


Abstract
The widespread utilization of gene silencing techniques, such as antisense, is impeded by the poor cellular delivery of oligonucleotides (ONs). Rational design of carriers for enhanced ON delivery demands a better understanding of the role of the vector on the extent and time course of antisense effects. The aim of this study is to understand the effects of polymer molecular weight (MW) and ON backbone chemistry on antisense activity. Complexes were prepared between branched polyethyleneimine (PEI) of various MWs and ONs of phosphodiester and phosphorothioate chemistries. We measured their physico-chemical properties and evaluated their ability to deliver ONs to cells, leading to an antisense response. Our key finding is that the antisense activity is not determined solely by PEI MW or by ON chemistry, but rather by the interplay of both factors. While the extent of target mRNA down-regulation was determined primarily by the polymer MW, dynamics were determined principally by the ON chemistry. Of particular importance is the strength of interactions between the carrier and the ON, which determines the rate at which the ONs are delivered intracellularly. We also present a mathematical model of the antisense process to highlight the importance of ON delivery to antisense down-regulation.
INTRODUCTION
Antisense technology provides a simple and elegant means to regulate gene expression, either for therapeutic purposes or for studying gene function (1–3). With one FDA approved drug and approximately 20 candidates in various stages of clinical trials, there is growing promise for the success of this approach (4,5). Additionally, the excitement generated by the advent of siRNA has rejuvenated interest in gene silencing technologies in general (6). Advances in oligonucleotide (ON) synthesis have now made it relatively simple to modify the chemistry of these molecules. This has enabled the creation of ONs with diverse properties and has thus greatly widened their overall utilization as specific mediators of gene silencing.
With the growing progress in functional genomics, there is an increasing need for the routine application of gene silencing tools such as antisense in basic research. Despite the conceptual simplicity, utilization of antisense as a routine tool for in vitro studies is greatly impaired by the poor cellular delivery of these molecules. Delivery of ONs in a stable form and relevant dose to the appropriate target site of action remains a considerable challenge to date (7,8). The reduced cellular entry and rapid degradation of these molecules in the presence of cellular nucleases calls for the utilization of carrier molecules. Various types of carriers, including polymers, lipids and peptides (9–13) of diverse chemistries, have been tested for their effectiveness as DNA delivery vectors. However, substantial optimization is generally required to make these carriers work for the particular application at hand. The large variations in vector effectiveness among cell types, as well as the relatively high cytotoxicity of the currently available carriers, continue to fuel the demand for more rationally designed carrier systems (14).
To improve further the design of DNA carriers, extensive research is being conducted to identify cellular barriers to carrier mediated delivery of ONs (15). Efforts are directed towards determining structure–property relationships that relate carrier properties to antisense effectiveness. However, it remains somewhat unclear what factors ultimately dictate the effects observed. Of particular importance is the fact that much of the design principles developed for plasmid DNA carriers are often used interchangeably for antisense ONs or siRNA. Although this may work in certain cases, the small size of ONs (10–22 bases) and variations in their backbone chemistry and structure endow them with unique properties that alter their interactions with carriers in comparison to plasmid DNA, and hence influence the design criteria for ON carriers. There are several examples in literature that point to such differences in terms of ON structure, ON chemistry and sequence composition (16–19). Hence, there is a need for systematic investigations of the interactions of carriers with ONs, both at the molecular level as well as at the cellular level, to acquire a mechanistic understanding of their cellular processing.
The goal of the present study is to understand the role of the vector, i.e. the properties of both the carrier and the ON, on the extent and dynamics of the antisense effect. For our study, we chose the cationic polymer, polyethyleneimine (PEI), as the model polymer. Our rationale for doing so is 2-fold. First, PEI is a well characterized carrier molecule (20). It has been utilized extensively for the delivery of plasmid DNA due to its high charge density and endosomolytic activity. Several reviews are available detailing the effectiveness and mechanism of action of PEI and its various modifications for the delivery of DNA to a range of cell types (21–23). There are fewer studies focusing specifically on the delivery of ONs using PEI. Second, we wish to determine the optimal PEI molecular weight (MW) for delivery of ONs of various chemistries. Specifically, we utilized five MWs of the branched form of the polymer, in combination with phosphodiester (PO) and phosphorothioate (PS) ON backbone chemistries. While a range of alternative chemistries is now available, PO and PS ONs are still utilized most often for in vitro work. We studied the polymer–ON interactions and further evaluated their efficacy in delivering active antisense ONs to cells. We demonstrate that the observed antisense effect is not determined solely by the carrier properties, but by the particular combination of polymer and ON properties. Of particular importance is the strength of interactions between the carrier and the ON, which determines the rate at which the ONs are delivered intracellularly.
METHODS
Materials
A 20-mer anti-GFP sequence identified previously (24,25) as an effective inhibitor of pd1EGFP expression (5′-TTG TGG CCG TTT ACG TCG CC -3′) and a scrambled control (5′- TTG CTT GTA CCG TGC GTG CC -3′) were utilized in the study. The phosphodiester, phosphorothioate and fluorescently tagged (5′ Cy5 end modified) forms of these sequences were obtained from Integrated DNA Technologies (Coralville, IA, USA). Stock solutions were prepared by reconstituting each pellet in water to a final concentration of 100 μM. Branched PEI of molecular weights 1.2 kDa (Cat# 6088), 10 kDa (Cat# 19850) and 70 kDa (Cat# 00618) was purchased from Polysciences, Inc. (Warrington, PA). Additionally, PEI of molecular weight 25 kDa (Cat# 408727) and 600–1000 kDa (Cat# 3880) was purchased from Sigma. Stock solutions at a concentration of 10 residue mM (0.43 mg/ml) were prepared in water and the pH adjusted to 7.0 using HCl. OliGreen, a fluorescent dye that binds strongly to single-stranded DNA, was obtained from Molecular Probes (Eugene, OR, USA). Heparin sodium salt was obtained from Sigma (Cat# H4784). Unless stated otherwise, all cell culture products were obtained from Invitrogen (Carlsbad, CA, USA).
PEI/ON complex formation
PEI/ON complexes were prepared at desired PEI/ON charge ratios by mixing equal volumes of PEI (of varying concentrations) and ONs in PBS. The samples were vortexed briefly, and the solutions were then incubated at room temperature for 10–15 min to ensure complex formation. Experimental evidence (stabilization of fluorescence corresponding to free ON) confirmed that this time was sufficient for complex formation. The PEI/ON charge ratios were calculated on a molar basis. The complexes were prepared at a final ON concentration of 10 μg/ml (approximately 1.64 μM) unless stated otherwise.
Detection of free ON using OliGreen
Complexes between PEI and ONs were prepared at various charge ratios as described above. One hundred microliters of each complex solution was transferred to a 96 well (black-walled, clear-bottom, non-adsorbing) plate (Corning, NY, USA). A total of 100 μl of diluted OliGreen reagent (1:100 in TE buffer at pH 8) was then added to all samples for free ON detection. Fluorescence measurements were made after a 3–5 min incubation using a Cytofluor (Applied Biosystems, CA, USA), at excitation and emission wavelengths of 485 and 520 nm, respectively, and a voltage gain of 55. All measurements were corrected for background fluorescence from a solution containing TE buffer and diluted OliGreen reagent.
