

Abstract
It has recently been demonstrated that the C-terminal deletion mutant of recombinant human carbonic anhydrase IV (G267X CA IV) converts the normally glycosylphosphatidylinositol-anchored enzyme into a soluble secretory form which has the same catalytic properties as the membrane-associated enzyme purified from human tissues. We have determined the three-dimensional structure of the secretory form of human CA IV by x-ray crystallographic methods to a resolution of 2.8 Å. Although the zinc binding site and the hydrophobic substrate binding pocket of CA IV are generally similar to those of other mammalian isozymes, unique structural differences are found elsewhere in the active site. Two disufide linkages, Cys-6–Cys-11G and Cys-23–Cys-203, stabilize the conformation of the N-terminal domain. The latter disulfide additionally stabilizes an active site loop containing a cis-peptide linkage between Pro-201 and Thr-202 (this loop contains catalytic residue Thr-199). On the opposite side of the active site, the Val-131–Asp-136 segment adopts an extended loop conformation instead of an α-helix conformation as found in other isozymes. Finally, the C terminus is surrounded by a substantial electropositive surface potential, which is likely to stabilize the interaction of CA IV with the negatively charged phospholipid headgroups of the membrane. These structural features are unique to CA IV and provide a framework for the design of sulfonamide inhibitors selective for this particular isozyme.
Keywords: protein crystallography, zinc enzyme, carbon dioxide hydration
There are seven genetically distinct isozymes of mammalian carbonic anhydrase, designated I–VII (1, 2, 3), each of which catalyzes the reversible hydration of carbon dioxide (H2O + CO2 ⇄ HCO3− + H+) by a zinc-hydroxide mechanism (4, 5, 6). The three-dimensional structures of human isozyme I (7), human isozyme II (8, 9), bovine isozyme III (10), and murine isozyme V (11) reveal a common fold dominated by a β-sheet superstructure. In each mammalian isozyme of known structure, the catalytically obligatory zinc ion resides on one face of the β-sheet at the bottom of a 15-Å-deep, conical active site cleft. Zinc is liganded by His-94, His-96, His-119, and hydroxide ion with tetrahedral geometry; Thr-199 accepts a hydrogen bond from zinc-bound hydroxide ion. Despite overall structural homology, local structural differences in the active sites of isozymes I, II, III, and V give rise to 103-fold differences in catalytic activity. Of these isozymes, human carbonic anhydrase II (CA II) is currently the most thoroughly characterized by enzymological and x-ray crystallographic methods (4, 5, 6).
Only one mammalian carbonic anhydrase isozyme is known to be membrane associated, and this isozyme is designated carbonic anhydrase IV (CA IV) (12). Like CA II, CA IV is a high-activity isozyme which catalyzes the CO2 hydration reaction at a rate near that of diffusion control (13). Native CA IV isolated and purified from human kidney is a nonglycosylated 35-kDa protein that is posttranslationally cleaved and modified at the C terminus with a glycosylphosphatidylinositol (GPI) tail (13, 14), a common membrane-anchoring motif (15, 16). The CA IV isozyme contains two disulfide linkages which contribute to its remarkable stability upon solubilization in 5% sodium dodecyl sulfate (SDS) (12, 17). The full-length cDNA for human CA IV has been isolated from a λgt10 kidney cDNA library and expressed in COS cells (18). The deduced amino acid sequence includes an 18-residue signal sequence, a 260-residue segment exhibiting 30–36% sequence identity with other human carbonic anhydrase isozymes, and an additional 27-residue segment at the C terminus containing a 21-residue hydrophobic domain. The C-terminal domain likely specifies the association of CA IV with the membrane and contains a signal for posttranslational cleavage and transfer to the GPI anchor (18). Removal of the C-terminal domain results in a fully active, soluble, secretory form of CA IV with catalytic properties identical to those of GPI-anchored CA IV (19).
Both CA IV and CA II play important roles in the function of the human kidney, and their cellular locations in the nephron are quite distinct. The cytosolic isozyme CA II is found in certain cells of renal tubules and collecting ducts, where it participates in proximal and distal urinary acidification (20, 21, 22, 23). Membrane-anchored isozyme CA IV is located in the apical plasma membranes of the brush border in two segments of the nephron: the proximal convoluted tubule and the thick ascending limb of Henle (24). The CA IV isozyme plays a role in modulating equilibrium pH in the lumen (25), and it plays a dominant role in bicarbonate reabsorption from the lumen (24, 26, 27). The identification of CA IV in the basolaterial plasma membranes of the proximal tubule and thick ascending limb also suggests a role for this isozyme in bicarbonate plasma transport (24).
