptor–mediated signaling, in conjunction with hypoxia, also caused neointimal pulmonary artery lesions. More recently, work by Rabinovitch and colleagues10 found that 5% of transgenic mice overexpressing S100A4/Mts 1 developed plexogenic lesions at the level of the respiratory bronchiole and alveolar duct. Despite these excellent studies, further insights into the mechanisms leading to the development of neointimal proliferation in PAH are needed.
The endothelins (ET) are a family of isopeptides with potent vasoactive properties. ET-1 levels are elevated in PAH and correlate with disease severity.11 The actions of ET-1 are dependent on activation of at least 2 receptor subtypes: ETA and ETB. ETA receptors are located on smooth muscle cells and mediate vasoconstriction and smooth muscle proliferation.12 In contrast, ETB receptors are present on both ECs and VSMCs in the rat and human pulmonary circulations.13 Stimulation of endothelial ETB receptors causes vasodilation through release of NO and prostacyclin and also functions to remove ET-1 from the circulation.14,15 Stimulation of ETB receptors on smooth muscle causes vasoconstriction in the rat lung.16
We have previously shown that the pulmonary vasculature of the transgenic sl/sl rat is deficient in the ETB receptor.17 This animal exhibits an exaggerated pulmonary vasopressor response to acute hypoxia and exogenous ET-1 infusion.17 In addition, deficiency of the ETB receptor in the pulmonary circulation predisposes rats to the development of chronic hypoxic PAH but does not lead to neointimal lesions in this model.18 The purpose of this study was to examine the role of the ETB receptor in the pathogenesis of severe MCT-induced pulmonary hypertension and to determine whether ETB receptor deficiency was associated with development of neointimal lesions by comparing the development of chronic pulmonary hypertension to MCT in transgenic control (+/+) and sl/sl rats.
Methods
Animal Model of PH
The Institutional Animal Care and Use Committee of the University of Colorado Health Sciences Center approved this study. Fifty-three transgenic control (+/+) and ETB-deficient (sl/sl) rats were studied. The colony was established with the use of founder animals provided by Dr M. Yanagisawa.19 The genotype of each animal was confirmed by polymerase chain reaction of genomic DNA with the use of standard techniques, as previously described.19 Male rats received a single subcutaneous injection of MCT (60 mg/kg) at 4 to 6 weeks of age.
Hemodynamic Measurements
Hemodynamic study of conscious catheterized rats was performed in 16 animals (MCT+/+, n=6; MCTsl/sl, n=5; and after being raised in a hyperbaric chamber shortly after birth to compensate for the mild hypoxia of Denver’s altitude by simulating sea level oxygen tension, MCTsl/sl hyperbaric, n=5). After 4 weeks of MCT treatment, the animals were anesthetized with ketamine/xylazine for placement of catheters in the right carotid artery, main pulmonary artery, and jugular vein. The catheters were filled with heparinized saline, sealed, and tunneled subdermally to the back of the neck and were then exteriorized and enclosed in a small plastic container. After 24 hours of recovery in room air, conscious rats were placed in a small plastic chamber that was flushed continuously with 21% oxygen during hemodynamic measurements of cardiac output, PAP, and systemic blood pressure, as previously described.17 Total pulmonary resistance (TPR) was calculated as mean PAP/cardiac output (mm Hg/L per minute). PaO2, PaCO2, and pH were measured with the use of a clinical blood gas analyzer (Radiometer).
Right Ventricular Hypertrophy
Immediately after the animals were euthanized with an overdose of pentobarbital, the heart was resected in MCT+/+ (n=12), MCTsl/sl (n=10), and MCT hyperbaric (n=6) rats, and the atria were removed to the plane of the atrioventricular valves. The free wall of the right ventricle (RV) was then dissected free of the left ventricle (LV) and septum (LV+septum). The RV and LV+septum were weighed, and the RV/LV+septum ratio was calculated.17,18
Histology and Morphometric Analysis of Pulmonary Arteries
To evaluate changes in the pulmonary vasculature, MCT+/+ and MCTsl/sl rat lungs (n=10 for each group) were fixed for histology by tracheal instillation of 4% buffered paraformaldehyde under constant pressure (30 cm H2O). The trachea was ligated after sustained inflation, and the lungs were excised and immersed in 4% buffered paraformaldehyde overnight. Paraformaldehyde-fixed lung tissue was cut into 5-μm-thick sections, placed in 70% ethanol, and embedded in paraffin. Paraffin sections (5 μm thick) were mounted and stained with hematoxylin and eosin or Verhoeff von Gieson (VVG) stains. Representative sections were coded and evaluated in a blinded manner for measurements of wall thickness of pulmonary arteries 30 to 100 μm in diameter located at the level of the terminal bronchiole, respiratory bronchiole, or alveolar duct. The wall thickness of VVG-stained vessels was measured at every 90° of circumference and averaged.20 The presence of neointimal lesions was measured in 20 vessels <50 μm from each slide. The percentage and presence of neointimal lesions were calculated as follows: (neointimal lesion−positive vessel count)/20×100.21 The arterial-to-alveolar ratio was determined.22
Immunohistochemistry
Immunohistochemistry studies were performed on MCT+/+ and MCTsl/sl lung tissue with the use of anti–factor VIII–related antigen (1:1000, rabbit polyclonal, Dako), anti–smooth muscle-α actin (1:200, mouse monoclonal, Dako), anti-human VEGF (1:200, mouse monoclonal, Santa Cruz), and anti-chicken TN-C (1:500, Chemicon). Staining was graded +, ++, or +++ for weak, moderate, and strong staining, respectively. For detection of proliferating cell nuclear antigen (PCNA) (1:100, Dako), samples were incubated with a biotinylated anti-mouse IgG prepared according to the manufacturer’s instructions (Vector). For PCNA, all immunoassayed sections were exposed to diaminobenzidine diluted in 50-mmol/L Tris buffer containing hydrogen peroxide and lightly counterstained with eosin for PCNA. All others were counterstained with hematoxylin.