Determination of particle size using dynamic light scattering
Complexes were prepared by mixing PEI and ON solutions (final ON concentration of 50 μg/ml, charge ratio of 10:1 in PBS), and immediately analyzed using a Brookhaven Particle Size Analyzer (Holtsville, NY, USA) for 15 min with readings taken at an interval of every 3 min. Measurements were performed in triplicate.
PEI/ON dissociation studies
PEI/ON complexes (charge ratio 10:1, final ON concentration of 5 μg/ml ∼ 0.8 μM, volume 50 μl) were prepared as described previously and transferred to a 96 well (black walled, clear bottom, nonadsorbing) plate. One hundred microliters of diluted OliGreen reagent were added to each well and mixed manually with a multichannel pipet. Fifty microliters of heparin solution (at various concentrations prepared in TE buffer at pH 8) were then added to the wells, and the plate was maintained at 37°C. Fluorescence measurements were made at the end of 1 h from the time of heparin addition using the Cytofluor plate reader. The percentage of ONs released was calculated from the fluorescence (F) values as
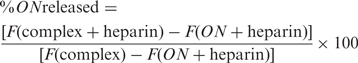 |
In each measurement, we corrected for changes in background fluorescence due to heparin addition alone. This change was found to be dependent on the heparin concentration (data not shown), and thus was measured and accounted for in each experiment.
Cell culture
Chinese hamster ovary (CHO-K1) cells (ATCC, Manassas, VA, USA) were maintained in F-12K medium (Kaighn's modification of Ham's F-12; ATCC, Manassas, VA) supplemented with 10% fetal bovine serum and penicillin–streptomycin solution. CHO-d1EGFP cells (CHO-K1 cells stably expressing a destabilized green fluorescent protein transgene) were previously produced by transfecting CHO-K1 cells with the 4.9 kb d1EGFP-N1 plasmid (BD Biosciences Clontech, Palo Alto, CA, USA), and maintained under constant selective pressure by G418 (500 μg/ml). All cell lines were cultivated in a humidified atmosphere containing 5% CO2 at 37°C.
Antisense experiments
CHO or CHO-d1EGFP cells were plated at a density of 1.5 × 105 cells/well in 12 well plates 18 h prior to transfection. Prior to introduction of ONs, cell culture medium in each well was replaced with 800 μl of OptiMEM(R) (reduced serum medium). Further, 200 μl of PEI/ON complex solution prepared in PBS was added to each well so the final ON concentration in each well (1 ml) was 300 nM. To measure the intracellular levels of ONs released from the complexes, cells were treated with complexes prepared with Cy5-ONs. At the end of the incubation period of 4 h, the transfection mixture was replaced with serum containing growth medium and maintained under normal growth conditions (5% CO2, humidified atmosphere, 37°C). Medium in the wells was replaced with fresh serum containing medium every 24 h. At various times, cells were assayed for antisense activity [green fluorescent protein (GFP) fluorescence] and/or ON levels (Cy5-ON fluorescence) by flow cytometry. As controls, cells were also exposed to only PEI (in the absence of ONs), only ONs (in the absence of PEI) as well as complexes prepared with scrambled ON sequences to check for sequence specificity. All times indicated are relative to when complexes were first introduced to the cells, which represents t = 0.
Flow cytometry
Replicate wells of cells were washed in PBS, detached with trypsin-EDTA, washed with serum-containing medium, pelleted by centrifugation for 5 min at 200 g, washed with PBS, resuspended in 500 μl of PBS and maintained on ice before being subjected to flow cytometry analysis. Ten thousand cells were analyzed on a FACSCalibur two-laser, four-color flow cytometer (BD Biosciences) for GFP fluorescence in FL1 (green) channel and Cy5-ON fluorescence in FL4 (far-red) channel. CellQuest software was used to acquire and analyze the results. Viable cells were gated according to their typical forward/side scatter characteristics. The flow cytometer was calibrated with fluorescent beads (CaliBRITE Beads, BD Biosciences) prior to each use to ensure comparable readings over the period of the study.
Mathematical model
We previously developed a detailed mathematical model describing the cellular events that an antisense ON undergoes in its attempt to reach and block its target in the cell (26). The model was based on mass action kinetic equations on the ON and took into account a set of compartments based on cellular location (e.g. cell membrane, endosome, cytoplasm) and molecular state (e.g. free, hybridized or degraded). Here, we present a simplified version that describes the intracellular delivery of ONs and the subsequent antisense response. The model highlights the significance of ON release from polymer/ON complexes to the overall nature of antisense down-regulation.
Transfer of ONs from outside the cell to the intracellular space is described as a single process. For the sake of simplicity, all intermediate steps such as cellular uptake, endosomal escape and release from PEI/ON complexes are lumped into this single process. Antisense ONs outside the cell and inside the cell are each assumed to degrade by a first-order process.
By solution of the governing mass-action kinetic equations (Appendix), the dynamics of intracellular ON levels (Ai) can be represented by
| 1 |
Experimental results (Cy5-ON fluorescence) were fit to Equation (1) using a non-linear equation solver, and the parameters α, β and γ were estimated. A combination of these parameters, γ(β − α), is equal to the initial rate of intracellular uptake/release of ONs (Appendix, Equation A4).
Antisense ONs released from PEI/ON complexes intracellularly are then capable of binding to the target mRNA to elicit an antisense response. We neglect all other events such as non-target interactions or protein binding. Further, we assume the ON-mRNA hybridization to be in rapid equilibrium, denoted by the equilibrium constant K. The total mRNA from the target gene can therefore be present in the unbound or hybridized form. It can be shown (Appendix) that, following perturbation of the steady-state with an antisense ON, the amounts of mRNA and protein, relative to steady-state, μ and π respectively, are given by
| 2 |
| 3 |
In these equations, the GFP mRNA and protein degradation rates (λ and δ respectively) utilized in the model were 0.069 and 0.69 h−1, respectively. Given the Ai(t) profiles separately measured and fit to Equation 1, only the equilibrium constant K remains as an adjustable parameter. Equations (2) and (3) were solved simultaneously for the objective of minimizing the error associated with K. A more detailed explanation of the methods employed to solve the model and fit its parameters is provided in the Appendix.
RESULTS
First, we tested the ability of PEI of molecular weights ranging from 1.2 K to 600 K to form complexes with ONs, as a function of the PEI/ON charge ratio. We measured the amounts of free (unbound) DNA in solution using a dye, OliGreen, which fluoresces upon binding to single-stranded DNA (9,16). As shown in Figure 1, the fluorescence levels decrease with increasing PEI/ON charge ratios. At higher charge ratios (⩾5 : 1 for PO ONs or ⩾2 : 1 for PS ONs), only residual amounts of free (unbound) ONs are detected in solution, indicating complex formation. All PEI MWs behave similarly in their ability to bind with ONs, except that complex formation is highly inefficient for PEI/PO complexes at the lowest MW PEI (1.2 K). In fact, even at the highest charge ratio (20 : 1), there is no significant binding between PEI (MW 1.2 K) and PO ONs. Therefore, PEI MW 1.2 K was excluded from all further studies. For all PEI MWs, the complexation curve is shifted to lower charge ratios for PS ONs relative to PO ONs, indicating a greater affinity for the PEI-PS ON interaction.