The CA IV isozyme is also localized on the luminal surface of pulmonary endothelial cells (28, 29). Here, CA IV functions to catalyze the dehydration of serum bicarbonate to yield CO2, which then readily diffuses across the capillary endothelial surface to pass out of the lung upon expiration (12). The CA IV proteins isolated and purified from lung and kidney are essentially identical enzymes (14). Interestingly, CA IV is also found in the endothelial cells of an ocular capillary bed, the choriocapillaris (30), implicating this isozyme in addition to CA II as a pharmaceutical target of CA inhibitors used to regulate intraocular pressure (25, 30).
Here, we report the x-ray crystallographic structure determination of the secretory form of human CA IV expressed in Chinese hamster ovary cells (17). This secretory form contains enzyme residues 1–259, using the numbering scheme established for CA II. The actual sequence identity between CA IV and CA II based on superposition and alignment of their tertiary structures is 33% (Fig. 1).§ The CA IV isozyme exhibits CO2 hydration activity roughly comparable to that of CA II with kcat/Km = 5 × 107 M−1·s−1 and bicarbonate dehydration activity ≈6-fold faster than that of CA II with kcat/Km = 7 × 107 M−1·s−1 (T. T. Baird and C. A. Fierke, personal communication). To date, CA IV and CA II are the only two carbonic anhydrase isozymes known to have evolved close to catalytic “perfection” (31), which is remarkable in view of their relatively early evolutionary divergence (3). Hence, the three-dimensional structure of CA IV is critical not only for understanding the biological function of this novel membrane-anchored zinc enzyme but also for understanding the structural basis of diffusion-controlled catalysis maintained over the course of pronounced evolutionary drift in comparison with CA II. Additionally, the structure of CA IV might provide insight on design strategies for isozyme-specific inhibitors.
Figure 1.
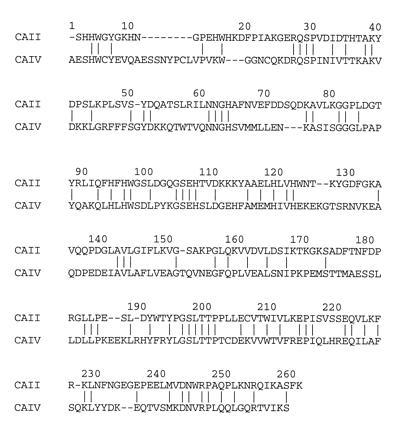
Sequence alignment of CA IV and CA II based on superposition of their three-dimensional structures. Relative to CA II, CA IV contains 8 insertions and 3 deletions; the overall sequence identity is 33%. The CA IV numbering scheme used in this work is based on this alignment; inserted residues are indicated by the number of the residue preceding the insertion and the suffix A, B, C, etc.
MATERIALS AND METHODS
The 30-kDa secretory form of human CA IV was expressed in Chinese hamster ovary cells and purified as described (17) prior to crystallization by the hanging-drop vapor diffusion method. Typically, 5 μl of protein solution (5–8 mg/ml CA IV solubilized in 50 mM Tris sulfate, pH 8.0) was equilibrated against 5 μl of 25% polyethylene glycol (Mr 3350)/100 mM sodium acetate (pH 5.1)/2 mM benzamidine/5 mM n-octyl β-d-glucopyranoside at 4°C. Parallelepipedons grew within a few days, the largest of which had typical dimensions of 0.5 mm × 0.4 mm × 0.2 mm. Diffraction data were collected from these crystals on an R-Axis IIc image plate detector using Cu-Kα radiation (λ = 1.5418 Å) generated by a Rigaku RU-200HB rotating anode operating at 50 mV and 100 mA. Crystals belonged to space group C2 with unit cell parameters a = 89.5 Å, b = 47.7 Å, c = 141.0 Å, and β = 106.3° (data reduction statistics are recorded in Table 1). Self-rotation function calculations were consistent with two molecules per asymmetric unit, yielding a packing density VM = 2.4 Å3/Da (32).
Table 1.
Data collection and refinement statistics
| Parameter | Value |
|---|---|
| Number of crystals | 2 |
| Number of measured reflections | 46,250 |
| Number of unique reflections | 13,626 |
| Maximum resolution, Å | 2.8 |
| Minimum resolution, Å | 20.0 |
| Rmerge* | 0.080 |
| Completeness of data, % | 95 |
| Number of reflections used in refinement (>2σ) | 12,067 |
| Number of reflections in Rfree test set | 650 |
| Rcryst† | 0.197 |
| Rfree‡ | 0.252 |
| Number of nonhydrogen protein atoms§ | 4,089 |
| Number of solvent molecules included in refinement | 34 |
| rms deviation from ideal bond lengths, Å | 0.007 |
| rms deviation from ideal bond angles, ° | 1.4 |
| rms deviation from ideal dihedral angles, ° | 24.8 |
| rms deviation from ideal improper angles, ° | 1.2 |
Rmerge for replicate reflections, R = Σ |Ih − 〈Ih〉|/Σ〈Ih〉; Ih = intensity measured for reflection h; 〈Ih〉 = average intensity for reflection h calculated from replicate data.
