

Abstract
A small globular protein, the third repeat of the c-Myb DNA-binding domain, which is composed of 54 amino acid residues, was engineered so as to understand the structural uniqueness of native proteins. This small protein has three α-helices that form a helix-turn-helix structure, which is maintained by the hydrophobic core with three Ile residues. One of the mutant proteins, with two of the buried Ile (Ile-155 and Ile-181) substituted with Leu residues, showed multiple conformations, as monitored by heteronuclear magnetic resonance spectroscopy for 13C- and 15N-labeled proteins. The increase in the side-chain conformational entropy, caused by changing the Ile to a Leu residue on an α-helix, could engender the lack of structural uniqueness. In native proteins, the conformations of not only the β-branched side chains, but also those of the neighboring bulky side chains, can be greatly restricted, depending upon the local backbone structure.
Keywords: c-Myb DNA-binding domain, NMR
Considerable efforts of de novo protein design have been made so as to invent a new amino acid sequence that will fold into a desired tertiary structure. These exercises of design and synthesis have shown that a unique structure is very difficult to attain. Most synthesized proteins in aqueous solutions are stable and globular, with a pronounced amount of secondary structure as designed, but they usually lack definite organization of the side chains (1, 2). These features are similar to those in the molten globule state of native proteins (3). Structured peptides from random sequence libraries also show similar features (4). However, Dill et al. (5) discriminate these states that exist only in the artificial peptides as the “gemisch” states, as compared with those of the molten globules, because a peptide in the gemisch state has multiple ground-state conformations without any uniquely folded structure, which can always be attained by a native protein in an appropriate environment.
What determines structural uniqueness in native proteins? There are two different aspects of the determinants of structural uniqueness: (i) positive factors and (ii) negative factors in the folded structure. The positive contributions to the folded structure are the formation of specific combinations of side chains in the hydrophobic core (5, 6, 7, 8), hydrogen bonds (9), and disulfide bonds (10) between specific residues. A negative factor for the determination of unique structure is the negative role of the hydrogen bonds, so that a protein folds to avoid leaving any uncoupled hydrogen bond donor or acceptor in its interior, due to the significant electrostatic self-energy (11, 12, 13).
In this paper, we propose another negative factor, namely, the decrease in the side-chain conformational entropy, which can suppress the structural diversity and can destabilize the intermediate folding state. The side-chain conformational entropy has been analyzed empirically and theoretically (14, 15, 16), and it is understood to have important thermodynamic roles in protein folding (17). Moreover, it is well known that β-branched amino acids can have only restricted side-chain conformations in α-helices, because of steric effects (18), and thus oppose α-helix formation, due to the reduced conformational entropy (14). However, in the folded native protein, the orientations of most side-chains in the hydrophobic core are well fixed, independent of their backbone structure, and the conformational entropy is small enough. Therefore, the β-branched amino acids on an α-helix could contribute to suppressing the structural diversity, rather than to destabilizing the folded structure. In fact, Ile and Val residues are frequently observed in the helical sequences of native proteins, although Leu residues are found more frequently than Ile or Val (18). The function of this negative factor has been suggested and was used in the actual de novo design of helices (1, 19), but it has never been shown that an engineered protein exhibited novel structural diversity when a β-branched amino acid on the hydrophobic side of an α-helix was substituted with Leu.
The DNA-binding domain of the c-Myb protooncogene product consists of three repeats (R1, R2, and R3) of 51 or 52 amino acid residues. The tertiary structures of the individual free repeats and the complex structure of R2R3 with bound DNA were determined by the NMR-distance geometry method (20, 21). The most stable third repeat, R3 (residues 142-193; ref. 22), has three helices (residues 149-162, 166-172, and 178-187), forming a helix-turn-helix structure maintained by the hydrophobic core, which includes three Ile residues, Ile-155, Ile-169, and Ile-181, on the three different helices, respectively. In the present study, these Ile residues were changed to Leu residues, to focus on the role of a β-branched amino acid in an α-helix in causing structural uniqueness.
MATERIALS AND METHODS
Plasmids and Site-Directed Mutagenesis.
A DNA fragment encompassing the third repeat in the DNA-binding domain of c-Myb was amplified by PCR, using pact-c-myb (23) as the template and two synthetic primers, to generate an NcoI site and a BamHI site at the 5′ and 3′ ends of the amplified fragment, respectively. After digestion with NcoI and BamHI, the DNA fragment was cloned into pAR2156NcoI (24) to yield the expression plasmid, pRP3. An additional Met-Glu- sequence was introduced at the N terminus of R3. Site-directed mutagenesis was performed by two-step PCR, as described by Higuchi (25).
Protein Expression and Purification.