Barium/Gelatin Angiograms
Barium angiography was performed in +/+, MCT+/+, and MCTsl/sl rat lungs (n=4 for each group). After the animals were euthanized, a thoracotomy was rapidly performed, and heparin was injected into the RV to prevent blood from clotting in the lungs. A tracheostomy was performed, and the lungs were inflated with air through a gavage needle inserted into the tracheostomy. Blood was flushed from the lungs with heparinized saline through a catheter inserted through the wall of the RV into the main pulmonary artery. A heated solution of gelatin and barium was infused into the main pulmonary artery catheter at 74 mm Hg pressure for 3 to 4 minutes. The main pulmonary artery was ligated under pressure, and the lungs were inflation-fixed with formalin at constant pressure (20 cm H2O) for at least 24 hours. The barium-filled arterial structure in the lungs was imaged by x-ray radiography.23
Northern Blot Analysis
Total RNA was purified from MCT+/+ and MCTsl/sl rat lungs (n=4 for each group) with the use of the GenElute Total RNA kit (Sigma). Twenty micrograms of total RNA per lung were analyzed by standard Northern blot and hybridization techniques with the use of cDNA probes. Rat preproET-1, ETA, ETB, and ECE-1 cDNA probes were labeled with α[32P] dCTP with the use of random primed labeling (RTS Random Primer DNA Labeling System; Gibco BRL). An 18s rRNA oligonucleotide was labeled with the use of terminal deoxytransferase and α[32P] dCTP. After hybridization, blots were washed in low and high stringency conditions. Imaging and quantification of mRNA signals were performed with a Molecular Dynamics Storm 860 PhosphorImager. Normalization to 18s rRNA levels was used in quantification of mRNA signals.17,18
Lung ET Content
ET-1 peptide was measured in lung samples from MCT+/+ and MCTsl/sl rat lungs (n=5 for each group) with a commercially available ELISA kit (BioMedica). Results were normalized to lung protein content.24
Western Blot Analysis
Western blot analysis was performed according to previously published techniques with the use of a monoclonal antibody to VEGF24 or a monoclonal antibody to TN-C.25 Densitometry was performed with a scanner and NIH Image software (National Institutes of Health).
Gelatin Substrate Zymography
Tissue extracts were prepared by homogenizing MCT+/+ and MCTsl/sl rat lungs (n=4 for each group) at 4°C in protein lysis buffer. Homogenates were centrifuged at 5000 rpm for 5 minutes and stored at −70°C. Sixty-microgram samples were mixed 1:1 in 2× sample buffer (Invitrogen) and then electrophoresed under nonreducing conditions in a 10% agarose gel containing 1 mg/mL of gelatin (Invitrogen). After electrophoresis, the gel was incubated in reducing buffer for 30 minutes. The gel was then incubated in developing buffer at 37°C for 16 hours. After staining with Coomassie blue, gelatin-degrading enzymes were identified by their ability to clear the substrate at their respective molecular weights. To determine whether matrix metalloproteinases (MMPs) in lung tissue are equivalent in molecular size and activity to MMP-9 and MMP-2, a purified MMP-9/MMP-2 standard (Sigma) was also separated on substrate gels.25
Statistical Analysis
All results are represented as mean±SEM. Comparisons were made with the use of 1- or 2-way ANOVA with Fisher’s protected least significant difference post hoc test, with P<0.05 accepted as significant (Statview).
Results
Exaggerated PAH in ETB Receptor–Deficient Rats
To determine whether ETB receptor deficiency affects the development of PAH in rats exposed to MCT, a number of parameters of PAH were assessed in both control (+/+) and ETB receptor–deficient (sl/sl) rats. Four weeks after MCT treatment, no differences in body weight were detected between the study groups (Table). However, RV weight and RV hypertrophy, as determined by the ratio of RV/LV+septum weight, were higher in sl/sl than +/+ animals (Table). To determine whether the mild hypoxia of Denver’s altitude contributed to the degree of PAH, 5 sl/sl animals were raised as pups in a hyperbaric chamber simulating sea level atmospheric pressure and treated with MCT at 6 weeks of age. There were no significant differences between MCTsl/sl animals and MCTsl/sl animals raised in the hyperbaric chamber. The pulmonary artery medial wall thickness was greater in the sl/sl lung than the +/+ lung. Furthermore, there was a marked increase in the medial wall thickness of MCTsl/sl animals and MCTsl/sl animals raised in a hyperbaric chamber (Table). Neointimal lesions were only seen in MCTsl/sl lungs. During the breathing of room air (21% oxygen), the mean PAP was higher in the MCTsl/sl than in the MCT+/+ animals (Table). Cardiac output was lower and TPR was 3-fold greater in MCTsl/sl than MCT+/+ animals. Aortic pressure did not differ between the 2 groups (Table). As well, arterial blood gases were not different between the study groups, with the exception of PaCO2 (Table). Collectively, these studies show that PAH is exaggerated in ETB receptor–deficient rats.
Body Weight, RV Hypertrophy, Morphometric Changes, and Hemodynamic and Blood Gas Variables During Normoxia
| MCT+/+ (n=6) | MCTsl/sl (n=5) | MCTsl/sl Hyperbaric (n=5) | |
|---|---|---|---|
| Body weight, RV hypertrophy, and morphometric changes | |||
| Body weight, g | 313±3 | 270±18 | 255±21 |
| RV, g | 0.20±0.04 | 0.29±0.04* | 0.25±0.01 |
| RV/LV+septum | 0.39±0.03 | 0.61±0.06* | 0.49±0.05* |
| Wall thickness, μm | 11.6±0.2 | 24.9±0.7* | 22.9±0.4* |
| Arterial/alveolar ratio | 6.85±0.75 | 2.50±0.27* | 2.60±0.28* |
| Neointimal lesions, % | 0.0±0.0 | 30.0±6.1* | 18.8±5.5* |
| Hemodynamic and blood gas variables during normoxia | |||
| Mean PAP, mm Hg | 28±2 | 41±7* | 39±4* |
| Cardiac output, mL/min | 64±4 | 29±3* | 30±7* |
| TPR, mm Hg/(L·min) | 438±41 | 1418±236* | 1677±539* |
| Mean aortic pressure, mm Hg | 114±2 | 109±5 | 123±9 |
| pH, units | 7.30±0.04 | 7.30±0.02 | 7.39±0.06 |
| PaCO2, mm Hg | 30±1 | 24±3* | 24±1* |
| PaO2, mm Hg | 67±2 | 65±8 | 75±7 |
Values are mean±SEM.