Crystallographic R factor, Rcryst = Σ| |Fo| − |Fc| |/Σ|Fo|; |Fo| and |Fc| are the observed and calculated structure factors, respectively, for those reflections not included in the Rfree test set.
Free R factor, Rfree = Σ| |Fo| − |Fc| |/Σ|Fo| for 650 reflections included in the test set which were excluded from refinement.
In asymmetric unit (two protein molecules; in molecule B the Lys-124–Glu-138 segment is disordered and is not included in the final model).
The initial electron density map of CA IV was obtained by molecular replacement techniques using AMoRe (33) as modified for the CCP4 suite of programs (34). Structure factor amplitudes with |Fo| > 3σ in the 15.0- to 3.5-Å range were used in rotation function calculations. The Patterson integration radius used for the search was 23 Å and the angular step size was 2.5° in β. The initial search probe was a polyalanine model derived from the refined 1.54-Å resolution structure of CA II (9) less 30 residues at the N terminus. This model was placed in an artificial unit cell with dimensions a = b = c = 100 Å and α = β = γ = 90°. The cross-rotation search yielded a 7σ peak with α = 291.4°, β = 70.8°, and γ = 283.1°; a 4σ peak at α = 15.4°, β = 76.2°, and γ = 77.8° was the second-highest solution. These two rotation solutions yielded the best translation function solutions with peak heights of 5.7σ and 4.5σ, respectively. These two solutions corresponded to a rotation through a noncrystallographic dyad.
Rigid body refinement using 15.0- to 3.5-Å data yielded a crystallographic R factor of 0.49. Strict noncrystallographic symmetry (NCS) constraints were employed at this stage. Following one round of positional refinement, electron density maps were generated with Fourier coefficients 2|Fo| − |Fc| and |Fo| − |Fc| and phases calculated from the polyalanine model. Maps were NCS-averaged using rave (35). Approximately 80% of the main and side-chain atoms of CA IV were fit into these initial maps. Iterative rounds of simulated annealing refinement (36) and model building improved the quality of the maps and the protein model. The NCS constraints were gradually released into heavily weighted NCS restraints as judged by the Rfree criterion (37, 38). The current model contains all but 4 residues at the N terminus. The crystallographic R factor for the final model is 0.197 (Rfree = 0.252) with excellent stereochemistry (Table 1). A total of 87% and 13% of backbone φ/ϕ conformations are within the most favored and additionally allowed regions of the Ramachandran plot, respectively, as assessed with procheck (39).
RESULTS AND DISCUSSION
The overall fold of CA IV is similar to that of CA II and is dominated by a central, eight-stranded β-sheet superstructure.§ Although the two molecules of CA IV in the asymmetric unit (designated A and B) were refined with heavily weighted NCS restraints and therefore exhibit generally similar structural features (rms deviation = 0.02 Å), an important difference is noted in the Lys-124–Glu-138 region. In molecule A, this segment is a fully ordered, extended loop; in molecule B, this segment is not visible and is therefore not included in the final model. Interestingly, a large fraction of CA IV identified in urinary membranes has undergone proteolytic nicking (23), and the location of proteolytic susceptibility in the secretory form of CA IV was identified as the Arg-129–Asn-130 linkage (17). Since nicked CA IV is detected in redissolved crystals by gel electrophoresis (data not shown), it is possible that substantial cleavage of the Arg-129–Asn-130 linkage in molecule B may result in the disordering of the Lys-124–Glu-138 region, as well as other neighboring loop regions, as reflected by weak electron density and/or elevated thermal B factors.
Active Site Structure.
Many features in the immediate active site are conserved between CA IV and CA II. This is particularly noteworthy, since these two isozymes are currently the only two known to have evolved close to catalytic perfection (31) despite pronounced sequence divergence (3). The two isozymes share a conserved metal binding site: protein ligands to zinc consist of the Nɛ atoms of His-94 and His-96 and the Nδ atom of His-119. The electron density map of CA IV reveals a substantial peak corresponding to a fourth, nonprotein ligand (Fig. 2). When this electron-rich peak is modeled as a water molecule, its thermal B factor decreases to the minimum value of 2 Å2 set in refinement. Therefore, interpretation as a single water molecule is unsatisfactory. However, when this peak is modeled as sulfate, refinement proceeds satisfactorily. Sulfate was present during crystallization at pH 5.1 and is known to inhibit CA IV at low pH values (T. T. Baird and C. A. Fierke, personal communication). Moreover, the refined position of sulfate corresponds to that observed in the complex of sulfate with the Gln-92 → Leu variant of CA II (40). At higher pH in the absence of sulfate the nonprotein ligand is a hydroxide ion, which is the nucleophile in the CO2 hydration reaction catalyzed by CA II (9) and CA IV (T. T. Baird & C. A. Fierke, personal communication).