Figure 1.

Effect of PEI molecular weight on complex formation of PEI with (A) PO and (B) PS ONs. Complexes were prepared in PBS at a final ON concentration of 10 μg/ml. Fluorescence corresponds to free (unbound) DNA in solution, detected using a commercially available dye OliGreen that fluoresces upon binding to single-stranded DNA. Data represent mean ± SD (n = 3).
In order to characterize further the PEI/ON complexes, particle sizes were estimated in the form of mean hydrodynamic diameter using dynamic light scattering. Complexes prepared at various PEI/ON charge ratios in PBS were subjected to particle size measurements for 15 min at a regular interval of 3 min. For charge ratios below 10 : 1, complexes were found to aggregate, as indicated by the rapid increase in particle size (data not shown). Although the OliGreen binding assay indicated PEI-ON association at these charge ratios, stable, submicron sized complexes were formed only at a charge ratio of 10: 1 or above. At a charge ratio of 10 : 1, only 10 K/PO complexes displayed particle aggregation, with the particle diameter increasing from 200 to 450 nm within 15 min. Irrespective of the PEI MW and ON chemistry, all other particles were stable in the presence of salt and maintained a mean diameter of approximately 200 nm (Figure 2). All further studies were therefore performed at a charge ratio of 10 : 1.
Figure 2.

Mean hydrodynamic diameter (nm) of complexes (charge ratio 10: 1, final ON concentration 50 μg/ml in PBS) of PEI with (A) PO and (B) PS ONs. Data represent mean ± SD (n ≥ 3).
For effective cellular delivery, polycation–ON complexes should be of an appropriate strength to withstand encounters with other macromolecular species while entering the cell and during intracellular trafficking but also to dissociate (unpackage) the ON at some point to allow recognition of the target mRNA. We probed the strength of the PEI/ON interactions by studying the dissociation behavior of these complexes upon exposure to heparin sulfate as a competitive binding agent (16). Complexes were prepared at a PEI/ON charge ratio of 10 : 1, after which heparin was added to dissociate the complexes, releasing ONs into solution. OliGreen was used to measure the amount of ONs released, following subtraction due to the minor effect of heparin on OliGreen fluorescence. The dose response of ON release with varying amounts of heparin was determined, using release at the end of 1 h as the measurement. Increasing amounts of ONs were released with higher doses of heparin for PEI/ON complexes of both ON chemistries, with several notable characteristics (Figure 3). For PEI/PO complexes, the data revealed a threshold heparin concentration above which most of the ONs were released from the complexes. There was no significant effect of the PEI molecular weight on the release of PO ONs from PEI. On the other hand, PEI MW plays a more definitive role on PEI/PS interactions. Higher amounts of PS ONs were released with increasing PEI MW (Single factor analysis of variance (ANOVA) test, p = 0.0005, for PEI/PS complexes challenged with heparin at 20 μg/ml), indicating reduced strength of binding between PEI of higher MW and PS ONs. Furthermore, a comparison of the release profiles for the two ON chemistries indicates enhanced strength of binding (lesser release) between PEI and PS ONs as compared to PO ONs.
Figure 3.

Heparin dose response of the release of (A) PO and (B) PS ONs from PEI/ON complexes. Complexes (charge ratio 10:1, final ON concentration 5 μg/ml in PBS) were treated with heparin at various concentrations and maintained at 37°C for 1 h. ONs released from the complexes were detected using OliGreen as in Figure 1. Data represent mean ± SD (n = 3).
Having studied the physico-chemical properties of PEI/ON complexes, we tested the effectiveness of these polymers in delivering anti-d1EGFP ONs to CHO cells stably expressing the d1EGFP transgene (9). Cells were treated with complexes of PEI (various MWs) and ONs (PS and PO backbone chemistries) for 4 h, and subjected to flow cytometry at each of several times over a 72 h time period. Under all conditions, >90% of the cells were gated as live. The fluorescence levels indicated in Figure 4 are normalized to the green fluorescence of time-matched, untreated CHO cells that stably express the d1EGFP transgene.
Figure 4.

Dynamics of GFP down-regulation. CHO-d1EGFP cells were treated with PEI/Cy5-anti-GFP ON complexes prepared with (A) PO and (B) PS ONs at a final ON concentration of 300 nM for 4 h under serum-free conditions. At various times, 10 000 cells were analyzed for GFP fluorescence using flow cytometry. GFP fluorescence values are normalized to untreated control cells (100%). Data represent mean ± SD (n = 6).
Antisense ONs delivered with PEI produced transient reductions in average GFP levels, with as much as 80% reduction observed 8 h from when cells are treated with PEI/ON complexes under the best conditions. The time scale over which antisense effects were observed varied distinctly with the ON chemistry. As shown in Figure 4A, PO ONs delivered with PEI produced a rapid and brief response. GFP levels declined steeply, with as much as 60% of the fluorescence lost in the first 4 h, when ONs are delivered with PEI MW 25 K. The maximum down-regulation was observed close to the 8 h time point, shortly after which the GFP levels rise swiftly and return to base level by 48 h from administration. In comparison, PS ONs delivered with PEI exhibited a more gradual antisense response that was sustained for a longer duration (Figure 4B). The antisense effects were maximal between 8 and 24 h, and only gradually returned to base level over a 72 h time period. These differences in the onset of down-regulation between PO and PS ONs are more distinct at the earlier time points such as 4 h.
Whereas ON chemistry determined primarily the dynamics of antisense effects, the molecular weight of PEI used to deliver the ONs influenced strongly the extent of down-regulation observed over the same time scale. PO ONs delivered with intermediate MW PEI (25 K) produced the highest levels of GFP down-regulation, while lower levels of down-regulation were recorded when PO ODNs were delivered with all other MWs (10 K, 70 K and 600 K). Interestingly, for PS ONs, the effect of the carrier MW was more pronounced and quite different from that of PO. Almost no down-regulation was observed when PS ONs were delivered with the PEI of MW 10 or 25 K. In contrast, almost 80% inhibition was obtained when PS ONs were delivered with higher MW PEI (70 and 600 K).
A number of controls were utilized to evaluate the contribution of ON labeling dye, free polymer and ON sequence on GFP down-regulation (Figure 5). When cells were treated with ONs in the absence of PEI, no antisense effects (i.e. decrease in GFP expression) were observed, highlighting the need for a carrier. The carrier itself exhibited minimal non-specific effects. We also exposed cells to complexes of PEI and scrambled anti-GFP sequences to verify the specificity of the anti-GFP sequences. In order to segregate any false effects due to inefficient/incomplete ON delivery, we used the particular PEI MW that provided maximum intracellular ON levels for each of the backbone chemistries, i.e. MW 25 K for PO ONs and MW 70 K for PS ONs. In all cases, the anti-GFP sequences exhibited significantly greater down-regulation than the scrambled ones (Single factor ANOVA test, p < 0.01 for complexes of PEI with PO and PS ONs), providing evidence for reasonable sequence specificity of antisense inhibition.
Figure 5.