P<0.05 vs MCT+/+.
Neointimal Proliferation in ETB Receptor–Deficient Rat Lungs
In its more severe form, PAH is characterized by the development of occlusive neointimal lesions. To determine whether PAH induced by a combination of ETB receptor deficiency and MCT treatment results in neointimal formation, the morphology of control and sl/sl rat lungs treated with MCT was assessed. Hematoxylin and eosin staining of MCT+/+ and MCTsl/sl rat lungs revealed medial hypertrophy. Neointimal proliferation was noted in 19% to 30% of small pulmonary arteries <50 μm in diameter in MCTsl/sl rats (Table). Approximately 60% of sl/sl rats treated with MCT developed neointimal lesions. Neointimal proliferation was not seen in MCT+/+ lungs, in larger pulmonary arteries from MCTsl/sl animals, or in sl/sl animals older than 3 months treated with MCT (data not shown). VVG staining revealed a well-defined internal elastic lamina in MCT+/+ rat pulmonary arteries. In contrast, the internal elastic lamina was less apparent in MCTsl/sl small pulmonary arteries with neointimal proliferation (Figure 1).
Figure 1.
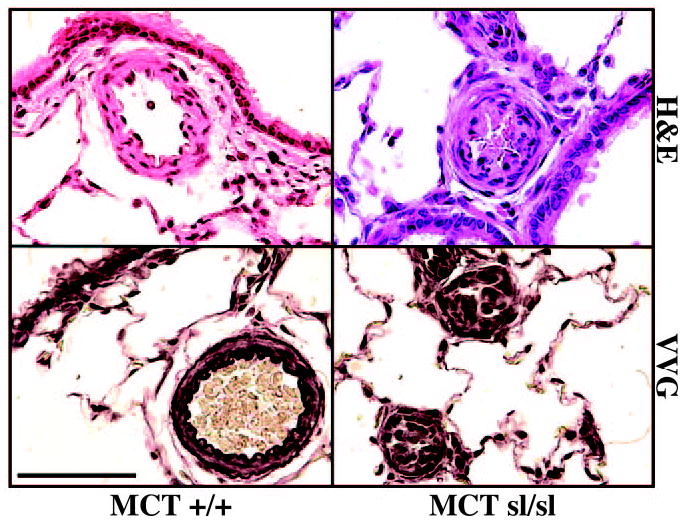
Hematoxylin and eosin (H&E) staining of MCT+/+ rat lung revealed medial hypertrophy of small pulmonary arteries. VVG staining revealed a well-defined internal elastic lamina. In contrast, near occlusion of small pulmonary arteries in the MCTsl/sl rat lung is shown. VVG staining of MCTsl/sl rat lung of plexogenic lesions failed to reveal a well-defined internal elastic lamina. Bar=50 μm.
Next, to assess the extent of neointimal occlusion, barium angiography was performed to evaluate filling of small pulmonary arteries. MCT+/+ rat lungs revealed a nonhomogeneous blush phase and diminished filling of small pulmonary arteries in comparison with normal rat lungs (Figure 2). Furthermore, MCTsl/sl lungs infused with barium showed even less filling of distal pulmonary arteries than normal and MCT+/+ lungs.
Figure 2.
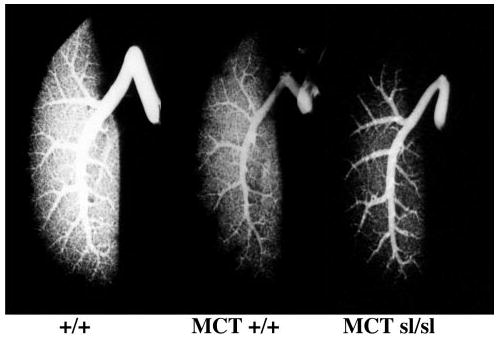
Barium angiograms of control rat lung (+/+), MCT+/+ rat lung, and MCTsl/sl rat lung. Note progressive decrease in filling of distal pulmonary vasculature in MCT+/+ and MCTsl/sl lung.
Characterization of Neointimal Lesions in ETB Receptor–Deficient Rats
To begin to characterize the cellular composition of neointimal lesions in MCTsl/sl lungs, immunohistochemistry was performed. Factor VIII–related antigen was expressed in the neointima of occluded MCTsl/sl lung arteries, indicating the presence of ECs (Figure 3). In addition, smooth muscle-α actin–positive cells were evident in the neointimal lesions, suggesting the presence of VSMCs and/or reactive fibroblasts (Figure 3). Thus, neointimal lesions in MCTsl/sl lungs pheno-copy those observed in human PAH.1–4
Figure 3.
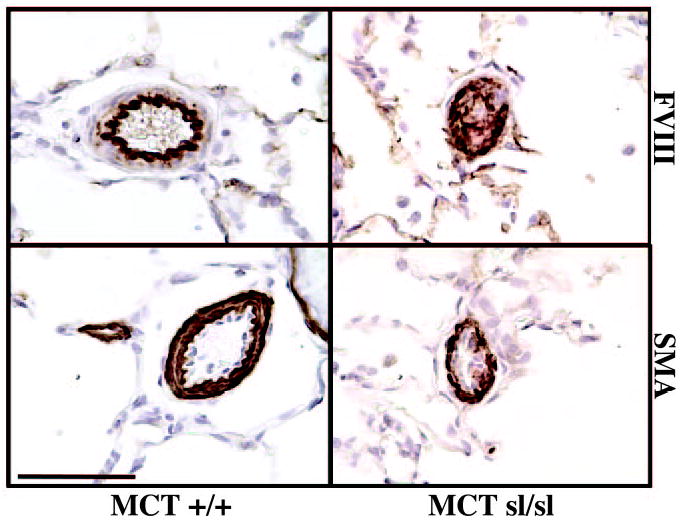
Immunohistochemical staining of MCT+/+ and MCTsl/sl rat lung. Factor VIII (FVIII) staining of MCT+/+ rat lung revealed a well-defined endothelial layer. Smooth muscle α-actin (SMA) staining of MCT+/+ lungs identified VSMCs in vessel media. In MCTsl/sl rat lung, FVIII and SMA expression was detected within occlusive plexogenic lesions, indicating the presence of EC and VSMC markers. Bar=50 μm.