Figure 2.
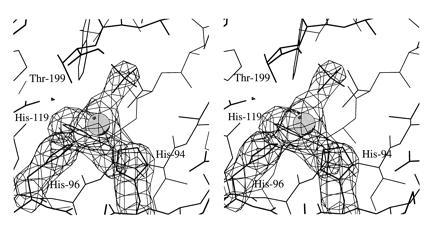
Omit map of the zinc binding site of CA IV contoured at 4σ; refined atomic coordinates are superimposed and selected active site residues are labeled. Zinc is tetrahedrally coordinated by His-94, His-96, His-119, and a sulfate oxygen. The hydrophobic substrate binding pocket is visible in the upper right background.
The substrate binding pocket is similarly conserved between CA IV and CA II, as is the catalytic proton shuttle (41, 42), His-64 (data not shown). This shuttle residue resides in the “out” conformation (i.e., directed away from the active site) in the CA IV structure determined at pH 5.1, which is similar to that observed in CA II at low pH (43). It is possible that at higher pH values, His-64 adopts the “in” conformation (i.e., directed toward the active site). The conformational mobility of this residue accompanies its role as a proton shuttle in catalysis in CA II (44), and the same may be true for His-64 of CA IV.
Despite structural similarities in their immediate active site clefts, significant structural differences are found in the greater active site clefts of CA IV and CA II. In particular, CA IV contains two disulfide linkages, which contribute to its exceptional stability to solubilization in 5% SDS (12, 17). As correctly predicted in solution studies (17), the disulfides are found in the N-terminal region linking Cys-6–Cys-11G and Cys-23–Cys-203. The latter disulfide stabilizes an important polypeptide loop in the active site (Figs. 3 and 4). This loop contains a catalytic residue, Thr-199, which hydrogen bonds to and orients zinc-bound hydroxide for catalysis (45, 46, 47). This loop also contains a cis-peptide linkage between Pro-201 and Thr-202. Importantly, this is not the usual type of cis-peptide linkage involving proline residues: cis-Xaa-Pro linkages are much more commonly observed in proteins due to a 0.5-kcal/mol destabilization relative to the trans conformation (48, 49). A cis-Pro-Xaa linkage is substantially more destabilizing due to steric clashes between adjacent side chains.
Figure 3.
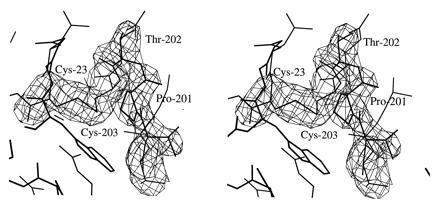
Omit map showing the Cys-23–Cys-203 disulfide linkage and the Pro-201–Thr-202 cis-peptide linkage (contoured at 4σ). Refined atomic coordinates are superimposed and selected active site residues are labeled.
Figure 4.
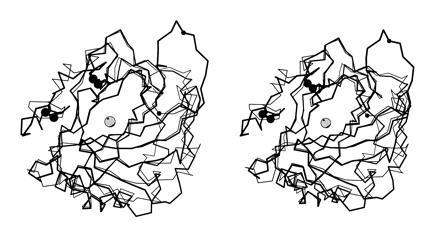
Least-squares Cα superposition of CA IV (thick bonds) and CA II (thin bonds). The active site zinc ion appears as a gray sphere, and disulfide linkages of CA IV are indicated by large black spheres. Note that although the overall folds of the two isozymes are generally similar, there are significant differences in the region of residue 131 (small black sphere). The extended conformation of the Arg-129–Asn-130 linkage in CA IV may enhance its susceptibility to proteolysis (17, 23).
To illustrate the energetic destabilization of a cis-Pro-Xaa linkage, consider that CA II has a cis-peptide linkage between Pro-201 and Pro-202; this linkage remains cis in the Pro-202 → Ala variant at a net cost of ≈5 kcal/mol destabilization (50). Although the Pro-201–Thr-202 cis-peptide linkage of CA IV must be similarly destabilizing, the adjacent disulfide linkage between Cys-23 and Cys-203 offsets this destabilization and buttresses the high-energy Pro-201–Thr-202 conformation. This yields an overall loop conformation similar to that of CA II (Fig. 4). Since nearby residue Thr-199 is important for catalysis, it is interesting that unfavorable local conformations are sometimes tolerated in protein functional sites to accommodate the precise geometric requirements of binding and catalysis (51).