The wild type or mutant plasmid was introduced with pLysS into Escherichia coli BL21(DE3) (26). Freshly precultivated cells were inoculated into growth medium containing 100 μg of ampicillin per ml and 20 μg of chloramphenicol per ml and were grown at 37°C. M9ZB (26) was used as the medium for the expression of nonlabeled proteins. M9 medium containing [15N]NH4Cl and/or [13C]glucose was used for the expression of uniformly 15N- and/or 13C-labeled proteins. When the culture reached an OD600 of ≈0.6, isopropyl β-d-thiogalactopyranoside was added to a final concentration of 2.0 mM. The cells were cultured at 22°C for another 12 h. The harvested cells were suspended in 150 mM phosphate buffer (pH 6.85) and were lysed by sonication at 4°C. After the cell debris was removed by centrifugation, ammonium sulfate was added to the supernatant to 60% saturation. After an incubation at 4°C for 1 h, the supernatant was dialyzed against 150 mM phosphate buffer containing 100 mM NaCl (pH 6.85) and was then applied to a P11 phosphocellulose column. The fractions that eluted with 150 mM phosphate buffer containing 0.8 M NaCl (pH 6.85) were combined and then applied to a Sephadex G-50 column.
The protein concentrations were determined from UV absorption at 280 nm in 150 mM phosphate buffer (pH 6.85) and were calculated by using the molar absorption coefficients of 1.70 × 104 M−1, 1.57 × 104 M−1, 1.43 × 104 M−1, and 1.52 × 104 M−1 for the wild type, I155L, I155L/I181L, and I155L/I181V proteins, respectively, on the basis of an amino acid analysis of each purified protein. Here, I155L designates the R3 mutant protein, in which Ile at position 155 is replaced by Leu, and other mutant proteins are abbreviated in the same manner. A slash indicates the combination of the simultaneous mutations.
The purity of the mutant R3s was confirmed in the HPLC profiles, as shown in Fig. 1.
Figure 1.
HPLC elution profiles of purified mutant R3s. Purified I155L (a), I155L/I181L (b), and I155L/I181V (c) were applied to a Cosmosil 5C4-300 column (Nacalai Tesque, Kyoto) and equilibrated with 0.1% trifluoroacetic acid. The proteins were eluted with a linear gradient of acetonitrile (0–60%) in 0.1% trifluoroacetic acid.
CD and NMR Measurements.
CD spectra were measured at 10°C in 5 mM sodium acetate buffer (pH 5.0) with a Jasco (Tokyo) spectropolarimeter (model J-600). The protein concentration was 0.1 mg/ml in a 0.2-cm optical path length cell.
All NMR spectra were measured on a Bruker (Tsukuba, Ibaraki) AMX-600 spectrometer (600.13 MHz for 1H, 60.82 MHz for 15N, and 150.91 MHz for 13C) with a probe temperature of 10°C. The proteins were dissolved to a final concentration of ≈3 mM in 90% H2O/10% 2H2O (50 mM C2H3COONa/0.4 mM NaN3, pH 5.0) for NMR measurements.
Assignments of the chemical shifts were performed using the following methods: double quantum filtered correlated spectroscopy (DQF-COSY; ref. 27), total correlated spectroscopy (TOCSY) (28), and nuclear Overhauser enhancement spectroscopy (NOESY; ref. 27), three-dimensional (3D) TOCSY-heteronuclear multiple quantum coherence spectroscopy (HMQC; ref. 29), 3D NOESY-HMQC (29, 30), 3D HNCA (31), and 3D HNCOCA (32).
The H2H exchange experiment at 10°C was performed as described below. The lyophilized, uniformly 15N-labeled protein was dissolved in precooled (10°C) 99.6% 2H2O (50 mM C2H3COONa/0.4 mM NaN3, pH 5.0) at a concentration of ≈3 mM. The first spectrum was recorded 30 min after the dissolution of the protein. Two-dimensional 1H-15N HMQC data were acquired utilizing 1024 complex data points over 8196 Hz in the 1H dimension and 128 increments over 1824 Hz in the 15N dimension. The total acquisition time for one spectrum was 50 min. The spectra were zero-filled to 1024 × 512 real points and were processed with a mild squared-sine-bell window function in both dimensions. The volume integrals of the cross-peaks were calculated for each spectrum using the Bruker program uxnmr, and the decays were fitted to a single exponential. The total data points were 24 until 72 h after the dissolution for I155L and 20 until 36 h after the dissolution for I155L/I181L and I155L/I181V, respectively. The protection factors of the amide protons were calculated as the ratio of exchange rates determined experimentally to that for the unfolded structure, reported by Bai et al. (33).