Controls for antisense experiment are described in Figure 4. Data represent treatment with single PEI MW (that delivered maximum PO or PS ONs), and at a single time point (at which maximum antisense inhibition was detected). The abbreviation ‘sc’ denotes scrambled sequence control, while ‘Cy-’ denotes ON was tagged at 5′ end with Cy5 dye. Data represent mean ± SD (n = 3).
In general, utilization of the d1EGFP transgene as the antisense target provides a simple means to capture the antisense down-regulation by measurement of GFP fluorescence (9,24,25). To detect simultaneously the presence of delivered, intracellular ONs, fluorescently (Cy5) end-tagged ONs were utilized in our experiments. Statistically indistinguishable levels of antisense inhibition were observed with dye-labeled vs. unlabeled ONs (Single factor ANOVA test, p > 0.05, for complexes of PEI with anti-GFP PO ONs, and for PEI with anti-GFP PS ONs), indicating that the label did not appreciably alter delivery or antisense behavior (Figure 5). Because Cy5 tagged ONs complexed to PEI do not fluoresce (data not shown), the ON fluorescence measured by flow cytometry corresponds to ONs released from PEI within cells. Absolute fluorescence levels are indicated in Figure 6 and are representative of data from several runs. The levels of intracellular PO ONs increase rapidly to a maximum at around 4 h after exposure (Figure 6A). PO ONs also disappear quickly from cells with negligible amounts detected after 48 h. Maximum levels of intracellular PO ONs are detected when delivered using the intermediate PEI MW of 25 K. In contrast, the levels of intracellular PS ONs delivered with PEI increase slowly, reaching a maximum between 8 and 24 h after treatment (Figure 6B). PS ONs are retained within cells for a longer duration, being cleared only after 72 h. It was interesting to note that very little ON was detected intracellularly when PEI of lower MW such as 10 K and 25 K were used as carriers for PS ONs. Higher MW PEI (70 K, 600 K) delivered the highest amounts of intracellular PS ONs.
Figure 6.

Dynamics of intracellular ON levels. CHO-d1EGFP cells were treated as described in Figure 4 with PEI/Cy5-anti-GFP ON complexes prepared with (A) PO and (B) PS ONs. Data represents raw geometric mean fluorescence levels of intracellular Cy5-tagged ONs captured simultaneously by flow cytometry in a separate channel, while cells are analyzed for their GFP fluorescence levels.
To quantify the relationship implied by Figures 4 and 6 between intracellular ON release and antisense inhibition, we integrated these measurements using a mathematical model based on mass-action kinetics. By fitting the results from the ON uptake experiments to a simple lumped uptake/release process with intracellular and extracellular degradation (Appendix, Figure A1(a) and Equation A3), we simulated the dynamics of intracellular ON levels shown in Figure 7A and B. A comparison of the model fit to the experimental results in Figure 6A and B shows that this model captures the dynamics observed in the intracellular release levels. Using a combination of parameters obtained from the model fit, we calculated the initial intracellular ON release rates (k1Ae0) for various PEI/ON complexes (Table 1). This one parameter captures the observed PEI MW and ON backbone dependences. Maximum ON release rates were obtained with intermediate PEI MW (25 K) for PO ONs, while the higher PEI MW (70K and 600K) release more PS ONs. This parameter also shows quantitatively that the initial uptake and release of PO ONs exceeds that of PS ONs.
Figure 7.

Model predictions of the dynamics of intracellular ON concentrations (A and B) and GFP down-regulation (C and D) in cells treated with (A and C) PEI/PO and (B and D) PEI/PS complexes, respectively.
Table 1.
Maximum intracellular ON release rates (arbitrary units) from PEI/ON complexes, computed using the fit model parameters (Equation 1)
| PEI MW | ON chemistry | |
|---|---|---|
| PO | PS | |
| 10 K | 111.6 | 15.1 |
| 25 K | 365.9 | 20.6 |
| 70 K | 273.7 | 59.4 |
| 600 K | 33.3 | 86.8 |
Using a mass action kinetic description of ON-mRNA hybridization (Appendix, Figure A1(b)) and coupling it to a description of translation (Appendix, Figure A1(c)), we also modeled the overall antisense dynamics. Estimates of GFP mRNA half-life from literature vary from 3–12 h (27–30), from which we utilized a half-life of 10 h for our calculations. The protein half-life was estimated as 1 h, which is the nominal value for the destabilized version of the enhanced d1EGFP. By incorporating these half-life estimates and the parameters from the ON uptake model, a single value of the ON-mRNA equilibrium constant (0.004 and 0.0084 for PO and PS ONs, respectively) was fit for each ON chemistry across all PEI MWs. As shown in Figure 7c and d, the GFP down-regulation data (Figure 4A and B) were captured reasonably well by the model. Although the maximum down-regulation is somewhat under-predicted, the model predicts accurately the trends with respect to PEI MW and the overall time scale of effects for each of the ON chemistries, in particular the delayed onset and sustained activities of PS versus PO backbones ONs.
DISCUSSION
PEI has garnered significant attention in recent times as a building block for creating effective DNA carriers and has been tested both in vitro and in vivo to target a large number of cell types (14,21,31–35). Several modifications have been proposed to take advantage of the unique buffering capabilities of this molecule, while reducing the toxicity associated with its use (36,37). However, most of these reports focus on delivering plasmid DNA for gene therapy applications. There are strikingly fewer systematic studies on the utilization of PEI for single-stranded DNA molecules. Insights obtained from PEI mediated plasmid DNA studies are generally extended for the application of delivering small DNA such as antisense ONs. However, as noted by us and other researchers (18,31,38), these generalizations may not hold, especially given the considerable differences in the size, structure and chemistry between ONs and plasmid DNA. In fact, these small DNA molecules (10–20 bases) are often observed to exhibit weaker electrostatic complexation with polycations due to the small number of charged units per ON molecules. While the linear form of PEI of MW 25 K is often touted as the most effective carrier for delivering plasmid DNA, we found it ineffective for both PO and PS ONs (data not shown). Hence, to specifically identify issues related to PEI mediated ON delivery, we performed a systematic study with a set of branched PEI MWs and ON chemistries.
Using the d1EGFP gene as an easily quantifiable antisense target, we screened various combinations of PEI MWs (1.2 K, 10 K, 25 K, 70 K and 600 K) and ON chemistries (PO and PS) for their ability to elicit an effective antisense response. For PO ONs, maximum antisense response was observed with intermediate MW PEI (25 K) as the carrier, while complexes of PS ONs with higher MW PEI such as 70 K and 600 K produced comparable levels of d1EGFP down-regulation. These particular PEI/ON combinations that achieved highest antisense response were also the ones that delivered the most ONs, i.e. maximum intracellular ON levels were recorded for these cases. Indeed, a monotonic relationship is apparent when the maximum antisense inhibition is plotted against the maximum ONs delivered for each combination of PEI MW and ON chemistry (Figure 8). Conditions under which no antisense inhibition was observed occurred because no ONs were delivered using those particular PEI/ON combinations. A correlation between intracellular levels of short interfering RNA and gene silencing has also been reported (39).
Figure 8.

