


Abstract
In absence of β-lactam antibiotics, BlaI and MecI homodimeric repressors negatively control the expression of genes involved in β-lactam resistance in Bacillus licheniformis and in Staphylococcus aureus. Subsequently to β-lactam presence, BlaI/MecI is inactivated by a single-point proteolysis that separates its N-terminal DNA-binding domain to its C-terminal domain responsible for its dimerization. Concomitantly to this proteolysis, the truncated repressor acquires a low affinity for its DNA target that explains the expression of the structural gene for resistance. To understand the loss of the high DNA affinity of the truncated repressor, we have determined the different dissociation constants of the system and solved the solution structure of the B. licheniformis monomeric repressor complexed to the semi-operating sequence OP1 of blaP (1/2OP1blaP) by using a de novo docking approach based on inter-molecular nuclear Overhauser effects and chemical-shift differences measured on each macromolecular partner. Although the N-terminal domain of the repressor is not subject to internal structural rearrangements upon DNA binding, the molecules adopt a tertiary conformation different from the crystallographic operator–repressor dimer complex, leading to a 30° rotation of the monomer with respect to a central axis extended across the DNA.
These results open new insights for the repression and induction mechanisms of bacterial resistance to β-lactams.
INTRODUCTION
The Bacillus licheniformis BlaI protein (BLBlaI, 128 amino acids) is a transcriptional repressor of the BlaP β-lactamase. This enzyme is a specific hydrolase of β-lactam antibiotics, induced in response to the presence of this class of antibiotic outside the cell (1). BLBlaI is homologous to Staphylococcus aureus BlaI (SABlaI, 126 amino acids) and MecI (SAMecI, 123 amino acids) regulators involved in the induction of BlaZ β-lactamase and resistant penicillin-binding protein 2a (SAPBP2a or SAMecA), respectively (2). BlaZ β-lactamase and resistant PBP2a are the main factors involved in staphylococcal β-lactam antibiotic resistance. The BlaI/MecI repressors are organized in two domains, an N-terminal domain (NTD) for DNA binding and a C-terminal domain (CTD) for repressor dimerization (3). The crystallographic 3D structures of SABlaI and SAMecI dimers in free and in complex with their DNA operators have been determined (4–6) and the 3D structure of BLBlaI N-terminal DNA-binding domain has been determined by heteronuclear nuclear magnetic resonance (NMR) spectroscopy (7). For the three repressors, the BlaI/MecI-NTD share a common fold composed of three α-helices and three β-strands typical of the winged helix regulator proteins. The SABlaI/SAMecI structures highlight dimers of two independent N-terminal DNA-binding domains and two intertwined C-terminal dimerization domains (Figure 1A). The BlaI/MecI repressors bind specifically to similar nucleic sequences composed of an imperfect dyad symmetry (24 to 30 bp) containing a central conserved palindrome: 5′-TACANNTGTA-3′ (8) (nucleotide one-letter code; N for any nucleotide). The study of BLBlaI dimerization and its interaction with its bla operator (BLOPbla) has shown that, at a concentration below the dissociation constant of BLBlaI dimer ([BLBlaI] <25 µM), the binding of one BLBlaI monomer to its operator leads to the binding of the second monomer with an infinite cooperativity (9). In this way, it has proved impossible to isolate a BLBlaI monomer bound to its DNA operator (Figure 1B). In addition, for the same repressor, it has been shown that the BLBlaI-NTD obtained by papain proteolysis retains its capacity to bind BLOPbla. However, its affinity for its DNA target becomes at least 500 to 1000 times lower as determined by DNAseI footprinting experiments (3).
Figure 1.

Dimeric interaction of the BlaI/MecI repressor with its DNA operator. (A) Representation of the S. aureus dimeric MecI repressor (ribbon) in interaction with the bla operator (PBD ID 1SAX). C-terminus of MecI (dimerisation domain) is colored in pink and N-terminus (DNA binding domain) in blue. (B) Representation of the two potential parallels pathways represented for the B. licheniformis BlaI repressor with the bla operating sequence.
In S. aureus and B. licheniformis, the genes encoding for β-lactam resistance (blaZ, blaP and mecA) form a divergon with the blaI/blaR1 and mecI/mecR1 operons, respectively (10,11). blaR1/mecR1 encodes a penicillin receptor essential for the induction of the gene of resistance. The bla/mec operators are located in the intergenic DNA sequence between the blaP/blaZ/mecA gene and the blaI/mecI-blaR1/mecR1 operon. In presence of β-lactam antibiotics, the BlaR1/MecR1 receptor is acylated. The resulting activated receptor launches a cytoplasmic signal which inactivates BlaI/MecI repressor.
In S. aureus, the SABlaI inactivation is achieved by the proteolysis of the peptide bond linking residues 101 and 102, giving rise to SABlaI-NTD dissociated from SABlaI-CTD (12). The truncated SABlaI-NTD is a monomer, presents a lower affinity for its DNA operator and is released into the cytoplasm. For BLBlaI repressor, the presence of a coactivator (13) generated by the activation of the BlaR1 repressor has been postulated. The binding of the coactivator to BLBlaI would then result in a decreased affinity of BLBlaI repressor for its DNA target (14).
To better understand the mechanisms of the binding of the BlaI/MecI repressors on the DNA, and their inactivation during the induction of BlaP/BlaZ/MecA proteins, we solved the solution structure of the low affinity BLBlaI-NTD/1/2OP1blaP complex ([BLBlaI-NTD]/[1/2OP1blaP]). The dissociation constants of the SAMecI in complex with the B. licheniformis operator OP1 of blaP (OP1blaP) and in complex with the S. aureus operators of blaZ (OPblaZ) and mecA (OPmecA) have been determined. NMR spectroscopy allowed us to estimate the dissociation constants of the SAMecI-NTD and BLBlaI-NTD for the semi-operating sequence of the B. licheniformis blaP gene OP1 (1/2OP1blaP) and for the semi-operating sequence of the S. aureus mecA gene (1/2OPmecA).
MATERIALS AND METHODS
Sample preparation
The pET22b was used as vector for the overproduction of the His-tagged SAMecI protein (SAMecI-His6). The SAMecI coding sequence was amplified by PCR from the S. aureus ATCC 43300 genomic DNA using Taq polymerase (Promega) and the following oligonucleotides as primers: 5′-GAG-CAT-ATG-GAT-AAT-AAA-ACG-TAT-GAA-ATA-TCA-TC-3′ and 5′-CTC-GAG-TTT-ATT-CAA-TAT-ATT-TCT-CAA-TTC-TTC-TA-3′ purchased from EUROGENTEC, S.A., Belgium (http://eurogentec.com). The fragment generated corresponds to the SAMecI coding sequence within the restriction sites for NdeI and XhoI and was cloned into the pCR4 TOPO (Invitrogen) vector to generate the pCR4-mecI. The identity of the sequence was verified before the pCR4-mecI was digested with NdeI and XhoI enzymes and cloned into the pET22b to generate the pCIP451 which contains the MecI coding sequence with a polyhistidine tag at its carboxy-terminal end.