RESULTS
R3 was specifically expressed in E. coli, and three Ile residues, Ile-155, Ile-169, and Ile-181, were individually or simultaneously replaced with Leu via site-directed mutagenesis. The Ile-181 residue was also replaced by Val simultaneously with the substitution of Ile-155 by Leu. Of the resulting eight mutants’ R3s, I169L, I181L, I169L/I181L, and I155L/I169L/I181L showed a low level of expression in E. coli cells, and poor solubilities in aqueous solutions. Therefore, no further physicochemical analyses of these four mutants were performed.
The far-UV CD spectrum of I155L was almost identical to that of the wild type at 10°C. The mutants I155L/I169L, I155L/I181L, and I155L/I181V showed helical contents of 87%, 75%, and 90% of the wild type’s, respectively, assuming that the helical content is proportional to the far-UV CD value at 222 nm. The heat stability of each mutant was investigated by monitoring the far-UV CD with increasing temperature at pH 5.0, and reversible unfolding was observed with melting temperatures (Tm) of 59.6°C, 52.1°C, 49.0°C, and 37.6°C for the mutants I155L, I155L/I169L, I155L/I181L, and I155L/I181V, respectively. The Tm of the wild type under the same conditions was 55.9°C, slightly lower than that of I155L.
The tertiary structure of the mutant I155L was determined by the NMR-distance geometry method, using 440 distance restraints and 47 dihedral angle restraints interpreted from the cross-peak intensities of the NOESY spectra and the 3JHNα coupling constants, using the procedure described previously (21). An averaged structure of the final 20 structures without any violation to the experimental restraints is shown in Fig. 2. The root mean square deviations (rmsd) of the distance and dihedral angle restraints are 0.0089 ± 0.0007 Å and 0.191° ± 0.056°, respectively, for the 20 structures. The rmsd between the individual structures and the mean coordinates are 0.54 ± 0.10 Å for the backbone atoms and 1.17 ± 0.09 Å for all the heavy atoms, respectively, for the residues from Trp-147 to Trp-185.
Figure 2.
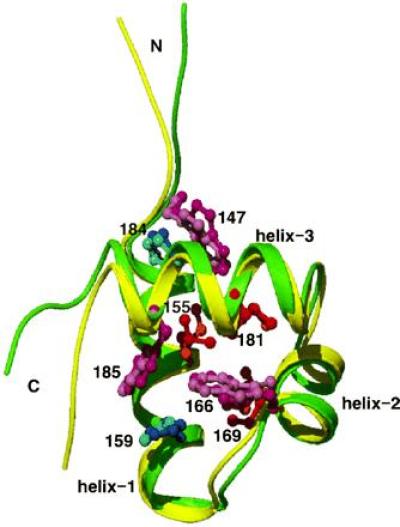
The refined average structure of the 20 I155L mutant structures determined by NMR-distance geometry method. The backbone is shown in green. Side-chain atoms of Trp and His residues are shown in purple and dark blue, respectively. Side-chains of Leu-155, Ile-169, and Ile-181 are shown in red. The backbone of the superimposed wild-type structure (21) is shown in yellow, and side chains of Ile, Trp and His are shown in orange, pink, and light blue, respectively. The characters N and C indicate the N and C termini, respectively, and several residue numbers are indicated. The figure was drawn with the program raster3d (34). The coordinates for the average and the 20 I155L mutant structures were submitted to the Protein Data Bank.
The backbone structure is very similar to the wild type, as shown in Fig. 2. In fact, the rmsd between the individual mutant structures and the wild-type structure (Protein Data Bank accession no. 1MBJ; ref. 21) are 1.00 ± 0.12 Å for the backbone atoms and 1.85 ± 0.13 Å for all heavy atoms, respectively, from Trp-147 to Trp-185, except for residue 155. Although the overall structure of I155L is very similar to that of the wild type, both the indole ring of Trp-147 and the imidazole ring of His-184 tilt ≈30° from those of the wild type. This is due to the absence of the δ-methyl group in the side chain of the residue at position 155 of I155L, which directly contacts the indole ring of Trp-147 in the wild-type protein, as indicated in Fig. 2.
Fig. 3 shows the sequential and short-range nuclear Overhauser enhancement (NOE) connectivities in the I155L, I155L/I169L, I155L/I181L, and I155L/I181V mutants, observed at 10°C. The profiles of I155L (Fig. 3a) and I155L/I169L (Fig. 3b) are almost identical to that of the wild type (21). The intense dNN(i, i+1), dαN(i, i+3), dαN(i, i+4), and dαβ(i, i+3) NOE signals, specific to a helical structure, were observed in residues 149-162, 166-172, and 178-187. When Ile was additionally replaced with Leu at position 181, however, the characteristic NOEs of the helical structure disappeared, especially in the carboxyl half of helix-3 (Fig. 3c). Since the analysis of structural diversity is our current purpose, position 181 in the mutant proteins is hereafter focused upon, specifically in the mutant I155L. As shown in Fig. 3d, no such disappearance of the sequential connectivity was observed in I155L/I181V.