On the opposite side of the active site cleft from Pro-201, the polypeptide chain in the vicinity of residue 131 (valine in CA IV, phenylalanine in CA II) adopts a remarkably different conformation in the two isozymes (Fig. 4). In CA II, Phe-131 is found within a short α-helix; in CA IV, Val-131 is found within an extended loop in molecule A, and in molecule B this polypeptide segment is disordered. In molecule A, this loop makes a β-sheet-like interaction with the Glu-171–Ser-173 segment of molecule A in an adjacent asymmetric unit (this segment is located in the eighth strand of the core β-sheet superstructure). Since the pH of CA IV crystallization is 5.1, and since crystals do not form at higher pH values, it is conceivable that the conformation of the Val-131 extended loop reflects partial acid-denaturation of what might otherwise be an α-helix at higher pH values. If a helix → loop transition is facilitated at low pH, and if this transition facilitates proteolysis of the Arg-129–Asn-130 linkage, then this could reflect some type of regulatory mechanism in the acidic environment of the urinary tract (23).
Since certain CA II inhibitors target interactions with Phe-131 (52, 53, 54), structural differences in the residue 131 region may account for the 15-fold lower affinity of many CA II inhibitors toward CA IV (25). However, we must confirm this speculation with x-ray crystallographic studies of relevant enzyme–inhibitor complexes. Structural differences in the residue 131 region of the carbonic anhydrase active site may likewise be exploited in the design of inhibitors that bind more tightly to CA IV than to CA II. Furthermore, it is interesting to compare the residue 131 region among different isozymes. For example, although Tyr-131 is located in a short α-helix in murine mitochondrial carbonic anhydrase V, this helix undergoes an ≈2-Å segmental shift compared with the corresponding helix of CA II (11). In human carbonic anhydrase I (7), it does not. Structural differences in the residue 131 region among isozymes II, IV, and V may provide leads for the design of isozyme-specific inhibitors capable of binding with high affinity and isozyme selectivity.
Association with the Membrane.
As mentioned in the introduction, CA IV is the only known membrane-anchored carbonic anhydrase isozyme. It is found on the luminal surface of epithelial cells in specific segments of the renal tubule, colon, gallbladder, and epididymis, and on the luminal surfaces of capillary endothelial cells in pulmonary microvasculature, choriocapillaris, and other microcapillaries such as those of heart and muscle. Zhu and Sly (14) demonstrated that the principal mechanism for anchoring to membranes is through a GPI tail linked to the C terminus of the protein. The C terminus of CA IV is on the opposite side of the globular protein from the conical active site cleft, and the mode and orientation of CA IV–membrane association is optimal for the biological function of the enzyme. Its active site is oriented toward the lumen in these structures to be fully accessible for catalysis.
Although the recombinant secretory form of CA IV crystallized in the current study lacks the membrane-anchoring GPI tail, the attachment of this tail to the C terminus should not perturb the protein structure. A significant electropositive surface potential surrounds the C terminus that is generated by the positively charged side chains of Lys-37, Lys-39, Lys-42, Lys-43, Arg-46, Lys-187, Lys-188, Arg-189A, His-190, Arg-213, and Lys-258 (Fig. 5). None of these residues is compensated by hydrogen bonds to negatively charged groups on the protein surface. Only Lys-39 and Arg-213 are conserved in other carbonic anhydrase isozymes. Apart from Lys-258 and Arg-213, these positively charged residues are contributed from two distinct loop segments on the protein surface: Lys-37–Arg-46 and Lys-187–His-190, the latter representing a region of insertion in CA IV relative to all other human isozymes (2). We propose that these residues facilitate the interaction of CA IV with the negatively charged phosphate groups of the phospholipid membrane. Given the lateral mobility of the protein conferred by the GPI anchor (15, 16), a complementary electrostatic interaction with the protein surface would help stabilize the orientation of the protein on the membrane surface. A model of CA IV associated with the membrane is presented in Fig. 6.
Figure 5.
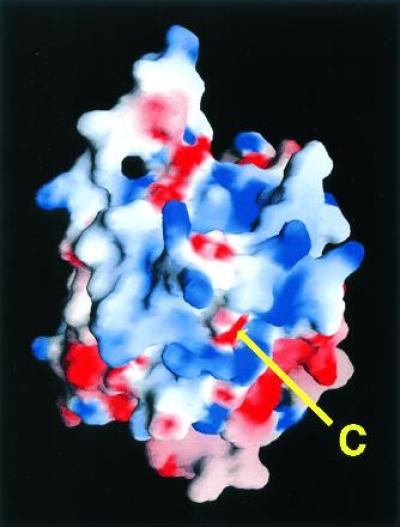
Electrostatic surface potential of CA IV calculated with grasp (55); the color scale ranges from −8 kT (red) to +8 kT (blue). Note the extensive positive electrostatic surface potential surrounding the C terminus. This feature reflects the adaptation of the carbonic anhydrase framework for GPI-anchoring and association with the membrane.
Figure 6.