Increased expression of angiogenic factors, such as VEGF, as well as induction of the pro-proliferative extracellular matrix protein TN-C, represents hallmarks of human PAH, and it is generally accepted that appearance of these proteins promotes accumulation of VSMCs and ECs within occlusive neointimal lesions.4,5,26 Accordingly, we determined whether cell proliferation, VEGF, and TN-C are increased in ETB receptor– deficient rats treated with MCT. Western analysis for VEGF did not reveal differences between MCTsl/sl and MCT+/+ lungs (not shown), but increased VEGF staining was noted in neointimal lesions (Figure 4). Staining for VEGF was strong (+++) in MCTsl/sl animals and was weak (+) in MCT+/+ animals. Immunohistochemistry for PCNA revealed the presence of an increased number of proliferating cells within neointimal lesions (Figure 4). In control rats maintained under normoxic conditions, extracellular TN-C protein was deposited throughout the lung parenchyma, as well as in smooth muscle of large and small vessels and in the smooth muscle layer of the airways (Figure 5A). In the sl/sl lung, smooth muscle positivity increased in all tissue compartments and was especially prominent in the large airway smooth muscle layer. In addition, TN-C positivity was apparent beneath the endothelium and in the thickened parenchyma, which showed highly heterogenous TN-C staining. In MCT-treated controls rats, TN-C was deposited throughout the parenchyma, beneath the airways, and in the smooth layers of pulmonary- and bronchiole-associated blood vessels. Submucosal positivity was also apparent. In the sl/sl rats treated with MCT, the expression of TN-C was greatly increased in blood vessels, including occluded small-resistance vessels (Figure 5A). Whole lung TN-C was increased in sl/sl lungs without MCT, suggesting a predisposition to PAH, but was not significantly increased after MCT (Figure 5B).
Figure 4.
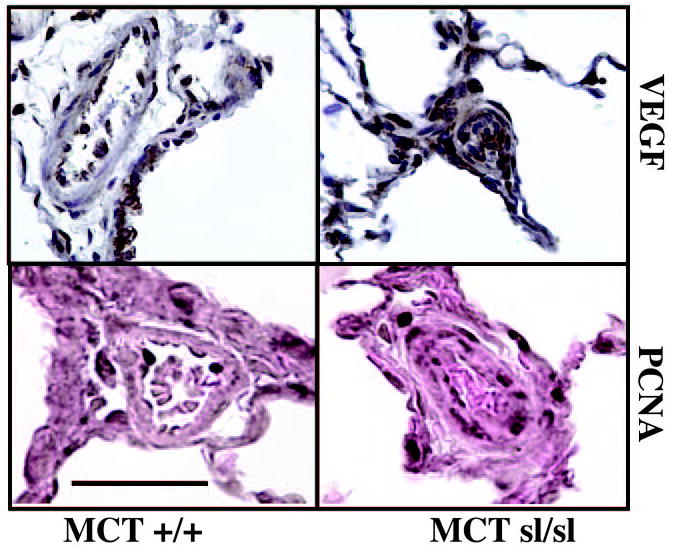
VEGF is expressed in alveoli and blood vessel wall of MCT+/+ rat lung. In plexogenic lesions in MCTsl/sl lungs, staining of the proangiogenic factor VEGF is noted. Immunohistochemical staining of MCT+/+ and MCTsl/sl rat lung for PCNA revealed PCNA-positive cells in EC layer of MCT+/+ group, whereas distribution of PCNA-positive cells in MCTsl/sl group was more widespread, being detected in the adventitial cell, medial cell, and EC layers. Bar=50 μm.
Figure 5.
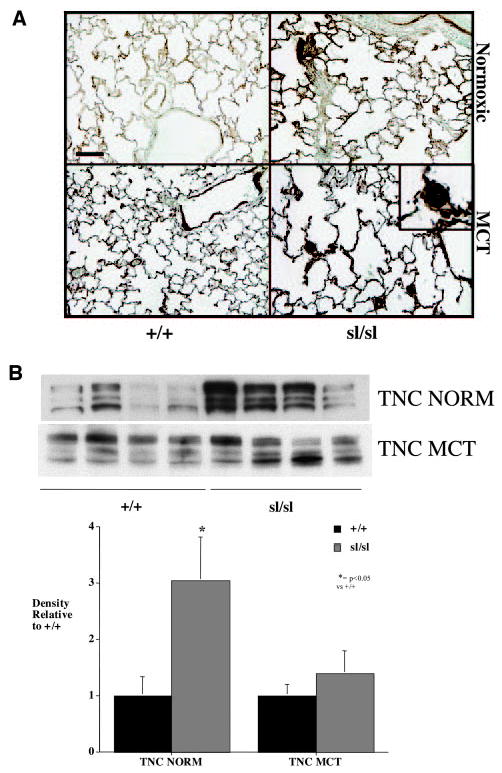
A, In control (+/+) rats, extracellular TN-C protein was deposited throughout the lung parenchyma, as well as in smooth muscle of large and small vessels and in smooth muscle layer of airways. In untreated sl/sl lung, smooth muscle TN-C positivity increased in all tissue compartments and was especially prominent in the large airway smooth muscle layer. In addition, TN-C positivity was apparent beneath the endothelium and in the thickened parenchyma, which showed highly heterogenous TN-C staining. In MCT-treated control rats, TN-C was deposited throughout the parenchyma, beneath the airways, and in smooth layers of pulmonary- and bronchiole-associated blood vessels. Submucosal positivity was also apparent. In sl/sl rats treated with MCT, expression of TN-C was greatly increased in blood vessels, including occluded small-resistance vessels (inset). B, Whole lung TN-C was increased in sl/sl lungs without MCT, suggesting a predisposition to pulmonary arterial hypertension, but was not significantly increased after MCT. NORM indicates normoxic. Bar=50 μm.