Figure 3.

Diagram of the sequential and short-range NOEs and the chemical shift indexes (36) observed for the mutant R3s from positions 145 to 190: I155L (a), I155L/I169L (b), I155L/I181L (c), and I155L/I181V (d). The abbreviations NN1, αN1, αN2, αN3, αN4, and αβ3 represent dNN(i, i+1), dαN(i, i+1), dαN(i, i+2), dαN(i, i+3), dαN(i, i+4), and dαβ(i, i+3), respectively. The thick, medium, and thin bars correspond to strong, medium, and weak NOEs, respectively. The downfield shifted 1Hα chemical shifts (negative ΔHα) and the upfield-shifted 13Cα chemical shifts (positive ΔCα) from the corresponding chemical shifts of a random coil are indicative of helical regions (35, 36). Helical regions in the amino acid sequence of I155L from the tertiary structure are boxed. Position 155 is indicated by an open triangle. Other substituted residues in the mutant R3s are indicated by filled triangles.
The two-dimensional 1H-15N HMQC spectra of the mutant R3s at 10°C are indicated in Fig. 4 a–c. Many minor peaks appeared in the spectrum of I155L/I181L (Fig. 4b). In contrast, the R3s that have a β-branched amino acid at position 181 (Fig. 4 a and c) lacked these minor peaks. With the aid of the 3D-NMR technique using I155L/I181L doubly labeled by 13C and 15N, the minor peaks were also assigned. Most of them were the backbone atoms of helix-3, and the traces of the major and minor species are indicated in Fig. 4d. The results clearly indicate that I155L/I181L has multiple conformations.
Figure 4.
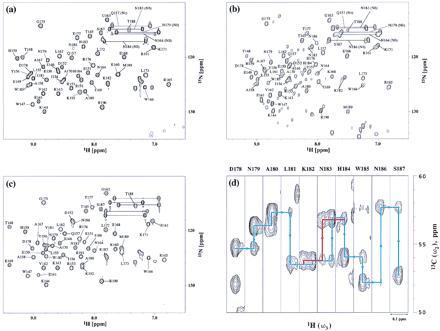
Expanded 1H-15N HMQC spectra in backbone amide regions of the uniformly 15N-labeled mutant R3s at 10°C in 50 mM C2H3COONa/0.4 mM NaN3, pH 5.0 (10% 2H2O): I155L (a), I155L/I181L (b), and I155L/I181V (c). The assigned amides are indicated by one-letter amino acid codes and residue numbers. The amides of Asn and Gln side chains are indicated by bars in the figures. An expanded 3D HNCA spectrum in the backbone region of the 13C- and 15N-labeled I155L/I181L at 10°C, pH 5.0 is shown in d. The traces of sequential assignment for the major and minor species are indicated by blue and red lines, respectively.
Fig. 5 shows the protection factors of I155L, I155L/I181L, and I155L/I181V from the H2H-exchange experiments. In the regions corresponding to helices, I155L showed high protection factors that were comparable to those of the wild type. I155L/I181L showed relatively low protection factors in helices-1 and -2, and the protection almost disappeared in helix-3 (red). The protection factors of the I155L/I181V mutant were relatively low as compared with those of I155L and the wild type, but the amide protons in helix-3 are still protected as well as in helices-1 and -2 (blue).
Figure 5.
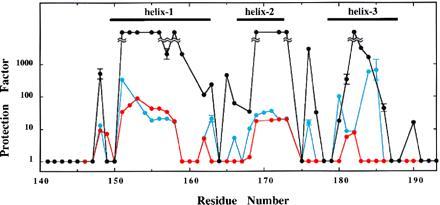
The protection factors of the mutant R3s from the H2H-exchange experiments at pH 5.0, 10°C. Black-, red-, and blue-filled circles indicate those of I155L, I155L/I181L, and I155L/I181V, respectively, with error bars. When the resonance signal of an amide proton decayed too quick to obtain the exchange rate, the protection factor is assumed to be one. In the case of I155L, several amide protons did not exchange under the current experimental condition. Helical regions of I155L are also indicated.