SAMecI-His6 samples were overexpressed in E. coli strain BL21(DE3). For the production of uniformly 15N-labeled samples, Luria-Bertani medium was replaced by a M9 minimal medium supplemented with 1.1 g/l 15NH4Cl, 2 mM MgSO4, 0.1 mM CaCl2 and 2 g/l glucose. Cells were grown at 37°C to a 600 nm absorbance of 0.6 and 0.5 mM IPTG was added for a 3-h induction period. Cells were then harvested by centrifugation, re-suspended in buffer A (20 mM NaH2PO4/Na2HPO4 buffer, 500 mM NaCl, pH 7.6) and disrupted by passage through an Inceltech disintegrator. The soluble fraction was separated by centrifugation at 40 000 g and loaded onto a NiPDC chelating column (2.6 × 10 cm2, Affliland) charged with 50 mM NiSO4 and equilibrated with buffer A. The SAMecI-His6 protein was eluted by a gradient of buffer B (250 mM imidazole, 500 mM NaCl, pH 8). The protein was dialyzed against buffer A and concentrated. The final yields of labeled and unlabeled purified protein were respectively 9 and 15 mg/l of cell culture. The isotopic labeling of 95% was determined by mass spectrometry.
The uniformly 13C/15N-labeled BLBlaI sample was prepared as described in Van Melckebeke et al. (7). The SAMecI-NTD protein was dialyzed into a 75 mM NaH2PO4/Na2HPO4 buffer, 200 mM KCl, 1 mM EDTA, 1 mM NaN3, pH 7.6 and was concentrated to 0.5 mM by ultrafiltration through a 5 kDa cut-off Amicon for further NMR analysis.
Unlabeled single-stranded DNA samples of NMR quality were chemically synthesized and purified by EUROGENTEC, S.A., Belgium (http://eurogentec.com). Freeze-dried samples were suspended in the buffers used for interaction studies. Single-stranded DNA were mixed in a 1:1 ratio, subsequently heated to 100°C and slowly cooled down at room temperature, in order to improve intermolecular arrangements. The mecA semi-operator sequence [5′-ATA-AGA-CTA-CAT-3′ and complementary strand 5′-ATG-TAG-TCT-TAT-3′] was designed according to previous papers results (7) and obtained at a 9 mM final concentration in 75 mM NaH2PO4/Na2HPO4 buffer, 200 mM KCl, 1 mM EDTA, 1 mM NaN3, pH 7.6. The OP1blaP half-dyad [5′-AAA-GTA-TTA-CAT-3′ and 5′-ATG-TAA-TAC-TTT-3′] was selected using former interaction results with BLBlaI-NTD and obtained at a 23 mM final concentration.
NMR spectroscopy
NMR experiments were performed on Varian Inova 600 and Inova 800 spectrometers, both equipped with a triple-resonance (1H, 13C, 15N) probe and shielded z-gradients. Furthermore, the Varian Inova 800 MHz spectrometer is equipped with a cooled probe. The temperature was set to 298 K. Proton chemical shifts were referenced with respect to an external DSS calibration. 13C and 15N chemical shifts were accordingly referenced indirectly using the 1H/X following ratios: 0.251449530 (13C) and 0.101329118 (15N). All experiments used the pulse sequences provided by the Varian Protein Pack (http://varianinc.com). Data processing and peak intensity measurements were performed using the NMRPipe program. Peak picking and spectra display were achieved using the NMRView software.
Affinity measurements
Electrophoretic mobility shift assays were carried out using an ALFexpress DNA sequencer as described in the literature (11). The CY5-labeled fluorescent double-stranded oligonucleotides used in these experiments are listed in Table 1 (15,16).
Table 1.
Oligonucleotides used in band-shift assays
| Operators | Oligonucleotide sequences |
|---|---|
| B. lichen. OP1blaP | 5′CY5-AAA GTA TTA CAT ATG TAA GAT TTA-3′ 3′-TTT CAT AAT GTA TAC TTA CTA AAT-5′ |
| S. aureus OPblaZ | 5′CY5-TAA AAA TTA CAA CTG TAA TAT CGG-3′ 3′-ATT TTT AAT GTT GAC ATT ATA GCC-5′ |
| S. aureus OPmecA | 5′CY5-ATA AGA CTA CAT TTG TAG TAT ATT-3′ 3′-TAT TCT GAT GTA AAC ATC ATA TAA-5′ |
The nucleotide sequences correspond to the blaP operator from B.lichen. (12), the blaZ and the mecA operators form S. aureus. Numbering used in this study for B. licheniformis BlaP start from the 5′ end (A1, A2, A3, …). The complementary strand is noted with a star (T1*, T2*, T3, …) and numbering starts from 3′ end.
For NMR interaction studies, 1H-15N HSQC spectra were collected along the titration of SAMecI and the BLBlaI truncated repressors with the 12 bp DNA. To limit dilution and favor sensitivity, low volumes of highly concentrated half-operators of mecA and blaP genes were added to each protein sample. 15N-labeled SAMecI-NTD and BLBlaI-NTD samples concentrations were set to 0.1 mM. Titration experiments led on 15N-labeled MecI-NTD (respectively 15N-labeled BLBlaI-NTD) with both unlabeled mecA and blaP semi-operating sequences were performed in a 75 mM (respectively 50 mM) NaH2PO4/Na2HPO4 buffer with 200 mM KCl, 1 mM EDTA, 1 mM NaN3 and 10% of D2O at a controlled pH of 7.6.
Data analysis and Kd calculation were performed with the titration script developed for the NMRView software (17). In each case, curves fitting were displayed by the Xmgrace software (http://plasma-gate.weizmann.ac.il/Grace/).
NMR structural restraints
1H, 13C and 15N assignment of the free BLBlaI-NTD protein has been previously reported and deposited in the BMRB (accession number 5873). Resonance assignment of the unlabeled 12 bp DNA of the OP1blaP semi-operating sequence was performed on a 1 mM sample in 50 mM of NaH2PO4/Na2HPO4, 200 mM KCl, 1 mM EDTA, 1 mM NaN3, pH 7.6 in 90%:10% H2O:D2O. A TOCSY spectrum with 80 ms mixing time and a NOESY spectrum with 150 ms mixing time were recorded on that sample. 1H-1H NOESY was also collected in 100% D2O with 150 ms mixing time. A classical homonuclear DNA assignment strategy was used (18). Due to the large number of overlap in H4′ and H5′/H5′′ regions, assignment and chemical-shift mapping were restricted to H1′, H2′, H3′, H5 and H6/H8 resonances. For the 12 bp 1/2OP1blaP assignment in the complex, we reiterated the procedure used for the free DNA but using only a 2D filtered NOESY experiment recorded on the [13C-15N BLBlaI-NTD]/[1/2OP1blaP] sample diluted in 100% 2H2O. The experiment was recorded on the 800 MHz spectrometer with a mixing time of 150 ms.
For the [BLBlaI-NTD]/[1/2OP1blaP] complex, the 1H, 13C and 15N protein resonances were not re-assigned ab initio. Assignment of the protein in complex was obtained by comparison of 1H-13C HSQC and a 1H-15N HSQC of the free and bound forms. Experiments were recorded with 2 mM samples and a [13C-15N BLBlaI-NTD]/[1/2OP1blaP] molar ratio of 1 in 100% D2O and in 90% H2O. To confirm this assignment, a 3D HC(C)H-TOCSY experiment was also recorded on the complex. The weak dependence of 13C chemical shifts to long distances variation and then to the complex formation has permitted to verify each corresponding residue assignment.