Relationship between antisense inhibition and intracellular ON levels. Each data point represents maximum down-regulation and maximum ON levels detected when the indicated PEI MW was used to deliver (A) PO and (B) PS ONs. For both sets, results correspond to data obtained 8 h after PEI/ON complexes were introduced to CHO-d1EGFP cells.
Why do different combinations of PEI MW and ON chemistry deliver different ON levels? Studies of various PEI MWs for delivery of plasmid DNA do not provide a clear view, as they have produced conflicting trends. In some cases, transfection efficiency was found to increase with PEI MW (40), while in others low MW PEI was effective as a gene delivery agent (35). The toxicity associated with very high MW PEI offsets its use as DNA carrier (41), making the less toxic low MW PEI a more attractive candidate for further improvement despite its lower net charge density. To date, very little mechanistic explanation for the differences in behavior of various MWs has been provided. Previous investigations of the cellular processing of PEI/DNA complexes suggests that complexes are taken up by binding with proteoglycans, such as syndecan, and further trafficked through the endocytic pathway (34,42). The similar size of all our complexes (200 nm) suggests a low probability of differences in the rate of internalization of the complexes. PEI is believed to enable escape of polyplexes from the endolysosomal pathway by a ‘proton-sponge’ effect, by which complexes and/or DNA are released into the cytoplasm (22,43). It is not clear where or how the DNA is ultimately released from the complex. Previous studies demonstrate no significant differences in the buffering capacity of PEI of various MWs in the relevant pH range of 5–7 (20). Some studies suggest the involvement of cytoplasmic proteins and other charged molecules in the competitive release of DNA from PEI/DNA complexes (44). Therefore, we hypothesize that at least part of the MW backbone effects could be due to the kinetics of complex dissociation (unpackaging) and that these could be evaluated in vitro using the heparin competition assay.
Indeed, the differences in PEI–ON interactions are reflected in the heparin competition assay, where we observe a MW dependence on the amount of ONs released from PEI/PS ON complexes. Specifically, in the presence of heparin, complexes made with higher MW release more PS ONs than complexes made with lower MW PEI. However, when we tested the strength of the PEI–PO ON interactions by competition with heparin, we did not find any significant differences among the various PEI MWs. As such, it appears that the MW influences delivery of PO ONs in a manner not captured by the heparin competition assay, perhaps determining where in the cell (e.g. endosome versus cytoplasm) the DNA gets released. Competition with heparin has been used in previous studies as a measure of the stability of carrier/DNA complexes. Our studies suggest that higher stability may correspond to very tight binding that inhibits the release of DNA from the complexes. There are several such examples in the literature, wherein polymers that bind DNA effectively do not release them intracellularly despite their buffering capabilities (36,45). Our results provide some insights for such observations and additionally suggest interplay of polymer architecture and ON chemistry in complex stability versus dissociation. Such differences in carrier performance based on ON chemistry have been observed by others (19). Similar to Dheur et al. (46), we found PEI 25 K ineffective for PS ONs, but in addition, we find MWs higher than 25 K (such as 70 K and 600 K) to be efficient in delivering PS ONs.
Apart from identifying higher PEI MWs as effective carriers for PS ONs, our results more significantly highlight the importance of the strength of the electrostatic interactions between the PEI and ONs, which ultimately dominates the rate and extent of DNA release. Both the PEI architecture and ON chemistry play a role in these interactions. The degree of protonation and the flexibility of the polymer chains are speculated to be significant. For example, fractured dendrimers that have more flexible chains are better at delivering plasmid DNA as compared to intact dendrimers (47). In our system, higher MW PEI probably possesses more flexible chains, which are able to interact with the heparin (in our non-cellular assay) and with unspecified species in cells, leading to release of ONs. The role of the ON backbone is somewhat less clear. Both ON chemistries are known to have similar charge densities due to the phosphate groups; however, phosphorothioates are known to be more hydrophobic than phosphodiesters. The PO and PS backbones differ only by a single atom: the non-bridging oxygen is replaced by sulfur in the PS backbone. Compared to oxygen, the sulfur atom has less electronegativity. Studies report that the lower charge density of the sulfur atom increases its polarizability, strengthening the interaction with lower charge density groups in proteins (48,49). This was reflected in our PEI–ON binding assay, in which PS ONs were found to bind to even the lowest MW of PEI (1.2 K). In contrast, 1.2 K PEI and PO ONs did not bind even at very high charge ratios. Furthermore, PEI–PS ONs displayed higher strength of interactions in the heparin competition assay. The manifestation of these molecular interactions on the cellular processing of the vectors is indeed quite dramatic, in that some complexes are apparently so tightly bound that they are incapable of releasing the ONs. As these interactions can be modulated by appropriate modifications to carrier and ON properties, they present a design opportunity for producing an effective antisense response.
We also observed distinctly different antisense dynamics dependent on ON chemistry. The overall antisense dynamics consists of two phases: (i) the initial onset of gene inhibition leading to maximum down-regulation and (ii) the duration for which the antisense effects last. The latter can be easily explained based on the known differences in the resistance of PO and PS ONs to nucleases. The rapid disappearance of PO effects is consistent with the susceptibility of PO ONs to intracellular nucleases. PS ONs are known to be more nuclease resistant and to survive in the cell for longer (50), leading to sustained antisense effects, which lasted for up to 72 h in our studies.
Based on our previous detailed kinetic modeling (26), we would expect, for equivalent delivery characteristics, substantially less silencing of gene expression for PO versus PS ONs based on the higher nuclease degradation rate of the former. The fact that we observed significant, albeit short-lived, silencing with some PO ONs suggests that they are being delivered to the cytoplasm at a faster rate than their PS counterparts. The dynamics of intracellular release and gene silencing support this view. For PO ONs, the onset of gene inhibition was more rapid with maximum inhibition observed at 8 h (Figure 4A). For PS ONs, the onset was somewhat slower, and antisense activity was greatest between 8 to 24 h from when cells were first exposed to PEI/ON complexes (Figure 4B). A more rapid intracellular release was also observed with the PO ONs (Figure 6), although the intracellular release measurements should be considered under the caveat that the measured intracellular fluorescence may include contributions from partially degraded ONs and free fluorescent tag in addition to delivered, fully intact ON. Overall, the results suggest that PEI/PS complexes take longer to release ONs. Our in vitro heparin competition assay results highlight these differences in strengths between PEI and the ONs of PO and PS chemistries (Figure 3). These results suggest the possibility that a mixture of PO and PS chemistries would possibly demonstrate both early and sustained antisense activity (51). We are currently testing this hypothesis by using PO backbones with varying degree of PS substitution.
Because of the rapidly changing levels of intracellular Cy5-ON fluorescence and d1EGFP fluorescence at early times and the discrete time points at which fluorescence in measured, it can be difficult to discern the differences in dynamics among some of the samples. Our mathematical model of the overall antisense process provides a useful framework for interpreting these measurements. By fitting experimental results to a relatively simple function, we were able to simulate the intracellular ON levels and calculate intracellular ON release rates from various PEI/ON complexes. The release rates account quantitatively for the variation in activity as a function of PEI MW and also demonstrate a markedly increased release rate for PO vs. PS ONs (Table 1). To capture the antisense dynamics, we simplified the overall antisense process by incorporating only critical features such as equilibrium ON-mRNA hybridization. The fact that, with the parameters calculated from the intracellular ON function and a single globally optimized parameter (ON-mRNA binding constant), we can fit all of the activity data with fidelity of the trends, is a further indication that intracellular release governs the activity to a considerable extent. Mechanistically this implies that downstream barriers such as trafficking, nucleo-cytoplasmic shuttling, association with proteins, and binding to non-target mRNA do not seem to be as significant in governing the dynamics and MW effects, though they could still be important in a manner that is not dependent on PEI MW. A similar approach has been used to identify rate-limiting steps in siRNA gene silencing in vitro and in vivo (52).