DISCUSSION
The motivation of this study is to investigate the origin of the structural uniqueness of native proteins. For this purpose, R3 of the c-Myb DNA-binding domain is a good, stable candidate with one hydrophobic core and without any disulfide bridges. Among the created mutant proteins, I155L/I181L shows significant evidence of multiple conformations. The minor peaks shown in Fig. 4 b and d reveal the remarkable absence of structural uniqueness. Gel-filtration chromatography confirmed the uniform peptide length of I155L/I181L and sedimentation equilibrium experiments revealed that I155L/I181L, as well as the other R3s, existed in a monomeric state at the high protein concentration used in the current NMR experiments (data not shown).
In the H2H-exchange experiment of I155L/I181L, the major peaks that correspond to the amide protons in the carboxyl half of helix-3 and the minor peaks were completely decayed at the first spectrum, indicating that the exchange of these protons to deuterons was complete within 30 min. A few amide protons in helix-3 remained to be exchanged in the major species. Helix-3 in the major species seems to have some tight structure and does not appear to have completely collapsed by the substitution of Leu for Ile-181. The chemical shift differences of the major backbone signals of I155L/I181L, ΔHα, and ΔCα, from those in the random coil (35, 36) are indicated in Fig. 3c. They show the tendency of helix-3 to adopt an α-helical structure in the major species. Since the chemical shift differences of the minor species did not differ much from the major ones, as shown in Fig. 4d, the backbone of the minor species is not thought to be very different from the major conformation.
When the temperature was gradually increased to 20°C, most of the minor peaks in Fig. 4b became broad, without any significant chemical shift change. Since we have not detected cross-peaks corresponding to the exchange process from the minor species to either the major or the minor species in HMQC-NOESY experiments with a 250-ms mixing time, the structural exchange among the multiple conformations must be very slow. The current results strongly suggest that I155L/I181L has several different conformations with similar low energies, which was predicted by Dill et al. (5) as the gemisch state.
We propose that one of the origins of the structural uniqueness of native proteins is the reduction of the conformational entropy of the side chains in the intermediate folding state, where only a few local backbone structures are constructed, if not tightly. The entropy decrease may also destabilize the putative gemisch state. In the case of an α-helix, β-branched amino acids could decrease the conformational entropy. The I155L and I155L/I181V proteins do not show any characteristics of multiple conformations, although the helical structure of the latter mutant is destabilized. In fact, the protection factors of I155L/I181V were relatively low, as compared with those of I155L and the wild type, as shown in Fig. 5. Since Tm of I155L/I181V is 22° lower than that of I155L, the increase in the exchange rate is well explained by the decrease of the overall structural stability. However, the amide protons in helix-3 are still protected to the same extent as those of helices-1 and -2, in contrast to those of I155L/I181L.
The question arises as to why a Leu residue at position 181 yields multiple conformations, but Leu residues at positions 155 and 169 do not. To understand the reason, the conformational entropies of the side chains of the three helices were taken into account, as follows.
The side-chain conformational entropy is defined as the Boltzmann sampling of states: S = −RΣspslnps, where ps is the probability of the combination of the side chains being in state s (16). Several methods have been proposed to compute ps by Monte Carlo sampling (14) and by mean-field theory (37). Here, we made an exhaustive search of the side-chain conformations for short helices with the fixed backbone structures, using the small-size rotamer library developed by Tanimura et al. (38), which is similar to that of Ponder and Richards (39). The conformational entropy was then obtained from the following equation at T = 300 K, accounting for the partition function Z for all combinations of rotamer states with the energy {Us}, as the sum of the interaction energies given by the all-atom parameters of the AMBER force field (40), excluding the nonbonded electrostatic interactions.
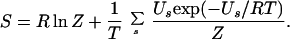 |
1 |
The side-chain conformational entropies were calculated for the individual three helices of both the wild-type and the engineered R3 sequences with Leu. Here, all the residues with relative accessibilities >60% in the folded structure were first replaced with Ala residues for simplicity (15). The probability of the ith side-chain taking the rth rotamer pir was calculated as follows:
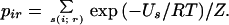 |
2 |
Here, ∑s(i; r) means the summation of only the states s, where residue has the ith rotamer. Then, the apparent residual entropy, Siapp = −RΣrpirlnpir, was also calculated for each ith residue. Using the conformational entropy values in the unfolded state SiU, estimated by Pickett and Sternberg (15), the entropy differences between the helical backbone structures and the unfolded states, ΔSiapp = SiU − Siapp, were calculated, and are shown in Fig. 6a. The differences of ΔSiapp between the wild-type and the Leu mutants for the three helices are shown in Fig. 6b.
Figure 6.