In order to obtain inter-molecular NOE restraints between the two partners of the [BLBlaI-NTD]/[1/2OP1blaP] complex, we collected isotopically doubly filtered 2D and 3D 13C/15N NOESY HSQC with mixing times set to 150 ms. A protein chemical-shift mapping was obtained by comparison of 1H-15N HSQC, methyl-selective 13C HSQC and 13C HSQC optimized for aromatics recorded on the free and bound forms of the protein. A chemical-shift mapping for the blaP half-operator was obtained by comparison of the 1H-1H NOESY recorded on the free DNA and the 2D filtered NOESY experiment recorded on the 1/1 [13C-15N BLBlaI-NTD]/[1/2OP1blaP] sample.
Docking procedure
Determination of the structure of molecular complexes from sparse NMR data is a difficult task, requiring the integration of local and long-range molecular plasticity (19). In order to allow the maximum available degrees of freedom we have determined the quaternary architecture of the BlaI repressor/DNA operator complex using a de novo approach starting from randomized coordinates. Experimental NOE collected on the 15N-13C BLBlaI-NTD free protein were used to fold the bound polypeptide (7). The use of these constraints is based on the observation that few chemical shifts change between the bound and free forms, showing that the fold of the two proteins is essentially the same. The 12 bp operator was constrained using distance restraints extracted from a canonical double-strand DNA. B-DNA standard angles (α, β, δ, γ, ε, ζ, ψ, υ0, υ1, υ2) were also included for each nucleotide to facilitate the DNA helix fitting.
Distances restraints extracted from intermolecular NOE were defined at 5 Å (one DNA proton correlated with one protein resonance). A thirdly set of structural data was introduced using chemical-shift mapping performed on the two molecules. Chemical-shift-derived distance restraints were created by combining ‘significant’ chemical-shifts variations identified on the polynucleotide 1H-1H NOESY with shifted residues in 15N-HSQC and 13C-methyl-selective-HSQC spectra, and including these constraints in an ambiguous manner (20,21). Distances inferred from these distance restraints were fixed at 10 Å in the docking simulation.
All calculations were performed using the program Discover with the AMBER4 force field (22). Simulated annealing was used to explore the conformational space for the structure determination and a restrained molecular dynamics calculation was used to refine each structure (23). Detailed initial conditions and physical characteristics of exploratory period have been reported previously (24).
RESULTS
Interaction between SAMecI and the DNA operators
The binding curves of SAMecI to its operator, OPmecA, and to the two β-lactamase operators, OP1blaP and OPblaZ, have been determined by band-shift assay. All show sigmoidal-binding curves as previously described for the interaction of BLBlaI repressor with its operators (9). So, as for BLBlaI, the binding parameters of SAMecI interaction include two equilibria and only the global dissociation constant Kd = Kd1. Kd2 can be obtained, where Kd1 and Kd2 are the dissociation constants of SAMecI dimer and SAMecI dimer-operator complex, respectively (Table 2).
Table 2.
Equilibrium parameters of the MecI-operator interactions
| Kd (10−15 M2) | Kd2(M) | |
|---|---|---|
| OPmecA | 1.7 ± 0.4 | 6.8 × 10−11 |
| OPblaZ | 1.4 ± 0.7 | 5.6 × 10−11 |
| OP1blaP | 4 ± 2 | 1.6 × 10−10 |
The values of Kd2 (Kd = Kd1.Kd2) were estimated by supposing that the value of Kd1 = 25 µM estimated for B. licheniformis BlaI dimer is the same for S. aureus MecI.
NMR titration of truncated monomeric NTD with cognate and crossed semi-operators
Titration of 15N-labeled BLBlaI or SAMecI truncated repressor with progressive amounts of unlabeled DNA half-dyads was performed by monitoring changes in 1H-15N HSQC spectra. Significant chemical-shift changes for correlation peaks in the 1H-15N HSQC spectra were observed when the 1/2OP1blaP and the 1/2OPmecA were added to protein samples. [DNA]/[Protein] molar ratios were varied from 0 to 10 for [BLBlaI-NTD]/[1/2OP1blaP], [BLBlaI-NTD]/[1/2OPmecA], [SAMecI-NTD]/[1/2OPmecA] and from 0 to 50 for [SAMecI-NTD]/[1/2OP1blaP]. The chemical-shift changes observed upon complexes formation reached a plateau over 7 [DNA]/[protein] molar ratios for [BLBlaI-NTD]/[1/2OP1blaP], [BLBlaI-NTD]/[1/2OPmecA], [SAMecI-NTD]/[1/2OPmecA] and a plateau over 30 for [SAMecI-NTD]/[1/2OP1blaP] (Figure 2). These results confirm the formation of stable intermolecular interactions with saturating DNA quantities. At any point during the titration, specific single correlation peaks were detected, suggesting that the truncated monomeric repressors/semi-operators complexes are in fast exchange regarding the NMR time scale. Only well-resolved correlation peaks with a chemical-shift cut-off equal or superior to 0.03 ppm were used in the four experiments. The affinity constants were calculated from a non-linear fit of the significant chemical-shift variations versus [DNA]/[protein] ratio using equation given by Morton et al. (25). Titration data were analyzed assuming that the observed chemical-shift perturbation is a weighted average between the two extreme values corresponding to the free (Δδ = 0) and the bound state (Δδ = Δδmax) so that:
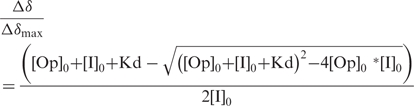 |
where [Op]0 and [I]0 are the total molar concentrations of DNA operator and protein. Statistical analysis using Monte-Carlo simulations were used to evaluate the uncertainty of the fitted parameters.
Figure 2.

Determination of dissociation constants using chemical shift titration obtained by NMR. Weighted sum of the 1H,15N chemical shift variation measured for the protein on 15N-HSQC is plotted as a function of the DNA/protein ratio for the different complexes. (A) B. licheniformis BlaI-NTD with the B. licheniformis 1/2-operator of the blaP gene and (B) with the S. aureus 1/2-operator of the mecAgene, (C) S. aureus MecI-NTD with the 1/2-operator of the mecA gene and (D) with the B. licheniformis 1/2-operator of the blaP gene. By fitting the curves, the different dissociation constants Kd were obtained: Kd1 [BLBlaI-NTD][1/2OP1blaP] = 190 ± 50 μM, Kd2 [BLBlaI-NTD][1/2OPmecA] = 170 ± 50 μM, Kd3 [SAMecI-NTD][1/2OP1blaP] = 860 ± 80 μM and Kd4 [SAMecI-NTD][1/2OPmecA] = 160 ± 60 μM.
Semi-operating sequences of the blaP and mecA genes titration curves of selected individual amino acid residues resulted in averaged binding constants of 190 ± 50 µM (Kd1), 170 ± 50 µM (Kd2) and 160 ± 60 µM for the [BLBlaI-NTD]/[1/2OP1blaP], the [BLBlaI-NTD]/[1/2OPmecA] and the [SAMecI-NTD]/[1/2OPmecA] complexes, respectively (Figure 2). For [SAMecI-NTD]/[1/2OP1blaP], the measured affinity constant reaches 860 ± 80 µM (Kd3).