Because recent studies indicate that the bioavailability of VEGF27 and the expression of TN-C5,25,26,28 rely on activation of specific MMPs, we next assessed MMP activity via gelatin zymography. MMP-9 activity was increased >4-fold in the MCTsl/sl lungs compared with MCT+/+ lungs (Figure 6), whereas MMP-2 activity did not differ between groups (Figure 6).
Figure 6.
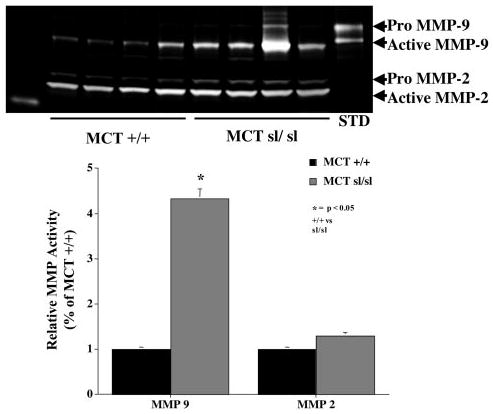
Gelatin substrate zymography reveals that MMP-9 activity was increased >4-fold in MCTsl/sl lungs compared with MCT+/+ lungs, whereas MMP-2 activity was not different between groups. Note that MMP-2 and MMP-9 are detected as both a higher-molecular-weight proform and a lower-molecular-weight active form. STD indicates recombinant MMP-9.
ETA and ETB Receptor Expression Is Altered With ETB Receptor Deficiency
The activity of ET-1 depends on binding to the ETA and ETB receptors as well as the balance of ET receptor number and activity.29,30 Recent studies have shown that blockade of the ETB receptor causes PAH in the ovine fetal lung 30 and predisposes to acute and chronic hypoxic PAH.17,18 To determine whether altered ET receptor expression contributes to MCT-induced PAH in the ETB receptor– deficient rat lung, expression of mRNA for pre-proET-1, the ETA receptor, and the ETB receptor was evaluated between MCT+/+ and MCTsl/sl lungs (Figure 7). MCTsl/sl animals had a 65±10% decrease in the mRNA for the ETB receptor and a 50±6% increase in the ETA receptor. Expression of the mRNA for pre-proET-1 and ECE-1 were not different. Whole lung ET-1 was not different between MCT+/+ (1.3±0.3 fmol/mL) and MCTsl/sl lungs (1.6±0.4 fmol/mL). These data suggest that alteration in the balance between ETA and ETB receptors contributes to PAH in this model.
Figure 7.
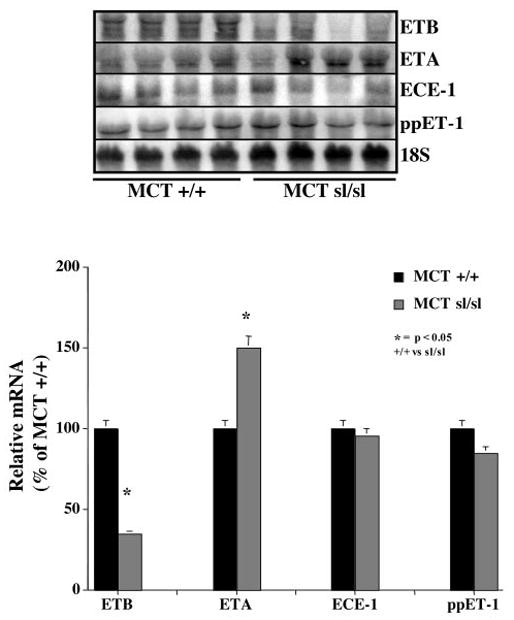
Northern blot analysis shows that MCTsl/sl animals have a 65±10% decrease in steady state levels of ETB receptor mRNA and a 50±6% increase in ETA receptor mRNA levels. In contrast, expression of mRNAs for pre-proET-1 (ppET-1) and ECE-1 did not change.
Discussion
A major finding of this study is that deficiency of the ETB receptor in the pulmonary circulation predisposes rats to the development of severe PAH. After treatment with the endothelial toxin MCT, the transgenic sl/sl rat developed exaggerated PAH characterized by elevated PAP and a markedly diminished cardiac output. In 60% of animals, morphometric evaluation revealed the presence of neointimal proliferation of small distal pulmonary arteries with cells expressing EC and VSMC markers. Barium angiography revealed diminished filling of small pulmonary arteries, which is consistent with a decreased arterial-to-alveolar ratio by morphometric evaluation. Moreover VEGF, a proangiogenic factor, and TN-C, a pro-proliferative extracellular matrix glycoprotein, were highly expressed in the neointimal lesions, which were composed of proliferating VSMCs and ECs. In addition, the activity of MMP-9, an upstream effector of VEGF and TN-C, was increased in MCT-treated sl/sl rat lungs. Possible mechanisms leading to these changes include alterations in expression of the ET receptors with diminished expression of the ETB receptor and increased expression of the ETA receptor. Furthermore, in the absence of MCT, the ETB receptor– deficient sl/sl lung increased production of TN-C. This study suggests that in pulmonary vessels, ETB receptor–mediated signaling counteracts the formation of neointimal lesions in MCT-induced pulmonary hypertension.
We have previously shown that the pulmonary vasculature of the transgenic sl/sl rat is deficient in the ETB receptor.17 This genetic model was produced by rescue of the spotting lethal rat, which is a naturally occurring rat strain that carries a 301-bp deletion in ETB, rendering the gene nonfunctional.19,31 Although the ETB-deficient rats lack expression of ETB driven by the endogenous promoter, they express ETB in adrenergic tissues, such as the adrenal medulla and sympathetic ganglion. The pulmonary circulation of the transgenic sl/sl rat lacks expression of the mRNA for the ETB receptor, lacks ET-1–mediated pulmonary vasodilation, exhibits an exaggerated pulmonary vasopressor response to acute hypoxia and exogenous ET-1 infusion, and is characterized by diminished NO production.17 In addition, deficiency of the ETB receptor in the pulmonary circulation predisposes rats to the development of chronic hypoxic pulmonary hypertension18 and hypoxia-induced pulmonary edema.24 In the present study we have shown that ETB receptor deficiency predisposes to severe PAH with neointimal proliferation of small pulmonary arteries.