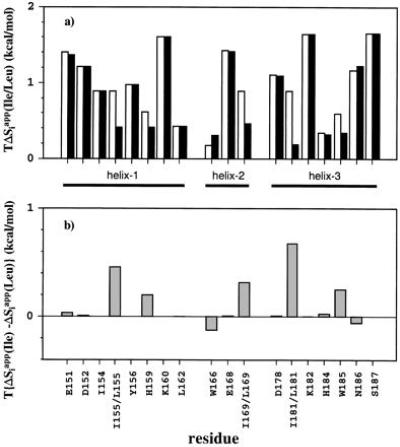
Differences of the apparent residual entropy between the helical backbone structures and the unfolded states (15) at T = 300 K in the three individual helices of R3 with Ile residues (open bars) and Leu residues (solid bars) (a). Differences between the values in a for Ile and Leu are shown in b.
It is evident that the conformational entropy difference of helix-3 is the largest among the three, when each inner Ile residue is replaced with Leu. Each Ile residue greatly contributes to a decrease in the conformational entropy of the associated helix, because an Ile residue in an α-helix has only one allowed rotamer on its χ1 angle (−60°; ref. 18). Moreover, the conformational entropies of His-159 and Trp-185, which are four residues apart from positions 155 and 181, respectively, are significantly decreased because of the strong steric interaction in the helix (41). Since Siapp neglects the cooperativity among residues, ΣiSiapp should be larger than the correct entropy calculated by eq. [1]. In fact, in the Leu-substituted helix-1 and helix-3, ΣiSiapp are 6.9 and 9.0 cal/mol per degree, whereas the correct S values from Eq. 1 are 6.5 and 7.6 cal/mol per degree, respectively. In helix-2, no such cooperativity was observed, and it retained the same value, 4.6 cal/mol per degree. The reason why the S value in the Leu-substituted helix-1 is smaller than that in helix-3 is mainly due to the fact that the Leu-155 side chain in helix-1 is more restricted than that of Leu-181. It is because position 155 is in the middle of the helix, and position 181 is at the N terminus of the helix.
Consequently, the conformation of the bulky side chain in helix-3 is fixed through the restricted conformation of the spacially neighboring β-branched amino acid, depending upon the local backbone structure. This provides a new insight into the importance of the side-chain conformational entropy in protein folding, because bulky side chains often have a key role in the unique packing of the hydrophobic core (7, 42). In fact, in the case of R3, Trp-185 is essential to core formation (43). If the conformations of such bulky side chains are determined by the local backbone structure, if not very precisely, the process to find the correct side-chain rotamers becomes much more rapid than that with the nonrestricted side-chain conformations that are independent of the local backbone structure. Since the rate-determining step in protein folding is considered to be finding the correct side-chain rotamers (6), the importance of the side-chain conformational entropy must be recognized. We are not quite sure at the moment whether this mechanism of fixing the side chains by dependence upon the local backbone structure is a general phenomenon in the protein folding process. However, we can point out that the β-branched amino acids, Val and Ile, participate in most of the experimentally observed local secondary structures formed at the beginning of the refolding process, as shown for cytochrome c (44), ribonuclease A (45), barnase (46), hen lysozyme (47), ubiquitin (48), T4-lysozyme (49), apomyoglobin (50), and chymotrypsin inhibitor 2 (51).
In conclusion, the results of this investigation provide evidence that a β-branched amino acid plays an important role in the formation of a unique protein structure. When we attempt to create an artificial protein that has a unique structure, like a native protein, it is essential to consider the side-chain conformational entropy, which depends upon the local backbone structure and the neighboring residues in space.
Acknowledgments
Plasmids pact-c-myb and pAR2156NcoI were kindly provided by Dr. Shunsuke Ishii, Tsukuba Life Science Center, The Institute of Physical and Chemical Research, Japan (RIKEN). We thank Dr. Kazuhiro Ogata (RIKEN) and Dr. Hidekazu Hiroaki (Biomolecular Engineering Research Institute) for discussions, and Ms. Miyo Sakai and Prof. Toshio Takagi (Osaka University) for the sedimentaion equilibrium analysis.
Footnotes
The publication costs of this article were defrayed in part by page charge payment. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. §1734 solely to indicate this fact.
Abbreviations: NOE, nuclear Overhauser enhancement; NOESY, NOE spectroscopy; 3D, three-dimensional; HMQC, heteronuclear multiple quantum coherence spectroscopy.
Data deposition: The atomic coordinates have been deposited in the Protein Data Bank, Chemistry Department, Brookhaven National Laboratory, Upton, NY 11973 (references 1IDY and 1IDZ).