Overall, the results presented here have important implications for the rational design of polymeric carriers and ON backbones used for antisense applications. We have demonstrated that the final antisense activity observed is determined not solely by either carrier or ON chemistry, but rather by the interplay of both factors. While the extent of down-regulation was determined primarily by the polymer MW, the dynamics were determined chiefly by the ON chemistry. Of particular importance is the strength of interaction between the carrier and the ON, which determines the rate at which the ONs are released intracellularly. From a practical standpoint, our results identify PEI MWs that are effective for delivering ONs of PO and PS chemistries. This approach should be useful in predicting and interpreting results for ONs of other chemistries as well, though it will be interesting to see to what extent the observed trends and correlations hold in other cell types. While PEI MW 25 K is generally considered the golden standard for plasmid DNA delivery, this is not true for the delivery of ONs. More strikingly, we find that the performance of a particular PEI MW is determined by the chemistry of the ON it is used to deliver. For example, PEI 25K was most effective in delivering PO ONs, but no PS ONs could be delivered with the same carrier. Modifications of polymers with targeting molecules such as ligands or PEG could affect the interactions with the ONs and hence influence the overall ON release dynamics. Thus, one should be able to control the onset and duration of antisense activity via biophysically guided selection of ONs and carriers.
Supplementary Material
ACKNOWLEDGEMENTS
We are grateful for financial support from the NIH (R01 GM65913) and Charles & Johanna Busch Memorial Fund. We also thank Zhuting Li, Neha Shah, Sandra Viriyayuthakorn, Jennifer Oddo, Mark Hwang and Lavanya Peddada for technical assistance. Funding to pay the Open Access publication charge was provided by NIH Grant R01 GM 65913.
Conflict of interest statement. None declared.
APPENDIX
Details of and solutions to the mathematical model
Figure A1.

Schematic representing processes taken into consideration for modeling (a) intracellular antisense ON levels, Ai, (b) mRNA levels, M and (c) protein levels, P.
Modeling of intracellular ON levels
Transfer of ONs from outside the cell (Ae) to the intracellular space (Ai) is described as a single process, represented by the first order kinetic rate constant k1. For the sake of simplicity, all intermediate steps such as cellular uptake, endosomal escape and release from PEI/ON complexes are lumped into this single process. Extracellular and intracellular ONs are assumed to each degrade by a first order process represented by the kinetic rate constants, kde and kdi respectively.
The differential equations describing the mass balances of extracellular and intracellular ONs are as follows:
| (A.1) |
| (A.2) |
Solving these differential equations subject to the initial conditions Ae(0) = Ae0 and Ai(0) = 0, we obtain
| (A.3) |
where
| (A.4) |
The combination of parameters γ(β − α) = k1Ae0 therefore represents the maximum rate of ONs released intracellularly. We fit our experimental data to Equation (A.3) using the fsolve function in MATLAB, and thus obtain best-fit estimates for α, β and γ for each combination of PEI MW and ON chemistry used.
Modeling of antisense activity
Antisense ONs released from PEI/ON complexes intracellularly (Ai) are then capable of binding to the mRNA (M) to elicit an antisense response. We neglect all other events such as non-target interactions or protein binding. Further, we assume the ON-mRNA hybridization to be in rapid equilibrium, denoted by the equilibrium constant K. The total mRNA (MT) from the target gene can therefore be present in the unbound form (M) or hybridized form (H). The constant Φ represents the rate of synthesis of mRNA, while the degradation of mRNA is described by a first order kinetic constant, λ. We assume that only unbound mRNA can be degraded. Similarly the synthesis and degradation of the protein (P) are represented by the first order kinetic rate constants ψ and δ, respectively.
The differential equations describing the mRNA and protein levels are:
| (A.5) |
| (A.6) |
The mass balance on the mRNA is:
| (A.7) |
Definition of the equilibrium constant K gives
| (A.8) |
Using (A.7) and (A.8), we obtain an expression for the fraction of unbound mRNA capable of translation into protein as,
| (A.9) |
Solving Equations (A.5) and (A.6) under steady-state conditions, we obtain the steady-state mRNA and protein levels:
| (A.10) |
Using these steady-state values, we can non-dimensionalize the equations as follows:
| (A.11) |
Combining with Equation (A.9), Equations (A.5) and (A.6) can be represented in dimensionless form as:
| (A.12) |
| (A.13) |
Incorporating Equation (A.3) into Equations (A.12) and (A.13) reduces these equations to a three parameter form. Estimates for λ (0.069 h−1) and δ (0.69 h−1), which denote the degradation rates of d1EGFP mRNA and protein respectively, were obtained from the literature. Our strategy for obtaining the dynamics of antisense activity was as follows: using a guess value for K, we simulated the dynamics of μ and π by solving the differential Equations (A.12) and (A.13) simultaneously using an ODE solver (ode15s) in MATLAB. By comparing these simulated dynamics to the experimental results, we computed the error at each data point. The overall error was calculated as the SSE (sum of squares of error) over all experimental time points for all PEI MW for each of the ON chemistries separately. A new K value was guessed, and the procedure was repeated until a minimum was achieved for the global error.