This study is of interest because few animal models of neointimal proliferation of small pulmonary arteries have been described, especially at the cellular and molecular levels. Pulmonary lesions in rats exposed to MCT in addition to a systemic to pulmonary artery shunt21 or pneumonectomy8 suggest the importance of vascular injury in addition to high flow for the development of neointimal lesions. However, hemodynamic stress is apparently not necessary to develop these lesions because hypoxia with blockade of the VEGF receptor 2 also causes neointimal proliferation of the pulmonary circulation.9 More recent studies have shown that genetic alterations may be sufficient to develop plexogenic arteriopathy because overexpression of S100A4/Mts 1, which confers a metastatic phenotype in tumor cells, caused plexogenic lesions in 5% of mice.10 Deficiency of the ETB receptor predisposes to neointimal proliferation of carotid arteries after balloon injury32 and to PAH in the fetal lamb.30 Our study also shows that genetic alteration of the ETB receptor with endothelial injury predisposes to plexogenic arteriopathy.
ETB receptor deficiency may predispose to exaggerated pulmonary hypertension and neointimal proliferation induced by MCT as a result of several mechanisms. The sl/sl rat pulmonary vasculature is characterized by diminished endothelial NO synthase protein and has decreased ability to produce NO. Furthermore, pulmonary arterial ET-1 levels are 8-fold higher in sl/sl than +/+ control animals.17 Further studies in hypoxic animals showed that prostacyclin synthase protein and prostacyclin production were decreased in the sl/sl rat lung.18 More recent studies showed increased VEGF protein in the sl/sl rat lung than in the +/+ lung.24 A recent study also found that MCT exaggerated the pulmonary hypertensive response in sl/sl animals,33 and a subsequent report revealed that the pulmonary hypertensive response was mediated largely through the ETA receptor.34 However, no neointimal lesions were seen in either of these 2 studies. The presence of neointimal lesions in our study may be due to a longer exposure to MCT or younger animals treated with MCT. The mild hypoxia at Denver’s altitude is likely not the primary factor because animals raised in a hyperbaric chamber and treated with MCT also developed neointimal lesions. In this study the extracellular matrix glycoprotein TN-C was markedly increased in both control sl/sl rat lungs and the neointimal lesions of MCT-treated rats. Thus, deficiency of the ETB receptor predisposes to pulmonary hypertension even in the absence of MCT treatment.
The neointimal lesions that form after MCT treatment of the ETB receptor– deficient rat share several features that are associated with human plexogenic arteriopathy. Both are characterized by intimal hyperplasia leading to occlusion of the arterial lumen. Cellular proliferation of ECs,2,35 VSMCs,6,7,36 and expression of specific extracellular matrix proteins such as TN-C3 are also shared by the present model and human PAH. Recent studies have suggested that VEGF may play a proangiogenic role in the development of plexogenic arteriopathy.4 Furthermore, the pro-proliferative TN-C is increased in plexogenic lesions of children with congenital heart disease,5 in patients with familial and sporadic forms of PAH (P.L. Jones, PhD, unpublished data, 2004), and in the present model. It is interesting that the internal elastic lamina was difficult to discern in occluded pulmonary vessels of MCT-treated sl/sl rats because recent studies have suggested that an endogenous vascular elastase may contribute to the proliferative lesions of plexogenic arteriopathy,28 yet this was not directly tested in this study. However, MMP-9, which is activated to a greater extent in MCT-treated sl/sl rats, has been shown to act as an elastase.37
ET receptor antagonists have been developed to treat PAH. Bosentan, a dual ET receptor antagonist, lowers PAP and vascular resistance in adults and children with PAH.38,39 Selective ETA receptor blockade with sitaxsentan is also effective in the treatment of PAH and has the potential advantage of maintaining ETB receptor–mediated ET clearance and NO release.40 However, the potential advantages of selective ETA receptor blockade remain incompletely understood. There is no clinical strategy to treat PAH with the use of selective ETB receptor antagonists, yet this study highlights the theoretical advantage of maintaining ETB receptor activity in PAH.
Acknowledgments
This work was supported in part by grants from the National Institutes of Health (HL-03823, Dr Ivy; HL-14985, Dr McMurtry; HL-68798 and NIH 2 P50 HL57144, Dr Jones), an American Heart Association, Desert/Mountain Affiliate Grantin-Aid (Dr Ivy), the Leah Bult Pulmonary Hypertension Fund (Dr Ivy), the Caitlyn Whitley Cardiology Research Fund (Dr Ivy), and the Giles Filley Memorial Award (Dr Jones).