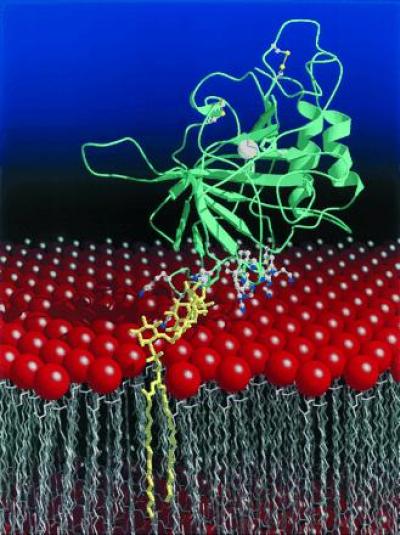
Cartoon of the CA IV–membrane interaction. The CA IV isozyme is anchored to the membrane by a GPI tail attached to its C terminus (yellow), which orients the enzyme active site toward the lumen for catalysis. This orientation is further stabilized by the interactions of 11 arginine, lysine, and histidine residues flanking the C terminus with the negatively charged phospholipid headgroups (red) of the membrane. The active site zinc ion appears as a white sphere, and the two disulfide linkages are indicated by bonded yellow spheres. The figure was prepared with molscript (56) and raster3D (57, 58).
Concluding Remarks.
The CA IV isozyme is unique as a membrane-anchored enzyme which has evolved to near-perfection despite pronounced evolutionary divergence from other human isozymes (3): the only other diffusion-controlled isozyme known to date, CA II, is related to CA IV by 33% sequence identity based on comparison of their three-dimensional structures. Conserved features in the active sites of these two isozymes include the hydrophobic substrate binding pocket, the zinc binding site, and a catalytic proton shuttle, His-64. However, CA IV has diverged from CA II in that the former isozyme is stabilized by two disulfide linkages in the N-terminal region. Additional structural differences are found in the residue 131 region of the two active sites. Finally, the CA IV structure is eminently adapted for optimal function in the membrane by the attachment of a membrane-anchoring group to the opposite side of the protein from the active site. This orients the active site of the enzyme away from the membrane so that it is fully exposed for efficient catalysis of CO2 hydration or HCO3− dehydration in the lumen.
Acknowledgments
We thank Prof. C.A. Fierke for helpful discussions, and we are grateful to C. A. Lesburg and D. Tobias for assistance preparing the figures. We thank the National Institutes of Health for Grants GM45614, DK40163, and GM34182 in support of this work. T.S. is the recipient of a Lynch Fellowship.
Footnotes
The publication costs of this article were defrayed in part by page charge payment. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. §1734 solely to indicate this fact.
Abbreviations: CA II, human carbonic anhydrase II; CA IV, human carbonic anhydrase IV; GPI, glycosylphosphatidylinositol; NCS, noncrystallographic symmetry.
Data deposition: The atomic coordinates have been deposited in the Protein Data Bank, Chemistry Department, Brookhaven National Laboratory, Upton, NY 11973 (reference 1ZNC). This information is embargoed for 1 year from the date of publication.
The residue numbering scheme for the CA IV crystal structure is based on its alignment with the CA II structure; inserted residues in CA IV relative to CA II are designated by the sequence number of the point of insertion and the suffix A, B, C, etc.
References
- 1.Tashian R E. Adv Genet. 1992;30:321–356. doi: 10.1016/s0065-2660(08)60323-5. [DOI] [PubMed] [Google Scholar]
- 2.Sly W S, Hu P Y. Annu Rev Biochem. 1995;64:375–401. doi: 10.1146/annurev.bi.64.070195.002111. [DOI] [PubMed] [Google Scholar]
- 3.Hewett-Emmett D, Tashian R E. Mol Phylogenet Evol. 1996;5:50–77. doi: 10.1006/mpev.1996.0006. [DOI] [PubMed] [Google Scholar]
- 4.Coleman J E. In: Zinc Enzymes. Bertini I, Luchinat C, Maret W, Zeppezauer M, editors. Boston: Birkhauser; 1986. pp. 49–58. [Google Scholar]
- 5.Silverman D N, Lindskog S. Acc Chem Res. 1988;21:30–36. [Google Scholar]
- 6.Christianson D W, Fierke C A. Acc Chem Res. 1996;29:331–339. [Google Scholar]