NMR structural determination
Considering the low affinity constant measured previously, we decided to investigate the solution structure of the [BLBlaI-NTD]/[1/2OP1blaP] complex using a sparse data approach. Structure prediction using a docking approach remains difficult because of the number and the variety of parameters that should be taken into account. Recent advances in the field (21) take advantage of structural restraints from experimental interaction data (biochemical and/or biophysical) to determine the relative position of the molecular partners. Such approaches have also been applied to the determination of the quaternary structure of protein/DNA complexes (26). Our de novo docking protocol has been performed as follows.
To investigate the structure of the complex between the 13C/15N-labeled BLBlaI protein with the unlabeled blaP semi-operating sequence, we collected two filtered NOESY experiments. The 2D isotopically filtered NOESY performed in 2H2O allowed us to observe three NOE. To verify these constraints, we collected an additional 3D NOESY 13C HSQC experiment in H2O, resulting in one additional inter-molecular contact. These four correlations have been assigned ambiguously with a tolerance value of 0.05 ppm for the protein and 0.03 ppm for the DNA.
To complement the four NOE distance constraints, a chemical-shift mapping has been carried out for both macromolecular partners as follows. For the protein, we compared 1HN and 15N (backbone amide), and 1HC and 13C methyl chemical shifts assigned for the free and bound form of the molecules, measured in 1HN-15N and 1HC-13C methyl-selective and aromatic-selective HSQC, respectively. Indeed, methyl group and aromatic chemical-shifts perturbations were judged to be good additional probes to precisely localize the interaction site. Concerning the nucleic acids, only the sugar H1′, H2′/H2′′, H3′ and the base H6/H8 protons were used for chemical-shift mapping due to the overlap problems observed in the other spectral regions.
The chemical-shifts variations resulting from complex formation allowed us to make an inventory of each individual amino acid or nucleotide potentially involved in the interaction. We only considered chemical variations larger than 0.8 ppm for the weighted sum of the 1H and 13C, or 1H and 15N chemical-shift variations of the protein with respect to the gyro-magnetic ratios, and larger than 0.09 ppm for the proton chemical-shift variation measured for the blaP half operator. In both cases, peaks that disappeared were incorporated in the docking procedure. For the blaP DNA operator, the majority of perturbations concerned nucleotides in the vicinity of the T8ACA11/A8*TGT11* motif namely the thymines 5, 7, 8 and 6*, 9*, 11* on the complementary strand, the adenines 6, 9, 7*, 8* and 12* and the guanines 4 and 10* (nucleotides nomenclature is described in Table 1 legend). For the protein, larger perturbations are observed for the N-terminal part, the H2 and H3 helix and around the wing.
The structure calculation started from random coordinates of the entire system, and used 1513 experimental intra-molecular NOE of the free form BLBlaI-NTD protein and simulated distance restraints for a standard B-helix (613 intra-residue distances and 52 inter-residue distances). Classical dihedral angles for DNA subunits were also included, namely, 22 α, 24 β, 23 γ, 23 δ, 22 ε, 22 ζ, 24 χ, 24 υ2, 24 υ1 and 24 υ0. The use of experimental NOE from the free form of the molecule is based on the lack of drastic shift between 1H-15N HSQC of the unbound and the bound protein, indicating that no significant structural rearrangement occurs. NMR dihedral angles were also calculated using TALOS software and incorporated in the calculation. Four intermolecular NOE were associated with 11 and 24 additional ambiguous distance restraints proceeding from 13C methyl-selective HSQC experiments and from 15N HSQC spectra, respectively.
Structure of the complex
Ten lowest energy structures from 250 calculations of the [15N-13C BLBlaI-NTD]/[1/2OP1blaP] have been generated using a de novo driven docking (Figure 3). As expected, the global fold of the NTD repressor domain is not modified by the complex formation. Pairwise RMSD calculated on the NTD backbone atoms between the monomeric complexed BLBlaI structure and free BLBlaI-NTD, SAMecI/OPblaZ and SABlaI/OPblaZ is 1, 1.6 and 1.5 Å, respectively. The typical structural arrangements of the WHP family protein is conserved without violations in the structure calculation file. The three α-helices H1 (9–20), H2 (26–36), H3 (41–54) and the three stranded β-sheets S1 (23–25), S2 (57–62), S3 (65–70) are packed following the sequence H1–S1–H2–H3–W1–S2–S3. The wing motif W1 consists of a short loop (residues 63 and 64). Three-dimensional structures of the free and bound forms of the BLBlaI-NTD are very close except for the more dynamic residues located in the N- and C-terminals extremities of the protein, namely M1 to I4 and Y77 to S82, respectively. Moreover, it should be noticed that restricted conformational modification occurred for the residues of the Wing (G63 and R64) and for few residues surrounding this motif (E62, V65 and F66).
Figure 3.

Ten lowest energy structures obtained by de novo docking of the Bacillus licheniformis BlaI-NTD in interaction with the blaP semi-operating sequence. (A) Front view and (B) back view of the structure ribbon representation. Root mean square deviation has been calculated to 0.7Å for all heavy atoms of the 10 structures. The 12 bp DNA of blaP half operator are shown in orange. H3 helix is inserted into the major groove and the minor groove is overhung by the wing motif.
In the complex, the B-DNA form is conserved in the final lowest energy structures. The position of the bases is less well-defined at both ends of the molecule than in the central part, possibly due to a lack of conformational restraints. Concaveness observed for the complete operator for SABlaI and SAMecI is not observed for the half operator (Figure 4). However, alignment of our 12 bp operator with the common base of the full MecI operator shows a very similar conformation (Figure 4B). The absence of a detectable kink could be due to the reduced length of the half-operator used in the NMR study.
Figure 4.

Front view of the superposition of the structure of the B. licheniformis BlaI-NTD in interaction with the blaP semi-operating sequence and the S. aureus MecI dimer in complex with the blaP operator (1SAX). (A) Alignment of the two complexes using [BLBlaI-NTD]/[1/2OP1blaP] and one of the [NTD]/[1/2OP] of SAMecI/SAOPmecA. Secondary structures of the monomer BLBlaI-NTD conserved a similar position relative to the major and minor groove of the DNA. (B) Alignment of the two structures using only the blaP semi-operating sequence and the corresponding 12 bp DNA sequence of the SAOPmecA. Monomer BLBlaI-NTD has twisted by 30 degrees relative to the sequence of the operator.
To analyze the orientation of the protein relative to the DNA, we have aligned our structure with the two X-ray structures of SABlaI and SAMecI using a superposition of the conserved DNA sequence. Compared to the two crystallographic structures, the relative orientation of the monomeric protein with respect to the DNA helix shows a 30° rotation along the long axis of the DNA and a translation of 3 Å (Figure 4B). To compare the stability of the different protein/DNA complexes, analysis of the protein/DNA interaction using LIGPLOT software (27) is presented in Table 3. Details of the nominative protein/DNA contacts observed in our structure are presented in supplementary Figure S1.
Table 3.