References
- 1.Rabinovitch M, Haworth SG, Castaneda AR, Nadas AS, Reid LM. Lung biopsy in congenital heart disease: a morphometric approach to pulmonary vascular disease. Circulation. 1978;58:1107–1122. doi: 10.1161/01.cir.58.6.1107. [DOI] [PubMed] [Google Scholar]
- 2.Tuder RM, Groves B, Badesch DB, Voelkel NF. Exuberant endothelial cell growth and elements of inflammation are present in plexiform lesions of pulmonary hypertension. Am J Pathol. 1994;144:275–285. [PMC free article] [PubMed] [Google Scholar]
- 3.Rabinovitch M. Pathobiology of pulmonary hypertension: extracellular matrix. Clin Chest Med. 2001;22:433–449. doi: 10.1016/s0272-5231(05)70282-3. [DOI] [PubMed] [Google Scholar]
- 4.Tuder RM, Chacon M, Alger L, Wang J, Taraseviciene-Stewart L, Kasahara Y, Cool CD, Bishop AE, Geraci M, Semenza GL, Yacoub M, Polak JM, Voelkel NF. Expression of angiogenesis-related molecules in plexiform lesions in severe pulmonary hypertension: evidence for a process of disordered angiogenesis. J Pathol. 2001;195:367–374. doi: 10.1002/path.953. [DOI] [PubMed] [Google Scholar]
- 5.Jones PL, Cowan KN, Rabinovitch M. Tenascin-C, proliferation and subendothelial fibronectin in progressive pulmonary vascular disease. Am J Pathol. 1997;150:1349–1360. [PMC free article] [PubMed] [Google Scholar]
- 6.Tanaka Y, Schuster DP, Davis EC, Patterson GA, Botney MD. The role of vascular injury and hemodynamics in rat pulmonary artery remodeling. J Clin Invest. 1996;98:434–442. doi: 10.1172/JCI118809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Botney MD. Role of hemodynamics in pulmonary vascular remodeling: implications for primary pulmonary hypertension. Am J Respir Crit Care Med. 1999;159:361–364. doi: 10.1164/ajrccm.159.2.9805075. [DOI] [PubMed] [Google Scholar]
- 8.Okada K, Bernstein ML, Zhang W, Schuster DP, Botney MD. Angiotensin-converting enzyme inhibition delays pulmonary vascular neointimal formation. Am J Respir Crit Care Med. 1998;158:939–950. doi: 10.1164/ajrccm.158.3.9710007. [DOI] [PubMed] [Google Scholar]
- 9.Taraseviciene-Stewart L, Kasahara Y, Alger L, Hirth P, Mc Mahon G, Waltenberger J, Voelkel NF, Tuder RM. Inhibition of the VEGF receptor 2 combined with chronic hypoxia causes cell death-dependent pulmonary endothelial cell proliferation and severe pulmonary hypertension. FASEB J. 2001;15:427–438. doi: 10.1096/fj.00-0343com. [DOI] [PubMed] [Google Scholar]
- 10.Greenway S, van Suylen RJ, Du Marchie Sarvaas G, Kwan E, Ambart-sumian N, Lukanidin E, Rabinovitch M. S100A4/Mts1 produces murine pulmonary artery changes resembling plexogenic arteriopathy and is increased in human plexogenic arteriopathy. Am J Pathol. 2004;164:253–262. doi: 10.1016/S0002-9440(10)63115-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rubens C, Ewert R, Halank M, Wensel R, Orzechowski HD, Schultheiss HP, Hoeffken G. Big endothelin-1 and endothelin-1 plasma levels are correlated with the severity of primary pulmonary hypertension. Chest. 2001;120:1562–1569. doi: 10.1378/chest.120.5.1562. [DOI] [PubMed] [Google Scholar]
- 12.Ihara M, Fukuroda T, Saeki T, Nishikibe M, Kojiri K, Suda H, Yano M. An endothelin receptor (ETA) antagonist isolated from Streptomyces misakiensis. Biochem Biophys Res Commun. 1991;178:132–137. doi: 10.1016/0006-291x(91)91789-f. [DOI] [PubMed] [Google Scholar]
- 13.McCulloch KM, Docherty C, MacLean MR. Endothelin receptors mediating contraction of rat and human pulmonary resistance arteries: effect of chronic hypoxia in the rat. Br J Pharmacol. 1998;123:1621–1630. doi: 10.1038/sj.bjp.0701785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hasunuma K, Rodman DM, O’Brien RF, McMurtry IF. Endothelin 1 causes pulmonary vasodilation in rats. Am J Physiol. 1990;259:H48–H54. doi: 10.1152/ajpheart.1990.259.1.H48. [DOI] [PubMed] [Google Scholar]
- 15.Fukuroda T, Fujikawa T, Ozaki S, Ishikawa K, Yano M, Nishikibe M. Clearance of circulating endothelin-1 by ETB receptors in rats. Biochem Biophys Res Commun. 1994;199:1461–1465. doi: 10.1006/bbrc.1994.1395. [DOI] [PubMed] [Google Scholar]
- 16.Sato K, Oka M, Hasunuma K, Ohnishi M, Sato K, Kira S. Effects of separate and combined ETA and ETB blockade on ET-1-induced constriction in perfused rat lungs. Am J Physiol. 1995;269:L668 –L672. doi: 10.1152/ajplung.1995.269.5.L668. [DOI] [PubMed] [Google Scholar]
- 17.Ivy D, McMurtry IF, Yanagisawa M, Gariepy CE, Le Cras TD, Gebb SA, Morris KG, Wiseman RC, Abman SH. Endothelin B receptor deficiency potentiates ET-1 and hypoxic pulmonary vasoconstriction. Am J Physiol. 2001;280:L1040–L1048. doi: 10.1152/ajplung.2001.280.5.L1040. [DOI] [PubMed] [Google Scholar]
- 18.Ivy DD, Yanagisawa M, Gariepy CE, Gebb SA, Colvin KL, McMurtry IF. Exaggerated hypoxic pulmonary hypertension in endothelin B receptor-deficient rats. Am J Physiol. 2002;282:L703–L712. doi: 10.1152/ajplung.00272.2001. [DOI] [PubMed] [Google Scholar]
- 19.Gariepy CE, Williams SC, Richardson JA, Hammer RE, Yanagisawa M. Transgenic expression of the endothelin-B receptor prevents congenital intestinal aganglionosis in a rat model of Hirschsprung disease. J Clin Invest. 1998;102:1092–1101. doi: 10.1172/JCI3702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Imamura M, Luo B, Limbird JN, Vitello AM, Oka M, Ivy DD, McMurtry IF, Garat CV, Fallon MB, Carter EP. Hypoxic pulmonary hypertension is prevented in rats with common bile duct ligation. J Appl Physiol. 2005;98:739–747. doi: 10.1152/japplphysiol.00556.2004. [DOI] [PubMed] [Google Scholar]
- 21.Okada K, Tanaka Y, Bernstein M, Zhang W, Patterson GA, Botney MD. Pulmonary hemodynamics modify the rat pulmonary artery response to injury: a neointimal model of pulmonary hypertension. Am J Pathol. 1997;151:1019–1025. [PMC free article] [PubMed] [Google Scholar]