- 7.Kannan K K, Ramanadham M, Jones T A. Ann NY Acad Sci. 1984;429:49–60. doi: 10.1111/j.1749-6632.1984.tb12314.x. [DOI] [PubMed] [Google Scholar]
- 8.Liljas A, Kannan K K, Bergsten P-C, Waara I, Fridborg K, Strandberg B, Carlbom U, Jarup L, Lovgren S, Petef M. Nat New Biol. 1972;235:131–137. doi: 10.1038/newbio235131a0. [DOI] [PubMed] [Google Scholar]
- 9.Håkansson K, Carlsson M, Svensson L A, Liljas A. J Mol Biol. 1992;227:1192–1204. doi: 10.1016/0022-2836(92)90531-n. [DOI] [PubMed] [Google Scholar]
- 10.Eriksson A E, Liljas A. Proteins Struct Funct Genet. 1993;16:29–42. doi: 10.1002/prot.340160104. [DOI] [PubMed] [Google Scholar]
- 11.Boriack-Sjodin P A, Heck R W, Laipis P J, Silverman D N, Christianson D W. Proc Natl Acad Sci USA. 1995;92:10949–10953. doi: 10.1073/pnas.92.24.10949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Whitney P L, Briggle T V. J Biol Chem. 1982;257:12056–12059. [PubMed] [Google Scholar]
- 13.Wistrand P J, Knuuttila K-G. Kidney Int. 1989;35:851–859. doi: 10.1038/ki.1989.63. [DOI] [PubMed] [Google Scholar]
- 14.Zhu X L, Sly W S. J Biol Chem. 1990;265:8795–8801. [PubMed] [Google Scholar]
- 15.Lisanti M P, Rodriguez-Boulan E, Saltiel A R. J Membr Biol. 1990;117:1–10. doi: 10.1007/BF01871561. [DOI] [PubMed] [Google Scholar]
- 16.Englund P T. Annu Rev Biochem. 1993;62:121–138. doi: 10.1146/annurev.bi.62.070193.001005. [DOI] [PubMed] [Google Scholar]
- 17.Waheed A, Okuyama T, Heyduk T, Sly W S. Arch Biochem Biophys. 1996;333:432–438. doi: 10.1006/abbi.1996.0412. [DOI] [PubMed] [Google Scholar]
- 18.Okuyama T, Sato S, Zhu X L, Waheed A, Sly W S. Proc Natl Acad Sci USA. 1992;89:1315–1319. doi: 10.1073/pnas.89.4.1315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Okuyama T, Waheed A, Kusumoto W, Zhu X L, Sly W S. Arch Biochem Biophys. 1995;320:315–322. doi: 10.1016/0003-9861(95)90015-2. [DOI] [PubMed] [Google Scholar]
- 20.Sly W S, Hewett-Emmett D, Whyte M P, Yu Y-S L, Tashian R E. Proc Natl Acad Sci USA. 1983;80:2752–2756. doi: 10.1073/pnas.80.9.2752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sly W S, Whyte M P, Sundaram V, Tashian R E, Hewett-Emmett D, Guibaud P, Vainsel M, Baluarte H J, Gruskin A, Al-Mosawi M, Sakati N, Olsson A. N Engl J Med. 1985;313:139–145. doi: 10.1056/NEJM198507183130302. [DOI] [PubMed] [Google Scholar]
- 22.Sly W S, Whyte M P, Krupin T, Sundaram V. Pediatr Res. 1985;19:1033–1036. doi: 10.1203/00006450-198510000-00017. [DOI] [PubMed] [Google Scholar]
- 23.Sato S, Zhu X L, Sly W S. Proc Natl Acad Sci USA. 1990;87:6073–6076. doi: 10.1073/pnas.87.16.6073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Brown D, Zhu X L, Sly W S. Proc Natl Acad Sci USA. 1990;87:7457–7461. doi: 10.1073/pnas.87.19.7457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Maren T H, Wynns G C, Wistrand P J. Mol Pharmacol. 1993;44:901–905. [PubMed] [Google Scholar]
- 26.Lucci M S, Tinker J P, Weiner I M, DuBose T D. Am J Physiol. 1983;245:F443–F449. doi: 10.1152/ajprenal.1983.245.4.F443. [DOI] [PubMed] [Google Scholar]
- 27.Brechue W F, Kinne-Saffran E, Kinne R K H, Maren T H. Biochim Biophys Acta. 1991;1066:201–207. doi: 10.1016/0005-2736(91)90187-d. [DOI] [PubMed] [Google Scholar]
- 28.Effros R M, Mason G, Silverman P. J Appl Physiol. 1981;51:190–193. doi: 10.1152/jappl.1981.51.1.190. [DOI] [PubMed] [Google Scholar]
- 29.Fleming R E, Crouch E C, Ruzicka C A, Sly W S. Am J Physiol. 1993;265:L627–L635. doi: 10.1152/ajplung.1993.265.6.L627. [DOI] [PubMed] [Google Scholar]
- 30.Hageman G S, Zhu X L, Waheed A, Sly W S. Proc Natl Acad Sci USA. 1991;88:2716–2720. doi: 10.1073/pnas.88.7.2716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Albery W J, Knowles J R. Biochemistry. 1976;15:5631–5640. doi: 10.1021/bi00670a032. [DOI] [PubMed] [Google Scholar]