References
- 1.Tanaka T, Kuroda Y, Kimura H, Kidokoro S, Nakamura H. Protein Eng. 1994;7:969–976. doi: 10.1093/protein/7.8.969. [DOI] [PubMed] [Google Scholar]
- 2.Bryson J W, Betz S F, Lu H S, Suich D J, Zhou H X, O’Neil K T, DeGrado W F. Science. 1995;270:935–941. doi: 10.1126/science.270.5238.935. [DOI] [PubMed] [Google Scholar]
- 3.Ptitsyn O B. Adv Protein Chem. 1995;47:83–229. doi: 10.1016/s0065-3233(08)60546-x. [DOI] [PubMed] [Google Scholar]
- 4.Davidson A R, Lumb K J, Sauer R T. Nat Struct Biol. 1995;2:856–864. doi: 10.1038/nsb1095-856. [DOI] [PubMed] [Google Scholar]
- 5.Dill K A, Bromberg S, Yue K, Fiebig K M, Yee D P, Thomas P D, Chan H S. Protein Sci. 1995;4:561–602. doi: 10.1002/pro.5560040401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shakhnovich E I, Finkelstein A V. Biopolymers. 1989;28:1667–1680. doi: 10.1002/bip.360281003. [DOI] [PubMed] [Google Scholar]
- 7.Finkelstein A V, Nakamura H. Protein Eng. 1993;6:367–372. doi: 10.1093/protein/6.4.367. [DOI] [PubMed] [Google Scholar]
- 8.Harbury P B, Tidor B, Kim P S. Proc Natl Acad Sci USA. 1995;92:8408–8412. doi: 10.1073/pnas.92.18.8408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lumb K J, Kim P S. Biochemistry. 1995;34:8642–8648. doi: 10.1021/bi00027a013. [DOI] [PubMed] [Google Scholar]
- 10.Kuroda Y, Nakai T, Ohkubo T. J Mol Biol. 1994;236:862–868. doi: 10.1006/jmbi.1994.1194. [DOI] [PubMed] [Google Scholar]
- 11.Finkelstein A V, Ptitsyn O B. Prog Biophys Mol Biol. 1987;50:171–190. doi: 10.1016/0079-6107(87)90013-7. [DOI] [PubMed] [Google Scholar]
- 12.Privalov P L, Makhatadze G I. J Mol Biol. 1993;232:660–679. doi: 10.1006/jmbi.1993.1417. [DOI] [PubMed] [Google Scholar]
- 13.Nakamura H. Q Rev Biophys. 1996;29:1–90. doi: 10.1017/s0033583500005746. [DOI] [PubMed] [Google Scholar]
- 14.Creamer T P, Rose G D. Proc Natl Acad Sci USA. 1992;89:5937–5941. doi: 10.1073/pnas.89.13.5937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pickett S D, Sternberg M J E. J Mol Biol. 1993;231:825–839. doi: 10.1006/jmbi.1993.1329. [DOI] [PubMed] [Google Scholar]
- 16.Doig A J, Sternberg M J E. Protein Sci. 1995;4:2247–2251. doi: 10.1002/pro.5560041101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Makhatadze G I, Privalov P L. Protein Sci. 1996;5:507–510. doi: 10.1002/pro.5560050312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Richardson J S, Richardson D C. In: Prediction of Protein Structure and the Principles of Protein Conformation. Fasman G D, editor. New York: Plenum; 1989. pp. 1–98. [Google Scholar]
- 19.Raleigh D P, DeGrado W F. J Am Chem Soc. 1992;114:10079–10081. [Google Scholar]
- 20.Ogata K, Morikawa S, Nakamura H, Sekikawa A, Inoue T, Kanai H, Sarai A, Ishii S, Nishimura Y. Cell. 1994;79:639–648. doi: 10.1016/0092-8674(94)90549-5. [DOI] [PubMed] [Google Scholar]
- 21.Ogata K, Morikawa S, Nakamura H, Hojo H, Yoshimura S, Zhang R, Aimoto S, Ametani Y, Hirata Z, Sarai A, Ishii S, Nishimura Y. Nat Struct Biol. 1995;2:309–320. doi: 10.1038/nsb0495-309. [DOI] [PubMed] [Google Scholar]
- 22.Sarai A, Uedaira H, Morii H, Yasukawa T, Ogata K, Nishimura Y, Ishii S. Biochemistry. 1993;32:7759–7764. doi: 10.1021/bi00081a022. [DOI] [PubMed] [Google Scholar]
- 23.Nishina Y, Nakagoshi H, Imamoto F, Gonda T J, Ishii S. Nucleic Acids Res. 1989;17:107–117. doi: 10.1093/nar/17.1.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tanikawa J, Yasukawa T, Enari M, Ogata K, Nishimura Y, Ishii S, Sarai A. Proc Natl Acad Sci USA. 1993;90:9320–9324. doi: 10.1073/pnas.90.20.9320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Higuchi R. In: PCR Technology. Erlich H A, editor. New York: Stockton; 1989. pp. 61–88. [Google Scholar]