- 22.O’Blenes SB, Merklinger SL, Jegatheeswaran A, Campbell A, Rabinovitch M, Rebeyka I, Van Arsdell G. Low molecular weight heparin and unfractionated heparin are both effective at accelerating pulmonary vascular maturation in neonatal rabbits. Circulation. 2003;108(suppl I):II-161–II-166. doi: 10.1161/01.cir.0000087380.36688.4c. [DOI] [PubMed] [Google Scholar]
- 23.Le Cras TD, Markham NE, Tuder RM, Voelkel NF, Abman SH. Treatment of newborn rats with a VEGF receptor inhibitor causes pulmonary hypertension and abnormal lung structure. Am J Physiol. 2002;283:L555–L562. doi: 10.1152/ajplung.00408.2001. [DOI] [PubMed] [Google Scholar]
- 24.Carpenter T, Schomberg S, Steudel W, Ozimek J, Colvin K, Stenmark K, Ivy DD. Endothelin B receptor deficiency predisposes to pulmonary edema formation via increased lung vascular endothelial cell growth factor expression. Circ Res. 2003;93:456–463. doi: 10.1161/01.RES.0000090994.15442.42. [DOI] [PubMed] [Google Scholar]
- 25.Jones PL, Chapados R, Baldwin HS, Raff GW, Vitvitsky EV, Spray TL, Gaynor JW. Altered hemodynamics controls matrix metalloproteinase activity and tenascin-C expression in neonatal pig lung. Am J Physiol. 2002;282:L26–L35. doi: 10.1152/ajplung.00171.2001. [DOI] [PubMed] [Google Scholar]
- 26.Jones PL, Crack J, Rabinovitch M. Regulation of tenascin-C, a vascular smooth muscle cell survival factor that interacts with the alpha v beta 3 integrin to promote epidermal growth factor receptor phosphorylation and growth. J Cell Biol. 1997;139:279–293. doi: 10.1083/jcb.139.1.279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Engsig MT, Chen QJ, Vu TH, Pedersen AC, Therkidsen B, Lund LR, Henriksen K, Lenhard T, Foged NT, Werb Z, Delaisse JM. Matrix metalloproteinase 9 and vascular endothelial growth factor are essential for osteoclast recruitment into developing long bones. J Cell Biol. 2000;151:879–889. doi: 10.1083/jcb.151.4.879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cowan KN, Heilbut A, Humpl T, Lam C, Ito S, Rabinovitch M. Complete reversal of fatal pulmonary hypertension in rats by a serine elastase inhibitor. Nat Med. 2000;6:698–702. doi: 10.1038/76282. [DOI] [PubMed] [Google Scholar]
- 29.Ivy DD, Kinsella JP, Abman SH. Physiologic characterization of endothelin A and B receptor activity in the ovine fetal pulmonary circulation. J Clin Invest. 1994;93:2141–2148. doi: 10.1172/JCI117210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ivy DD, Parker TA, Abman SH. Prolonged endothelin B receptor blockade causes pulmonary hypertension in the ovine fetus. Am J Physiol. 2000;279:L758–L765. doi: 10.1152/ajplung.2000.279.4.L758. [DOI] [PubMed] [Google Scholar]
- 31.Gariepy CE, Cass DT, Yanagisawa M. Null mutation of endothelin receptor type B gene in spotting lethal rats causes aganglionic megacolon and white coat color. Proc Natl Acad Sci U S A. 1996;93:867–872. doi: 10.1073/pnas.93.2.867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Murakoshi N, Miyauchi T, Kakinuma Y, Ohuchi T, Goto K, Yanagisawa M, Yamaguchi I. Vascular endothelin-B receptor system in vivo plays a favorable inhibitory role in vascular remodeling after injury revealed by endothelin-B receptor-knockout mice. Circulation. 2002;106:1991–1998. doi: 10.1161/01.cir.0000032004.56585.2a. [DOI] [PubMed] [Google Scholar]
- 33.Nishida M, Okada Y, Akiyoshi K, Eshiro K, Takoaka M, Gariepy CE, Yanagisawa M, Matsumura Y. Role of endothelin ETB receptor in the pathogenesis of monocrotaline-induced pulmonary hypertension in rats. Eur J Pharmacol. 2004;496:159–165. doi: 10.1016/j.ejphar.2004.06.028. [DOI] [PubMed] [Google Scholar]
- 34.Nishida M, Eshiro K, Okada Y, Takaoka M, Matsumura Y. Roles of endothelin ETA and ETB receptors in the pathogenesis of monocrotaline-induced pulmonary hypertension. J Cardiovasc Pharmacol. 2004;44:187–191. doi: 10.1097/00005344-200408000-00007. [DOI] [PubMed] [Google Scholar]
- 35.Tuder RM, Cool CD, Yeager M, Taraseviciene-Stewart L, Bull TM, Voelkel NF. The pathobiology of pulmonary hypertension: endothelium. Clin Chest Med. 2001;22:405–418. doi: 10.1016/s0272-5231(05)70280-x. [DOI] [PubMed] [Google Scholar]
- 36.Rabinovitch M. Pathobiology of pulmonary hypertension: impact on clinical management. Semin Thorac Cardiovasc Surg Pediatr Card Surg Annu. 2000;3:63–81. doi: 10.1053/tc.2000.6507. [DOI] [PubMed] [Google Scholar]
- 37.Senior RM, Griffin GL, Fliszar CJ, Shapiro SD, Goldberg GI, Welgus HG. Human 92- and 72-kilodalton type IV collagenases are elastases. J Biol Chem. 1991;266:7870–7875. [PubMed] [Google Scholar]
- 38.Rubin LJ, Badesch DB, Barst RJ, Galie N, Black CM, Keogh A, Pulido T, Frost A, Roux S, Leconte I, Landzberg M, Simonneau G. Bosentan therapy for pulmonary arterial hypertension. N Engl J Med. 2002;346:896–903. doi: 10.1056/NEJMoa012212. [DOI] [PubMed] [Google Scholar]
- 39.Barst RJ, Ivy D, Dingemanse J, Widlitz A, Schmitt K, Doran A, Bingaman D, Nguyen N, Gaitonde M, van Giersbergen PL. Pharmacokinetics, safety, and efficacy of bosentan in pediatric patients with pulmonary arterial hypertension. Clin Pharmacol Ther. 2003;73:372–382. doi: 10.1016/s0009-9236(03)00005-5. [DOI] [PubMed] [Google Scholar]
- 40.Barst RJ, Langleben D, Frost A, Horn EM, Oudiz R, Shapiro S, McLaughlin V, Hill N, Tapson VF, Robbins IM, Zwicke D, Duncan B, Dixon RA, Frumkin LR. Sitaxsentan therapy for pulmonary arterial hypertension. Am J Respir Crit Care Med. 2004;169:441–447. doi: 10.1164/rccm.200307-957OC. [DOI] [PubMed] [Google Scholar]