- 32.Matthews B W. J Mol Biol. 1968;33:491–497. doi: 10.1016/0022-2836(68)90205-2. [DOI] [PubMed] [Google Scholar]
- 33.Navaza J. Acta Crystallogr A. 1994;50:157–163. [Google Scholar]
- 34.Collaborative Computational Project, Number 4. Acta Crystallogr D. 1994;50:760–763. [Google Scholar]
- 35.Kleywegt G J, Jones T A. In: From First Map to Final Model. Bailey S, Hubbard R, Waller D A, editors. Daresbury Laboratory: Warrington; 1994. pp. 59–66. [Google Scholar]
- 36.Brünger A T, Kuriyan J, Karplus M. Science. 1987;235:458–460. doi: 10.1126/science.235.4787.458. [DOI] [PubMed] [Google Scholar]
- 37.Brünger A T. Nature (London) 1992;355:472–475. doi: 10.1038/355472a0. [DOI] [PubMed] [Google Scholar]
- 38.Brünger A T. Acta Crystallogr D. 1993;49:24–36. doi: 10.1107/S0907444992007352. [DOI] [PubMed] [Google Scholar]
- 39.Laskowski R A, MacArthur M W, Moss D S, Thornton J M. J Appl Crystallogr. 1993;26:283–291. [Google Scholar]
- 40.Lesburg C A, Christianson D W. J Am Chem Soc. 1995;117:6838–6844. [Google Scholar]
- 41.Steiner H, Jonsson B-H, Lindskog S. Eur J Biochem. 1975;59:253–259. doi: 10.1111/j.1432-1033.1975.tb02449.x. [DOI] [PubMed] [Google Scholar]
- 42.Tu C K, Silverman D N, Forsman C, Jonsson B-H, Lindskog S. Biochemistry. 1989;28:7913–7918. doi: 10.1021/bi00445a054. [DOI] [PubMed] [Google Scholar]
- 43.Nair S K, Christianson D W. J Am Chem Soc. 1991;113:9455–9458. [Google Scholar]
- 44.Krebs J F, Fierke C A, Alexander R S, Christianson D W. Biochemistry. 1991;30:9153–9160. doi: 10.1021/bi00102a005. [DOI] [PubMed] [Google Scholar]
- 45.Merz K M. J Mol Biol. 1990;214:799–802. doi: 10.1016/0022-2836(90)90333-H. [DOI] [PubMed] [Google Scholar]
- 46.Krebs J F, Ippolito J A, Christianson D W, Fierke C A. J Biol Chem. 1993;268:27458–27466. [PubMed] [Google Scholar]
- 47.Liang Z, Xue Y, Behravan G, Jonsson B-H, Lindskog S. Eur J Biochem. 1993;211:821–827. doi: 10.1111/j.1432-1033.1993.tb17614.x. [DOI] [PubMed] [Google Scholar]
- 48.Stewart D E, Sarkar A, Wampler J E. J Mol Biol. 1990;214:253–260. doi: 10.1016/0022-2836(90)90159-J. [DOI] [PubMed] [Google Scholar]
- 49.MacArthur M W, Thornton J M. J Mol Biol. 1991;218:397–412. doi: 10.1016/0022-2836(91)90721-h. [DOI] [PubMed] [Google Scholar]
- 50.Tweedy N B, Nair S K, Paterno S A, Fierke C A, Christianson D W. Biochemistry. 1993;32:10944–10949. doi: 10.1021/bi00092a003. [DOI] [PubMed] [Google Scholar]
- 51.Herzberg O, Moult J. Proteins Struct Funct Genet. 1991;11:223–229. doi: 10.1002/prot.340110307. [DOI] [PubMed] [Google Scholar]
- 52.Baldwin J J, Ponticello G S, Anderson P S, Christy M E, Murcko M A, Randall W C, Schwam H, Sugrue M F, Springer J P, Gautheron P, Grove J, Mallorga P, Viader M P, McKeever B M, Navia M A. J Med Chem. 1989;32:2510–2513. doi: 10.1021/jm00132a003. [DOI] [PubMed] [Google Scholar]
- 53.Smith G M, Alexander R S, Christianson D W, McKeever B M, Ponticello G S, Springer J P, Randall W C, Baldwin J J, Habecker C N. Protein Sci. 1994;3:118–125. doi: 10.1002/pro.5560030115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Jain A, Whitesides G M, Alexander R S, Christianson D W. J Med Chem. 1994;37:2100–2105. doi: 10.1021/jm00039a023. [DOI] [PubMed] [Google Scholar]
- 55.Nicholls, A. (1993) grasp, Graphical Representation and Analysis of Surface Properties (Columbia Univ. Press, New York) Version 1.2.
- 56.Kraulis P J. J Appl Crystallogr. 1991;24:946–950. [Google Scholar]
- 57.Bacon D, Anderson W P. J Mol Graphics. 1988;6:219–220. [Google Scholar]
- 58.Merritt E A, Murphy M E P. Acta Crystallogr D. 1994;50:869–873. doi: 10.1107/S0907444994006396. [DOI] [PubMed] [Google Scholar]