Number of protein/DNA interactions detected using LIGPLOT software for the different dimeric complexes: S. aureus SAMecI/OPblaZ (1SAX), S. aureus SABlaI/OPblaZ (1SXD), S. aureus SAMecI/OPmecA (2D45) and for our monomeric B. licheniformis [BLBlaI-NTD]/[1/2OP1blaP]
| Secondary structure elements | SAMecI (1SAX) | SAB1aI (1XSD) | SAMecI (2D45) | BLBlaI-NTD low. ene. | BLBlaI-NTD Ensemble Average |
|---|---|---|---|---|---|
| N-TER | 2 | 0 | 0 | 1 | 0.8 |
| H1 | 1 | 0 | 1 | 1 | 0.6 |
| H2 | 1 | 0 | 0 | 3 | 2.3 |
| H3 | 6 (3) | 2 (1) | 6 (3) | 4 (3) | 6.7 (5.2) |
| WING | 2 | 0 | 2 | 2 | 1.7 |
| OTHER | 0 | 2 | 1 | 5 | 2.04 (0.4) |
| TOTAL | 12 (3) | 4 (1) | 10 (3) | 16 (3) | 14.5 (5.6) |
Number of interactions has been obtained for the NMR lowest energy structure. Average values are displayed for the 10 lowest energy structural ensemble. Values in parentheses correspond to the number of sequence specific H-bonds involving atoms of the DNA bases.
Two helices (H2 and H3), the wing motif and the N-terminal domain of the B. licheniformis BLBlaI-NTD have been reported to establish contacts with DNA. In our structure, the H3 helix (P41-K53) is deeply inserted in the DNA major groove, whereas the minor groove is close to the wing motif (G63-R64). For our NMR-based model, residue R64 of the wing motif (G63-R64) binds to the 3′ end of thymines 3* and 2*. Moreover, interaction is occasionally propagated for residues surrounding the wing motif i.e. F66 and H61 which make contacts with A6, T7 and C4*, T3* nucleotides, respectively. In the MecI/BlaI X-ray structures, the wing amino-acid F67 binds the DNA backbone in the opposite side to that observed in the NMR model (A7C and T8C equivalent to position G4 and T5 in our DNA sequence). As a consequence of the global rotation, contacts of the wing with the DNA are also rotated with respect to the ‘crystal wing’ position in SAMecI/OPbla.
The H3 helix residues (P41-K53) contact principally with nucleotides belonging to the T8ACA11/T8*GTA11* motif. T43, T46 and R50 privilege interactions with nucleotides T9*, G10* and T11*, respectively. Furthermore, K54 (and W39) anchors the position of H3 via interaction with A12* (and G10*). In this low affinity complex, Q45 plays a central role, forming hydrogen bonds network with nucleotides T7 and T8 instead of an interaction with conserved A6-T6* bases as observed for the SAMecI.
In our structure, H2 helix (T26-T36) also binds to the DNA via T26 and N27 that interact with A6. This interaction is well-conserved through different bacterial strains. E11 of the H1 helix and residue K3 of the N-terminal region may reinforce DNA recognition of this extremity. A parallel could be made with the A11 of the H1 residue of SAMecI which also contacts DNA.
DISCUSSION
Monomer pathway has a significant role in the repressor–DNA binding
In the past it has been assumed that in β-lactam resistance regulation system, B. licheniformis 749/I BlaI-WT interacts as a preformed dimer with its operator (15). The binding constants of the full-length SAMecI and BLBlaI repressors in interaction with palindromic operating sequences have been measured in the range of tens of nanomolar (9). Using chemical-shift mapping, we have determined affinity constants for the BLBlaI and SAMecI NTD with the different half DNA operators. For all the complexes, [SAMecI-NTD]/[1/2OPmecA], [SAMecI-NTD]/[1/2OP1blaP], [BLBlaI-NTD]/[1/2OP1blaP] and [BLBlaI-NTD]/[1/2OPmecA] binding constants are in the same range of hundreds of micromolar, that is, 100 times higher than for the dimeric repressors. The relatively low dimerization constant of 25 μM (9) observed for BLBlaI has suggested that both monomer and dimer pathways were possible. Now, taking into account our quantitative values of monomer-DNA dissociation constants and in vivo concentrations of BLBlaI, estimated to 2 µM (9), we can conclude that the monomer pathway contributes significantly to the BlaI repressor binding mechanisms in vivo.
In various Winged HTH dimeric repressor systems, a similar monomer–dimer equilibrium has been established (9,28). Two repression systems comparable to BlaI are known: LexA and Rep. The DNA binding constant of LexA-NTD (29) and Rep-NTD (30) are respectively about 1 and 20 µM, close to values determined for SAMecI-NTD and BLBlaI-NTD. Moreover, initial assays measuring the dimerization constant of the LexA repressor (29,31) reported a Kd of about 10–50 µM. The association constant of the full-length Rep repressor has been evaluated to 3–5 nM as well. When Rep monomers bind specific DNA sequences, the CTD is moved away, and stable repressor-operator contacts can be established. If the specificity is not high enough, the tail competes with DNA and prevents DNA/protein non-specific associations.
The monomer pathway could provide a method for rapid localization of the binding site involving sliding of the protein along the DNA. Theoretical considerations and experimental evidence of protein sliding along non-specific DNA sequences are now well documented (32–35). This kind of search could combine 1D sliding with 3D diffusion (36) driven by energetic differences for specific and non-specific DNA-binding. In our system, the localization speed of the correct DNA-binding site depends on the competition of the monomer pathway with the dimer pathway. Although strong cooperative effects in monomer association with DNA do not allow us to experimentally evaluate this process, we note that such a mechanism could play a role in this system. Monomers would be able to reach the adapted DNA motif using such diffusive processes, and this pre-recognition step would allow a simpler contact between the two CTD. This intermediate step might be useful for the protein to establish correct and strong contacts.
Corepression and adaptability
The presence of low affinity constants for the DNA binding of the monomeric form can be correlated with the adaptability of the repressors for different operators. For example, the dimeric LexA repressor tightly binds the different recA, uvrB, dinC and dinB DNA sequences. In the case of BlaI, the co-repression mechanism of homologous repressors in S. aureus has been demonstrated (37). The Kd values measured in this work demonstrate that the BLBlaI-NTD protein is able to bind to both cognate and crossed semi-operators in the same affinity range. Thus, it suggests that low affinity complex formation might be an essential intermediate relay in binding variable regulation sequences.
Contrary to BLBlaI-NTD with mecA half-operator, we have shown that the affinity for blaP semi-operating sequence is 10 times reduced for the SAMecI-NTD repressor. This parameter might corroborate observations performed on clinical S. aureus isolates (38,39). Indeed, it has been shown that the majority of oxacillin-resistant strains contain deleted or mutated mecI genes while blaI sequences remain intact. Thus, in S. aureus clinical isolates, the exclusive expression of MecI repressor appears to be a drawback in stress condition. Indeed, in this case, bacteria are unable to respond rapidly. The BlaI/BlaR1 system associated with the MecI/MecR1 machinery may enhance antibiotic resistance by improving capability to respond. Affinity of the monomer could reflect the capacity to form some intermediate states required during the induction process and the BlaI-NTD might play a crucial role thanks to its inter-operator adaptability.