REFERENCES
- 1.Lee LK, Roth CM. Antisense technology in molecular and cellular bioengineering. Curr. Opin. Biotechnol. 2003;14:505–511. doi: 10.1016/s0958-1669(03)00115-0. [DOI] [PubMed] [Google Scholar]
- 2.Dias N, Stein CA. Antisense oligonucleotides: basic concepts and mechanisms. Mol. Cancer Ther. 2002;1:347–355. [PubMed] [Google Scholar]
- 3.Opalinska JB, Gewirtz AMA. Nucleic-acid therapeutics: basic principles and recent applications. Nat. Rev. Drug Discov. 2002;1:503–514. doi: 10.1038/nrd837. [DOI] [PubMed] [Google Scholar]
- 4.Gleave ME, Monia BP. Antisense therapy for cancer. Nat. Rev. Cancer. 2005;5:468–479. doi: 10.1038/nrc1631. [DOI] [PubMed] [Google Scholar]
- 5.Dean NM, Bennett CF. Antisense oligonucleotide-based therapeutics for cancer. Oncogene. 2003;22:9087–9096. doi: 10.1038/sj.onc.1207231. [DOI] [PubMed] [Google Scholar]
- 6.Dallas A, Vlassov AV. RNAi: a novel antisense technology and its therapeutic potential. Med. Sci. Monit. 2006;12:RA67–RA74. [PubMed] [Google Scholar]
- 7.Lebedeva I, Benimetskaya L, Stein CA, Vilenchik MA. Cellular delivery of antisense oligonucleotides. Eur. J. Pharm. Biopharm. 2000;50:101–119. doi: 10.1016/s0939-6411(00)00088-6. [DOI] [PubMed] [Google Scholar]
- 8.Shi F, Hoekstra D. Effective intracellular delivery of oligonucleotides in order to make sense of antisense. J. Control. Release. 2004;97:189–209. doi: 10.1016/j.jconrel.2004.03.016. [DOI] [PubMed] [Google Scholar]
- 9.Lee LK, Williams CL, Devore D, Roth CM. Poly(propylacrylic acid) enhances cationic lipid-mediated delivery of antisense oligonucleotides. Biomacromolecules. 2006;7:1502–1508. doi: 10.1021/bm060114o. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zatsepin TS, Turner JJ, Oretskaya TS, Gait MJ. Conjugates of oligonucleotides and analogues with cell penetrating peptides as gene silencing agents. Curr. Pharm. Des. 2005;11:3639–3654. doi: 10.2174/138161205774580769. [DOI] [PubMed] [Google Scholar]
- 11.Oishi M, Hayama T, Akiyama Y, Takae S, Harada A, Yamasaki Y, Nagatsugi F, Sasaki S, Nagasaki Y, et al. Supramolecular assemblies for the cytoplasmic delivery of antisense oligodeoxynucleotide: polyion complex (PIC) micelles based on poly(ethylene glycol)-SS-oligodeoxynucleotide conjugate. Biomacromolecules. 2005;6:2449–2454. doi: 10.1021/bm050370l. [DOI] [PubMed] [Google Scholar]
- 12.Junghans M, Loitsch SM, Steiniger SC, Kreuter J, Zimmer A. Cationic lipid-protamine-DNA (LPD) complexes for delivery of antisense c-myc oligonucleotides. Eur. J. Pharm. Biopharm. 2005;60:287–294. doi: 10.1016/j.ejpb.2005.01.006. [DOI] [PubMed] [Google Scholar]
- 13.Davis ME, Pun SH, Bellocq NC, Reineke TM, Popielarski SR, Mishra S, Heidel JD. Self-assembling nucleic acid delivery vehicles via linear, water-soluble, cyclodextrin-containing polymers. Curr. Med. Chem. 2004;11:179–197. doi: 10.2174/0929867043456179. [DOI] [PubMed] [Google Scholar]
- 14.Gebhart CL, Kabanov AV. Evaluation of polyplexes as gene transfer agents. J. Control. Release. 2001;73:401–416. doi: 10.1016/s0168-3659(01)00357-1. [DOI] [PubMed] [Google Scholar]
- 15.Roth CM, Sundaram S. Engineering synthetic vectors for improved DNA delivery: insights from intracellular pathways. Annu. Rev. Biomed. Eng. 2004;6:397–426. doi: 10.1146/annurev.bioeng.6.040803.140203. [DOI] [PubMed] [Google Scholar]
- 16.Sundaram S, Viriyayuthakorn S, Roth CM. Oligonucleotide structure influences the interactions between cationic polymers and oligonucleotides. Biomacromolecules. 2005;6:2961–2968. doi: 10.1021/bm0502314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sirsi SR, Williams JH, Lutz GJ. Poly(ethylene imine)-poly(ethylene glycol) copolymers facilitate efficient delivery of antisense oligonucleotides to nuclei of mature muscle cells of mdx mice. Hum. Gene Ther. 2005;16:1307–1317. doi: 10.1089/hum.2005.16.1307. [DOI] [PubMed] [Google Scholar]
- 18.Meidan VM, Glezer J, Amariglio N, Cohen JS, Barenholz Y. Oligonucleotide lipoplexes: the influence of oligonucleotide composition on complexation. Bba-Gen. Subjects. 2001;1568:177–182. doi: 10.1016/s0304-4165(01)00216-1. [DOI] [PubMed] [Google Scholar]
- 19.Lucas B, Van Rompaey E, Remaut K, Sanders N, De Smedt SC, Demeester J. On the biological activity of anti-ICAM-1 oligonucleotides complexed to non-viral carriers. J. Control Release. 2004;96:207–219. doi: 10.1016/j.jconrel.2003.12.028. [DOI] [PubMed] [Google Scholar]
- 20.von Harpe A, Petersen H, Li Y, Kissel T. Characterization of commercially available and synthesized polyethylenimines for gene delivery. J. Control. Release. 2000;69:309–322. doi: 10.1016/s0168-3659(00)00317-5. [DOI] [PubMed] [Google Scholar]
- 21.Boussif O, Lezoualc’h F, Zanta MA, Mergny MD, Scherman D, Demeneix B, Behr JP. A versatile vector for gene and oligonucleotide transfer into cells in culture and in vivo: polyethylenimine. Proc. Natl Acad. Sci. USA. 1995;92:7297–7301. doi: 10.1073/pnas.92.16.7297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sonawane ND, Szoka, F.C., Verkman AS. Chloride accumulation and swelling in endosomes enhances DNA transfer by polyamine-DNA polyplexes. J. Biol. Chem. 2003;278:44826–44831. doi: 10.1074/jbc.M308643200. [DOI] [PubMed] [Google Scholar]
- 23.Neu M, Fischer D, Kissel T. Recent advances in rational gene transfer vector design based on poly(ethylene imine) and its derivatives. J. Gene Med. 2005;7:992–1009. doi: 10.1002/jgm.773. [DOI] [PubMed] [Google Scholar]
- 24.Helin V, Gottikh M, Mishal Z, Subra F, Malvy C, Lavignon M. Cell cycle-dependent distribution and specific inhibitory effect of vectorized antisense oligonucleotides in cell culture. Biochem. Pharmacol. 1999;58:95–107. doi: 10.1016/s0006-2952(99)00083-0. [DOI] [PubMed] [Google Scholar]
- 25.Lee LK, Dunham BM, Li Z, Roth CM. Cellular dynamics of antisense oligonucleotides and short interfering RNAs. Ann. NY Acad. Sci. 2006;1082:47–51. doi: 10.1196/annals.1348.061. [DOI] [PubMed] [Google Scholar]
- 26.Roth CM. Molecular and cellular barriers limiting the effectiveness of antisense oligonucleotides. Biophys. J. 2005;89:2286–2295. doi: 10.1529/biophysj.104.054080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Raj A, Peskin CS, Tranchina D, Vargas DY, Tyagi S. Stochastic mRNA synthesis in mammalian cells. PLoS. Biol. 2006;4:1707–1719. doi: 10.1371/journal.pbio.0040309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bollig F, Winzen R, Kracht M, Ghebremedhin B, Ritter B, Wilhelm A, Resch K, Holtmann H. Evidence for general stabilization of mRNAs in response to UV light. Eur. J. Biochem. 2002;269:5830–5839. doi: 10.1046/j.1432-1033.2002.03300.x. [DOI] [PubMed] [Google Scholar]