- 26.Studier F W, Rosenberg A H, Dunn J J, Dubendorff J W. Methods Enzymol. 1990;185:60–89. doi: 10.1016/0076-6879(90)85008-c. [DOI] [PubMed] [Google Scholar]
- 27.Wüthrich K. NMR of Proteins and Nucleic Acids. New York: Wiley Interscience; 1986. [Google Scholar]
- 28.Bax A, Davis D G. J Magn Reson. 1985;65:355–366. [Google Scholar]
- 29.Marion D, Driscoll P C, Kay L E, Wingfield P T, Bax A, Gronenborn A M, Clore G M. Biochemistry. 1989;28:6150–6156. doi: 10.1021/bi00441a004. [DOI] [PubMed] [Google Scholar]
- 30.Kay L E, Marion D, Bax A. J Magn Reson. 1989;84:72–84. [Google Scholar]
- 31.Kay L E, Ikura M, Tschudin R, Bax A. J Magn Reson. 1990;89:495–514. doi: 10.1016/j.jmr.2011.09.004. [DOI] [PubMed] [Google Scholar]
- 32.Bax A, Ikura M. J Biomol NMR. 1991;1:99–104. doi: 10.1007/BF01874573. [DOI] [PubMed] [Google Scholar]
- 33.Bai Y, Milne J S, Mayne L, Englander W. Proteins. 1993;17:75–86. doi: 10.1002/prot.340170110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bacon D, Anderson W F. J Mol Graphics. 1988;6:219–220. [Google Scholar]
- 35.Wishart D S, Sykes B D, Richards F M. J Mol Biol. 1991;222:311–333. doi: 10.1016/0022-2836(91)90214-q. [DOI] [PubMed] [Google Scholar]
- 36.Wishart D S, Sykes B D. J Biomol NMR. 1994;4:171–180. doi: 10.1007/BF00175245. [DOI] [PubMed] [Google Scholar]
- 37.Koehl P, Delarue M. J Mol Biol. 1994;239:249–275. doi: 10.1006/jmbi.1994.1366. [DOI] [PubMed] [Google Scholar]
- 38.Tanimura R, Kidera A, Nakamura H. Protein Sci. 1994;3:2358–2365. doi: 10.1002/pro.5560031220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ponder J W, Richards F M. J Mol Biol. 1987;193:775–791. doi: 10.1016/0022-2836(87)90358-5. [DOI] [PubMed] [Google Scholar]
- 40.Weiner S J, Kollman P A, Nguyen D T, Case D A. J Comput Chem. 1986;7:230–252. doi: 10.1002/jcc.540070216. [DOI] [PubMed] [Google Scholar]
- 41.Padmanabhan S, Baldwin R L. J Mol Biol. 1994;241:706–713. doi: 10.1006/jmbi.1994.1545. [DOI] [PubMed] [Google Scholar]
- 42.Burley S K, Petsko G A. Science. 1985;229:23–28. doi: 10.1126/science.3892686. [DOI] [PubMed] [Google Scholar]
- 43.Kanei-Ishii C, Sarai A, Sawazaki T, Nakagoshi H, He D-N, Ogata K, Nishimura Y, Ishii S. J Biol Chem. 1990;265:19990–19995. [PubMed] [Google Scholar]
- 44.Roder H, Elove G A, Englander W. Nature (London) 1988;335:700–704. doi: 10.1038/335700a0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Udgaonkar J B, Baldwin R L. Proc Natl Acad Sci USA. 1990;87:8197–8201. doi: 10.1073/pnas.87.21.8197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bycroft M, Matouschek A, Kellis J T, Jr, Serrano L, Fersht A R. Nature (London) 1990;346:488–490. doi: 10.1038/346488a0. [DOI] [PubMed] [Google Scholar]
- 47.Radford S E, Dodson C M, Evans P A. Nature (London) 1992;358:302–307. doi: 10.1038/358302a0. [DOI] [PubMed] [Google Scholar]
- 48.Briggs M S, Roder H. Proc Natl Acad Sci USA. 1992;89:2017–2021. doi: 10.1073/pnas.89.6.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lu J, Dahlquist F W. Biochemistry. 1992;31:4749–4756. doi: 10.1021/bi00135a002. [DOI] [PubMed] [Google Scholar]
- 50.Jennings J A, Wright P E. Science. 1993;262:892–896. doi: 10.1126/science.8235610. [DOI] [PubMed] [Google Scholar]
- 51.Otzen D E, Itzhaki L S, ElMasry N F, Jackson S E, Fersht A R. Proc Natl Acad Sci USA. 1994;91:10422–10425. doi: 10.1073/pnas.91.22.10422. [DOI] [PMC free article] [PubMed] [Google Scholar]