The dimerization of the repressor on the DNA involves a tertiary rearrangement of the DNA–monomer complex
In the monomer pathway mechanism, a first monomer binds to the DNA, exhibiting the monomer-DNA conformation described in this work. This structure is indeed stabilized by a large number of protein–DNA interactions that are systematically present in the dimer–DNA interface (Table 3). Indeed, the comparison between the position of the monomeric and dimeric repressor on the DNA gives a 30° rotation. In this conformational change, the overall position of the protein on the DNA remains the same, that is the helix H3 stays in the main groove, but it shifts from one base (Figure 5). In this model, the energetically favorable monomer–DNA interface is destabilized upon the binding of another monomer molecule onto the DNA–monomer complex. However, it is easy to interpret the increase of the global affinity by considering the gain in enthalpy and entropy due to the presence of two DNA–monomer interfaces and an additional dimerization interface. We note that some energy is stored in the two monomer–DNA interfaces when a dimer–DNA complex is formed, that can be potentially released if the dimerization domain is destabilized.
Figure 5.

Superposition of the S. aureus MecI and the monomeric unit of the B. licheniformis BlaI repressor. (A) Side view of the dimeric S. aureus MecI DNA binding domain (in green and blue) in complex with the bla sequence (PDB ID in orange). The monomeric BLBlaI obtained by de novo docking (in red) has been superimposed to MecI using only the conserved DNA recognition motif. The B. licheniformis BlaI DNA binding domains (in red) and the arrows point out the 30° rotation which is necessary to superpose the two monomeric BlaI-NTD and the two MecI-NTD domains. (B) Side view of the S. aureus MecI repressor (ribbon representation) in interaction with the 25 base-pair bla operating sequence. The monomeric units are shown in different colors (light blue and green). (C) Orientation of the dimeric S. aureus MecI repressor where each SAMecI-NTD unit has been superimposed on the [BLBlaI-NTD]/[1/2OP1blaP] complex. The 30° rotation, described in panel A, to superimpose the SAMecI and the monomeric BLBlaI-NTD creates a complete disruption of the dimerization domains in the case of a complete rigid molecule. This twist can be considered as a starting point for more complex structural modification following the induction process.
A monomerization of the repressors can explain the derepression mechanism
The values of the monomer–DNA affinities measured in this work (hundreds of micromolar) and the concentrations of repressors measured in the cell (2 µM) (9) show that monomers are not able to repress the genes. In the presence of β-lactam antibiotics, the induction process modifies the affinity of the protein repressor for its cognate DNA sequence. The lower affinity measured for the monomer to its DNA-binding domain could then be sufficient to explain the derepression of the gene. However, intermediate steps driving the release mechanism are not clearly understood.
Biochemical investigations performed on the blaR1/blaI/blaZ β-lactamase regulation system (15) proposed that proteolysis drives the signal transduction. Indeed, in Staphylococci, the BlaR1 penicillin receptor is a membrane protein containing a zinc metalloprotease motif. Acylation of the sensor-transducer via penicillin binding triggers the signaling mechanism leading to bla genes transcription. This event has been proposed to be the result of the cleavage of the dimeric S. aureus BlaI repressor between residues N101 and F102. Two induction models have been proposed for the bla operator transcription under β-lactams stress conditions (12,14). The first takes into account the fact that a fourth gene, as blaR2 in bla strains, encodes a key protein necessary for the CTD accessibility improvement (5,6,13). The second considers that an inductor produced in the cytoplasm in stress conditions can modulate the quaternary blocked conformation of the regulations elements. In this way, the inductor would be able to act as a proteolysis enhancer like in the TetR system (40). To sum up, the lower affinity of the repressor during the induction process could be the result of either a proteolysis of the repressor, leading to monomerization, or a structural change of the dimer that would place the two monomers in an unfavorable conformation, leading to a dimer–DNA dissociation constant almost equal to a monomer–DNA dissociation constant.
The energetically most favorable monomer–DNA conformation could represent a possible intermediate state induced in the inactivation process
For both schemes, the structural study of the monomeric BLBlaI-NTD bound to its DNA gives some information on a possible intermediate state induced in the inactivation process, and sheds light on the structural role of the C-terminal domain in the DNA-binding affinity.
In both crystal structures of the SAMecI and SABlaI in interaction with the bla and mec DNA sequence (5,6), the cleavage site is not easily accessible, suggesting that conformational changes may be necessary. Furthermore, it has been observed that two forms, open and closed, could co-exist, allowing structural adaptation between mec and bla operators with different inter-N-terminal domain distances (10,41).
This structural flexibility may be permitted by the high dynamics observed for the dimerization part of the repressors (7). Here, considering the structural differences between the monomeric and dimeric interactions of BlaI with DNA, we propose a model where the dimer placed on DNA plays the role of a tense spring that can be released by a modification of the dimerization domains of the repressors. This tense spring is composed of a translational component similar to the one observed between the open and closed form (6) and a supplementary rotational contribution of 30°. During this release, the monomer repressor would slide 30° inside the major groove of the DNA until it reaches its equilibrium conformation (as described in this article). Figure 5C displays the relative position obtained for the two CTDs in case of a complete rigid structure and illustrates an incompatibility between the twist of the two NTDs and conservation of an intact dimerization domain. This twist displacement could in practice modify the accessibility of the cleavage site by involving a modification of the secondary structure arrangement in the core of CTD and could result in breakage of covalent bonds in this region. This structural modification would be responsible for an irreversible separation of the two helices of the dimerization domain, substantiating the possible existence of an inactivation-state DNA-binding domain conformation. In the MexR repressor study (42), two conformations have been observed as well. On one hand, the open state exhibits optimal DNA-binding domains interspaces, which privileges the association with the operator. On the other hand, a modification in between the DNA-binding domains avoids repressor stowage on DNA. Furthermore, this sliding mechanism might be an explanation for a potential repressor ability to adjust contact between the closed and open form.
Recently published results (43) underline the fact that mobility in the tertiary arrangement might be a useful requirement for thin regulation processes, even in eukaryotic systems. Indeed, the homeodomain of the transcription factor Pdx1 is able to bind a 15-bp DNA promoter (with a consensus binding site) and adopt two slightly different conformations in the same asymmetric unit. Both complexes differ by a 2.4° rotation. Additional modifications such as DNA curvature and N-terminal local adjustments have been also raised by the author.
Similarly, major structural rearrangements controlled by small molecules have been highlighted for other systems as hormone receptors (44). For the FadR transcriptional regulator (45), interaction with DNA is disrupted by the acyl-CoA binding. Dramatic structural rearrangements happen which leads the DNA recognition helices being separated by 7.2 Å. Another repressor inactivation process has been published concerning bacterial resistance to antibiotic. Indeed, the TetR repressor is released in presence of tetracycline. This molecule associated with Mg2+ plays the role of the inducer. Inducer binding generates structural changes in the CTD. Thus, a pendulum-like motion increases the separation of the attached DNA-binding domains (40) by 3 Å.
These models give strong support to the hypothesis that an inducer or a blaR2 product participates to the initiation of C-terminal remodeling and propagation to the N-terminal parts (46). In presence of the effector, specific and optimized contacts between the N-terminal part and the DNA could be re-established by increasing the interspacing between the TACA/TGTA motifs. The differences in the length of the interspaces between the recognition motifs could limit the DNA-binding domains adaptation quality of SAMecI-WT to bla operator.
To conclude, the weak affinities obtained for the NTD and the monomer–DNA structure demonstrate the crucial role played by the CTD in an optimal positioning of the N-terminal units. When a dimer–DNA complex is formed, sliding of the repressor on the DNA creates a tense spring, which can be released by the structural modifications produced by an inductor during the induction process. The monomer–DNA structure described here may then be seen as an intermediate state during the monomer pathway repression scheme, and also during induction after destabilization of the dimerization domain.