- 29.Glaunsinger B, Chavez L, Ganem D. The exonuclease and host shutoff functions of the SOX protein of Kaposi's sarcoma-associated herpesvirus are genetically separable. J. Virol. 2005;79:7396–7401. doi: 10.1128/JVI.79.12.7396-7401.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Paschoud S, Dogar AM, Kuntz C, Grisoni-Neupert B, Richman L, Kuhn LC. Destabilization of interleukin-6 mRNA requires a putative RNA stem-loop structure, an AU-rich element, and the RNA-binding protein AUF1. Mol. Cell Biol. 2006;26:8228–8241. doi: 10.1128/MCB.01155-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Vinogradov SV, Bronich TK, Kabanov AV. Self-assembly of polyamine-poly(ethylene glycol) copolymers with phosphorothioate oligonucleotides. Bioconjug. Chem. 1998;9:805–812. doi: 10.1021/bc980048q. [DOI] [PubMed] [Google Scholar]
- 32.Brus C, Petersen H, Aigner A, Czubayko F, Kissel T. Efficiency of polyethylenimines and polyethylenimine-graft-poly (ethylene glycol) block copolymers to protect oligonucleotides against enzymatic degradation. Eur. J. Pharm. Biopharm. 2004;57:427–430. doi: 10.1016/j.ejpb.2004.02.004. [DOI] [PubMed] [Google Scholar]
- 33.Forrest ML, Meister GE, Koerber JT, Pack DW. Partial acetylation of polyethylenimine enhances in vitro gene delivery. Pharm. Res. 2004;21:365–371. doi: 10.1023/b:pham.0000016251.42392.1e. [DOI] [PubMed] [Google Scholar]
- 34.von Gersdorff K, Sanders NN, Vandenbroucke R, De Smedt SC, Wagner E, Ogris M. The internalization route resulting in successful gene expression depends on both cell line and polyethylenimine polyplex type. Mol. Ther. 2006;14:745–753. doi: 10.1016/j.ymthe.2006.07.006. [DOI] [PubMed] [Google Scholar]
- 35.Boletta A, Benigni A, Lutz J, Remuzzi G, Soria MR, Monaco L. Nonviral gene delivery to the rat kidney with polyethylenimine. Hum. Gene Ther. 1997;8:1243–1251. doi: 10.1089/hum.1997.8.10-1243. [DOI] [PubMed] [Google Scholar]
- 36.Gabrielson NP, Pack DW. Acetylation of polyethylenimine enhances gene delivery via weakened polymer/DNA interactions. Biomacromolecules. 2006;7:2427–2435. doi: 10.1021/bm060300u. [DOI] [PubMed] [Google Scholar]
- 37.Thomas M, Lu JJ, Ge Q, Zhang C, Chen J, Klibanov AM. Full deacylation of polyethylenimine dramatically boosts its gene delivery efficiency and specificity to mouse lung. Proc. Natl Acad. Sci. USA. 2005;102:5679–5684. doi: 10.1073/pnas.0502067102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Glodde M, Sirsi SR, Lutz GJ. Physiochemical properties of low and high molecular weight poly(ethylene glycol)-grafted poly(ethylene imine) copolymers and their complexes with oligonucleotides. Biomacromolecules. 2006;7:347–356. doi: 10.1021/bm050726t. [DOI] [PubMed] [Google Scholar]
- 39.Chiu Y.-L, Ali A, Chu C, Cao H, Rana TM. Visualizing a correlation between siRNA localization, cellular uptake, and RNAi in living cells. Chem. Biol. 2004;11:1165–1175. doi: 10.1016/j.chembiol.2004.06.006. [DOI] [PubMed] [Google Scholar]
- 40.Godbey WT, Wu KK, Mikos AG. Size matters: molecular weight affects the efficiency of poly(ethylenimine) as a gene delivery vehicle. J. Biomed. Mater. Res. 1999;45:268–275. doi: 10.1002/(sici)1097-4636(19990605)45:3<268::aid-jbm15>3.0.co;2-q. [DOI] [PubMed] [Google Scholar]
- 41.Fischer D, Li Y, Ahlemeyer B, Krieglstein J, Kissel T. In vitro cytotoxicity testing of polycations: influence of polymer structure on cell viability and hemolysis. Biomaterials. 2003;24:1121–1131. doi: 10.1016/s0142-9612(02)00445-3. [DOI] [PubMed] [Google Scholar]
- 42.Kopatz I, Remy JS, Behr JP. A model for non-viral gene delivery: through syndecan adhesion molecules and powered by actin. J. Gene Med. 2004;6:769–776. doi: 10.1002/jgm.558. [DOI] [PubMed] [Google Scholar]
- 43.Akinc A, Thomas M, Klibanov AM, Langer R. Exploring polyethylenimine-mediated DNA transfection and the proton sponge hypothesis. J. Gene Med. 2005;7:657–663. doi: 10.1002/jgm.696. [DOI] [PubMed] [Google Scholar]
- 44.Okuda T, Niidome T, Aoyagi H. Cytosolic soluble proteins induce DNA release from DNA—gene carrier complexes. J. Control Release. 2004;98:325–332. doi: 10.1016/j.jconrel.2004.05.004. [DOI] [PubMed] [Google Scholar]
- 45.Funhoff AM, van Nostrum CF, Koning GA, Schuurmans-Nieuwenbroek NM, Crommelin DJ, Hennink WE. Endosomal escape of polymeric gene delivery complexes is not always enhanced by polymers buffering at low pH. Biomacromolecules. 2004;5:32–39. doi: 10.1021/bm034041+. [DOI] [PubMed] [Google Scholar]
- 46.Dheur S, Dias N, van Aerschot A, Herdewijn P, Bettinger T, Remy JS, Helene C, Saison-Behmoaras ET. Polyethylenimine but not cationic lipid improves antisense activity of 3′-capped phosphodiester oligonucleotides. Antisense Nucleic A. 1999;9:515–525. doi: 10.1089/oli.1.1999.9.515. [DOI] [PubMed] [Google Scholar]
- 47.Tang MX, Redemann CT, Szoka F.C., Jr. In vitro gene delivery by degraded polyamidoamine dendrimers. Bioconjug. Chem. 1996;7:703–714. doi: 10.1021/bc9600630. [DOI] [PubMed] [Google Scholar]
- 48.Tan W, Lai JC, Miller P, Stein CA, Colombini M. Phosphorothioate oligonucleotides reduce mitochondrial outer membrane permeability to ADP. Am. J. Physiol. Cell Physiol. 2006;292:C1388–C1397. doi: 10.1152/ajpcell.00490.2006. [DOI] [PubMed] [Google Scholar]
- 49.Maksimenko AV, Gottikh MB, Helin V, Shabarova ZA, Malvy C. Physico-chemical and biological properties of antisense phosphodiester oligonucleotides with various secondary structures. Nucleos. Nucleot. 1999;18:2071–2091. doi: 10.1080/07328319908044865. [DOI] [PubMed] [Google Scholar]
- 50.Ghosh MK, Ghosh K, Cohen JS. Phosphorothioate-phosphodiester oligonucleotide co-polymers: assessment for antisense application. Anticancer Drug Des. 1993;8:15–32. [PubMed] [Google Scholar]
- 51.Bartlett DW, Davis ME. Insights into the kinetics of siRNA-mediated gene silencing from live-cell and live-animal bioluminescent imaging. Nucleic Acids Res. 2006;34:322–333. doi: 10.1093/nar/gkj439. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.