ACCESSION NUMBERS
Coordinates for the model of the complex have been deposited in the protein Data Bank (PDB, ID code 2P7C).
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
ACKNOWLEDGEMENTS
The authors thank Catherine Bougault and Pierre Gans for stimulating discussions and Dr Kevin Gardner at the UT Southwestern Medical Center in Dallas for providing scripts on the NMRView software. This work was supported by the ‘Fonds National de la Recherche Scientifique’ (FNRS), the ‘Communauté Française de Belgique’ (projet Tournesol) and the European Union FP6 Integrated Project EUR-INTAFAR (Project LSHM-CT-2004-512138) under the thematic priority Life Sciences, Genomics and Biotechnology for Health. Funding to pay the Open Access publication charges for this article was provided by EUR-INTAFAR.
Conflict of interest statement. None declared.
REFERENCES
- 1.Salerno A, Lampen J. Transcriptional analysis of beta-lactamase regulation in Bacillus licheniformis. J. Bacteriol. 1986;166:769–778. doi: 10.1128/jb.166.3.769-778.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Fuda CC, Fisher JF, Mobashery,S. Beta-lactam resistance in Staphylococcus aureus: the adaptive resistance of a plastic genome. Cell. Mol. Life Sci. 2005;62:2617–2633. doi: 10.1007/s00018-005-5148-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wittman V, Lin H, Wong H. Functional domains of the penicillinase repressor of Bacillus licheniformis. J. Bacteriol. 1993;175:7383–7390. doi: 10.1128/jb.175.22.7383-7390.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.García-Castellanos R, Marrero A, Mallorquí-Fernández G, Potempa J, Coll M, Gomis-Ruth F. Three-dimensional structure of MecI. Molecular basis for transcriptional regulation of staphylococcal methicillin resistance. J. Biol. Chem. 2003;278:39897–39905. doi: 10.1074/jbc.M307199200. [DOI] [PubMed] [Google Scholar]
- 5.García-Castellanos R, Mallorquí-Fernández G, Marrero A, Potempa J, Coll M, Gomis-Rüth F. On the transcriptional regulation of methicillin resistance: MecI repressor in complex with its operator. J. Biol. Chem. 2004;279:17888–17896. doi: 10.1074/jbc.M313123200. [DOI] [PubMed] [Google Scholar]
- 6.Safo M, Zhao Q, Ko T, Musayev F, Robinson H, Scarsdale N, Wang A, Archer G. Crystal structures of the BlaI repressor from Staphylococcus aureus and its complex with DNA: insights into transcriptional regulation of the bla and mec operons. J. Bacteriol. 2005;187:1833–1844. doi: 10.1128/JB.187.5.1833-1844.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Van Melckebeke H, Vreuls C, Gans P, Filée P, Llabres G, Joris B, Simorre J.-P. Solution structural study of BlaI: implications for the repression of genes involved in beta-lactam antibiotic resistance. J. Mol. Biol. 2003;333:711–720. doi: 10.1016/j.jmb.2003.09.005. [DOI] [PubMed] [Google Scholar]
- 8.Gregory PD, Lewis RA, Curnock SP, Dyke,K.G. Studies of the repressor (BlaI) of beta-lactamase synthesis in Staphylococcus aureus. Mol. Microbiol. 1997;24:1025–1037. doi: 10.1046/j.1365-2958.1997.4051770.x. [DOI] [PubMed] [Google Scholar]
- 9.Filée P, Vreuls C, Herman R, Thamm I, Aerts T, De Deyn P, Frère J, Joris B. Dimerization and DNA binding properties of the Bacillus licheniformis 749/I BlaI repressor. J. Biol. Chem. 2003;278:16482–16487. doi: 10.1074/jbc.M210887200. [DOI] [PubMed] [Google Scholar]
- 10.Sharma V, Hackbarth C, Dickinson T, Archer G. Interaction of native and mutant MecI repressors with sequences that regulate mecA, the gene encoding penicillin binding protein 2a in methicillin-resistant staphylococci. J. Bacteriol. 1998;180:2160–2166. doi: 10.1128/jb.180.8.2160-2166.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Filée P, Delmarcelle M, Thamm I, Joris B. Use of an ALFexpress DNA sequencer to analyze protein-nucleic acid interactions by band shift assay. Biotechniques. 2001;30:1044–1048. doi: 10.2144/01305rr03. [DOI] [PubMed] [Google Scholar]
- 12.Zhang H, Hackbarth C, Chansky K, Chambers H. A proteolytic transmembrane signaling pathway and resistance to beta-lactams in staphylococci. Science. 2001;291:1962–1965. doi: 10.1126/science.1055144. [DOI] [PubMed] [Google Scholar]
- 13.Cohen S, Sweeney HM. Constitutive penicillinase formation in Staphyloccocus aureus owing to a mutation unlinked to the penicillinase plasmid. J. Bacteriol. 1968;95:1368–1374. doi: 10.1128/jb.95.4.1368-1374.1968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Filée P, Benlafya K, Delmarcelle M, Moutzourelis G, Frère J, Brans A, Joris B. The fate of the BlaI repressor during the induction of the Bacillus licheniformis BlaP beta-lactamase. Mol. Microbiol. 2002;44:685–694. doi: 10.1046/j.1365-2958.2002.02888.x. [DOI] [PubMed] [Google Scholar]
- 15.Wittman V, Wong H. Regulation of the penicillinase genes of Bacillus licheniformis: interaction of the pen repressor with its operators. J. Bacteriol. 1988;170:3206–3212. doi: 10.1128/jb.170.7.3206-3212.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Clarke S, Dyke K. Studies of the operator region of the Staphylococcus aureus beta-lactamase operon. J. Antimicrob. Chemother. 2001;47:377–389. doi: 10.1093/jac/47.4.377. [DOI] [PubMed] [Google Scholar]
- 17.Johnson B. Using NMRView to visualize and analyze the NMR spectra of macromolecules. Methods Mol. Biol. 2004;278:313–352. doi: 10.1385/1-59259-809-9:313. [DOI] [PubMed] [Google Scholar]
- 18.Hilbers CW, Wijmenga SS. Encyclopedia of Nuclear Magnetic Resonance. 1996. Nucleic acids: spectra, structures and dynamics. John Wiley & Sons, Chichester, UK, pp.3346–3359. [Google Scholar]
- 19.Bonvin A. Flexible protein-protein docking. Curr. Opin. Struct. Biol. 2006;16:194–200. doi: 10.1016/j.sbi.2006.02.002. [DOI] [PubMed] [Google Scholar]
- 20.Nilges M. Calculation of protein structures with ambiguous distance restraints. Automated assignment of ambiguous NOE crosspeaks and disulphide connectivities. J. Mol. Biol. 1995;245:645–660. doi: 10.1006/jmbi.1994.0053. [DOI] [PubMed] [Google Scholar]
- 21.Dominguez C, Boelens R, Bonvin A. HADDOCK: a protein-protein docking approach based on biochemical or biophysical information. J. Am. Chem. Soc. 2003;125:1731–1737. doi: 10.1021/ja026939x. [DOI] [PubMed] [Google Scholar]
- 22.Pearlman D, Kollman P. Are time-averaged restraints necessary for nuclear magnetic resonance refinement? A model study for DNA. J. Mol. Biol. 1991;220:457–479. doi: 10.1016/0022-2836(91)90024-z. [DOI] [PubMed] [Google Scholar]
- 23.Blackledge M, Medvedeva S, Poncin M, Guerlesquin F, Bruschi M, Marion D. Structure and dynamics of ferrocytochrome c553 from Desulfovibrio vulgaris studied by NMR spectroscopy and restrained molecular dynamics. J. Mol. Biol. 1995;245:661–681. doi: 10.1006/jmbi.1994.0054. [DOI] [PubMed] [Google Scholar]
- 24.Cordier F, Caffrey M, Brutscher B, Cusanovich M, Marion D, Blackledge M. Solution structure, rotational diffusion anisotropy and local backbone dynamics of Rhodobacter capsulatus cytochrome c2. J. Mol. Biol. 1998;281:341–361. doi: 10.1006/jmbi.1998.1950. [DOI] [PubMed] [Google Scholar]
- 25.Morton C, Pugh D, Brown E, Kahmann J, Renzoni D, Campbell I. Solution structure and peptide binding of the SH3 domain from human Fyn. Structure. 1996;4:705–714. doi: 10.1016/s0969-2126(96)00076-7. [DOI] [PubMed] [Google Scholar]
- 26.van Dijk M, van Dijk A, Hsu V, Boelens R, Bonvin A. Information-driven protein-DNA docking using HADDOCK: it is a matter of flexibility. Nucleic Acids Res. 2006;34:3317–3325. doi: 10.1093/nar/gkl412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wallace A, Laskowski R, Thornton J. LIGPLOT: a program to generate schematic diagrams of protein-ligand interactions. Protein Eng. 1995;8:127–134. doi: 10.1093/protein/8.2.127. [DOI] [PubMed] [Google Scholar]
- 28.Mohana-Borges R, Pacheco A, Sousa F, Foguel D, Almeida D, Silva J. LexA repressor forms stable dimers in solution. The role of specific dna in tightening protein-protein interactions. J. Biol. Chem. 2000;275:4708–4712. doi: 10.1074/jbc.275.7.4708. [DOI] [PubMed] [Google Scholar]
- 29.Kim B, Little J. Dimerization of a specific DNA-binding protein on the DNA. Science. 1992;255:203–206. doi: 10.1126/science.1553548. [DOI] [PubMed] [Google Scholar]
- 30.Ilangovan U, Wojciak J, Connolly K, Clubb R. NMR structure and functional studies of the Mu repressor DNA-binding domain. Biochemistry. 1999;38:8367–8376. doi: 10.1021/bi990530b. [DOI] [PubMed] [Google Scholar]
- 31.Schnarr M, Pouyet J, Granger-Schnarr M, Daune M. Large-scale purification, oligomerization equilibria, and specific interaction of the LexA repressor of Escherichia coli. Biochemistry. 1985;24:2812–2818. doi: 10.1021/bi00332a032. [DOI] [PubMed] [Google Scholar]
- 32.Gowers D, Wilson G, Halford S. Measurement of the contributions of 1D and 3D pathways to the translocation of a protein along DNA. Proc. Natl Acad. Sci. USA. 2005;102:15883–15888. doi: 10.1073/pnas.0505378102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Iwahara J, Clore G. Detecting transient intermediates in macromolecular binding by paramagnetic NMR. Nature. 2006;440:1227–1230. doi: 10.1038/nature04673. [DOI] [PubMed] [Google Scholar]
- 34.Winter R, Berg O, von Hippel P. Diffusion-driven mechanisms of protein translocation on nucleic acids. 3. The Escherichia coli lac repressor–operator interaction: kinetic measurements and conclusions. Biochemistry. 1981;20:6961–6977. doi: 10.1021/bi00527a030. [DOI] [PubMed] [Google Scholar]
- 35.Winter R, von Hippel P. Diffusion-driven mechanisms of protein translocation on nucleic acids. 2. The Escherichia coli repressor–operator interaction: equilibrium measurements. Biochemistry. 1981;20:6948–6960. doi: 10.1021/bi00527a029. [DOI] [PubMed] [Google Scholar]
- 36.Slutsky M, Mirny L. Kinetics of protein-DNA interaction: facilitated target location in sequence-dependent potential. Biophys. J. 2004;87:4021–4035. doi: 10.1529/biophysj.104.050765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.McKinney T, Sharma V, Craig W, Archer G. Transcription of the gene mediating methicillin resistance in Staphylococcus aureus (mecA) is corepressed but not coinduced by cognate mecA and beta-lactamase regulators. J. Bacteriol. 2001;183:6862–6868. doi: 10.1128/JB.183.23.6862-6868.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rosato A, Kreiswirth B, Craig W, Eisner W, Climo M, Archer G. mecA-blaZ corepressors in clinical Staphylococcus aureus isolates. Antimicrob. Agents Chemother. 2003;47:1460–1463. doi: 10.1128/AAC.47.4.1460-1463.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rosato A, Craig W, Archer G. Quantitation of mecA transcription in oxacillin-resistant Staphylococcus aureus clinical isolates. J. Bacteriol. 2003;185:3446–3452. doi: 10.1128/JB.185.11.3446-3452.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Orth P, Schnappinger D, Hillen W, Saenger W, Hinrichs W. Structural basis of gene regulation by the tetracycline inducible Tet repressor-operator system. Nat. Struct. Biol. 2000;7:215–219. doi: 10.1038/73324. [DOI] [PubMed] [Google Scholar]
- 41.Clarke S, Dyke K. The signal transducer (BlaRI) and the repressor (BlaI) of the Staphylococcus aureus beta-lactamase operon are inducible. Microbiology. 2001;147:803–810. doi: 10.1099/00221287-147-4-803. [DOI] [PubMed] [Google Scholar]
- 42.Lim D, Poole K, Strynadka N. Crystal structure of the MexR repressor of the mexRAB-oprM multidrug efflux operon of Pseudomonas aeruginosa. J. Biol. Chem. 2002;277:29253–29259. doi: 10.1074/jbc.M111381200. [DOI] [PubMed] [Google Scholar]
- 43.Longo A, Guanga GP, Rose RB. Structural basis for induced fit mechanisms in DNA recognition by the Pdx1 homeodomain. Biochemistry. 2007;46:2948–2957. doi: 10.1021/bi060969l. [DOI] [PubMed] [Google Scholar]
- 44.He X, Chow D, Martick M, Garcia K, et al. Allosteric activation of a spring-loaded natriuretic peptide receptor dimer by hormone. Science. 2001;293:1657–1662. doi: 10.1126/science.1062246. [DOI] [PubMed] [Google Scholar]
- 45.van Aalten D, DiRusso C, Knudsen J. The structural basis of acyl coenzyme A-dependent regulation of the transcription factor FadR. EMBO J. 2001;20:2041–2050. doi: 10.1093/emboj/20.8.2041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Vreuls C, Filée P, Van Melckebeke H, Aerts T, De Deyn P, Llabrès G, Matagne A, Simorre J, Frère J, et al. Guanidinium chloride denaturation of the dimeric Bacillus licheniformis BlaI repressor highlights an independent domain unfolding pathway. Biochem. J. 2004;384:179. doi: 10.1042/BJ20040658. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}